REVIEW: Bioactive Amides of Fatty Acids
V. V. Bezuglov*, M. Yu. Bobrov, and A. V. Archakov
Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, Moscow, 117871 Russia; fax: (095) 335-7103; E-mail: vvbez@oxylipin.siobc.ras.ru* To whom correspondence should be addressed.
Received July 28, 1997
Amides of fatty acids are lipid bioregulators formed from long chain saturated and unsaturated fatty acids via amidation by the corresponding amines. Ethanolamides of fatty acids are the most well-studied species of this group; an alternative pathway for their biosynthesis includes hydrolysis of N-acylated phosphatidylethanolamines by phospholipase D. Ethanolamides of fatty acids bind to the cannabinoid receptors of the central nervous system (CB1) or peripheral tissues (CB2) and can be considered as endogenous ligands of these receptors. Their pharmacological properties are similar to that of cannabimimetics. Simple amides of fatty acids are also endogenous bioregulators acting like sleep-inducing (oleamide) or angiogenic factors (erucamide). A new group of bioregulators comprise the amides of fatty acids and biologically active amines (vanillinamine, dopamine, and serotonin).
KEY WORDS: anandamide, oleamide, erucamide, ethanolamides of fatty acids, amidohydrolase, cannabinoid receptor, N-acylphosphatidylethanolamine, serotonin, dopamine
Abbreviations: FA) fatty acids; LO) lipoxygenase; ANA) anandamide (arachidonylethanolamide); APE) N-acylphosphatidylethanolamine; CB) cannabinoid receptor; PEA) palmitoylethanolamine; PLA2, PLC, PLD) phospholipases A2, C, and D, respectively; PMSF) phenylmethylsulfonyl fluoride; THC) Delta9-tetrahydrocannabinol.
Amide derivatives of fatty acids (FA) are widespread in nature. They are
incorporated into ceramides [1], glycosphingolipids
[2], N-acylated lipids [3], and
bacterial lipoproteins [4]. N-Acylation by a
saturated FA is an important posttranslational modification of proteins
[5].
Amides of FA and primary amines (ethanolamine) attracted attention as bioregulators for the first time in 1957 when is was demonstrated that N-palmitoylethanolamine is an anti-inflammatory factor contained in the lipid fraction of soybean, peanut oil, and eggs yolk [6]. A second birth of interest in FA amides with primary amines is associated with the discovery of potent neuromodulatory effects of natural N-arachidonylethanolamide (anandamide) [7] and oleoylamine (oleamide) [8]. Bioeffector characteristics of these and similar derivatives and their biosynthesis and metabolism are reviewed in the present manuscript.
ETHANOLAMIDES OF FATTY ACIDS
Cannabinoid Receptors
The study of pharmacological properties of cannabis resulted in isolation and characterization of a group of compounds designated cannabinoids. Cannabinoids are ligands of the specific brain membrane receptor (central cannabinoid receptor or CB1) coupled to the Gi-protein [9]. The receptor includes seven transmembrane domains similar to the transmembrane domains of rhodopsin [10] and upon solubilization, it is still able to bind the substrate and interact with G-protein [11]. Human [12] and rat cannabinoid receptors [13] are 97% homologous (100% in transmembrane domains). Rat CB1 is detected by several days post partum [14] and is abundant in the brain, especially in hypothalamus, cortex, basal nuclei, cerebellum, and olfactory structures [15, 16]. It is important that the mRNA of CB1 was detected in the spleen, tonsils, and peripheral blood leukocytes by the polymerase chain reaction method [17].
Peripheral cannabinoid receptor (CB2) that was also discovered recently [18] has 44% homology with human brain CB1 protein. Similar to CB1, CB2 is coupled to adenylate cyclase via the Gi-protein [19, 20]. The receptor mRNA is the most abundant in the human spleen and tonsils. The level of mRNA is maximal in B cells and NK cells and is less abundant in monocytes and polymorphonuclear leukocytes; the lowest level was detected in CD8+ and CD4+ cells [21]. Cannabinoid-binding sites were detected in B cells of the spleen, lymph nodes, and Peyer patches [22] and in mast cells [23].
The exact physiological role of cannabinoid receptors is still unknown. Considering certain psychotropic effect of cannabinoids, CB1 can participate in the regulation of motility, memory, emotions, and pain sensitivity as well as in the regulation of vegetative functions of the body, whereas CB2 is apparently associated with modulation of immunocompetent cells.
The presence of cannabinoid receptors indicate that a natural mimetic agent exists in the organism, and its physiological effects are similar to that of exogenous cannabinoids. This agent was isolated from porcine brain [7] and identified as ethanolamide of arachidonic acid designated anandamide by the authors (from the Sanskrit "ananda" that means bliss) (Fig. 1). Anandamide competitively inhibited specific binding of radioactive cannabimimetics with synaptosomal membranes and induced dose-dependent inhibition of contraction of electrically stimulated mouse vas deferens; it also exhibited other pharmacological properties characteristic for psychotropic cannabinoids [24].
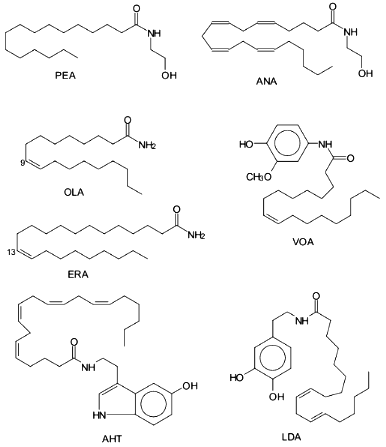
Later, two new ethanolamides of fatty acids were isolated from porcine brain including dihomo-gamma-linolenoylethanolamine and docosatetraenoylethanolamine, and they are also ligands of CB1 [25, 26]. It was demonstrated that two additional ethanolamides of polyunsaturated fatty acids (docosahexaenoic [27] and eicosatrienoic (20:3omega9) [28]) are endogenous cannabinoids.Fig. 1. Biologically active amides of fatty acids. ANA, anandamide; PEA, palmitoylethanolamine; OLA, oleamide; ERA, erucamide; AHT, arachidonyl-5-hydroxytryptamine; VOA, vanillinoleamide; LDA, linoleoyldopamine.
When cannabinoids interact with cells and tissues expressing CB, they inhibit stimulated (by forskolin or secretin) adenylate cyclase activity and opening of N-type calcium channels [29]; these processes are modulated by pertussis toxin [30] which blocks the dissociation of the alpha-subunit from the beta,gamma-subunits of the G-protein. In various cell models, it has been shown that ethanolamides of polyunsaturated FA also inhibit stimulated activity of adenylate cyclase [31, 32] and opening of N-type calcium channels [32, 33] and activate potassium outward current [34]. In CHO and AtT-20 cells expressing CB1 and CB2, stimulation of these receptors has similar effects on adenylate cyclase that is reversed by pertussis toxin. It is important that unlike CB1, stimulation of CB2 had no effect on ion channels [35]. Inhibition of adenylate cyclase was enhanced by addition of the specific inhibitor of serine amide hydrolases [36] (see below). These effects, similar to exogenous cannabinoids, were reversed by pertussis toxin [33]. CB-binding characteristics of various ethanolamides of FA depend on the length and structure of the fatty acid moiety (see the table). However, the data of different authors or in different test systems obtained with the very same compound can vary by several dozens of times. In most cases, the literature constants correspond to the displacement of radioactive cannabinoids from the CB preparation by the substances but not to the direct binding of anandamide and similar compounds to the receptor.
Binding of acylethanolamines (RCONHCH2CH2OH) to cannabinoid receptors
| RCO | Ki, nM | Test system | Agonist | Reference |
|---|---|---|---|---|
| CB1 | ||||
| 20:4omega6 | 52±1.8 | A | HU-243 | [7] |
| 20:4omega6 | 543±83 | B | CP-55,940 | [32] |
| 20:4omega6 | 37±3 | C | HU-243 | [31] |
| 20:4omega6 | >2000 | D | CP-55,940 | [37] |
| 20:4omega6 | 22±2 | D* | CP-55,940 | [37] |
| 20:4omega6 | 781±38 | B | CP-55,940 | [28] |
| 20:4omega6 | 5400±1600 | D | CP-55,940 | [38] |
| 20:4omega6 | 89±10 | D* | CP-55,940 | [38] |
| 20:4omega6 | 89 | A | CP-55,940 | [41] |
| 20:1omega9 | >10000 | D* | CP-55,940 | [38] |
| 20:1omega9 | >1000 | A | HU-243 | [39] |
| 20:2omega6 | 1500 | A | HU-243 | [39] |
| 20:3omega3 | >10000 | A | HU-243 | [39] |
| 20:3omega6 | 53.4±5.5 | A | HU-243 | [25] |
| 20:3omega6 | 598±264 | B | CP-55,940 | [32] |
| 20:3omega9 | 753±71 | B | CP-55,940 | [28] |
| 20:3omega9 | 680±48 | B | CP-55,940 | [35] |
| 20:5omega3 | 1470±500 | D* | CP-55,940 | [38] |
| 20:5omega3 | 162.3±13.6 | A | HU-243 | [39] |
| 22:4omega6 | 34.4±3.2 | A | HU-243 | [25] |
| 22:4omega6 | 848±102 | B | CP-55,940 | [32] |
| 22:6omega3 | 12200±500 | B | CP-55,940 | [32] |
| 18:2omega6 | >25000 | A | HU-243 | [39] |
| 18:3omega3 | inactive | A | HU-243 | [39] |
| 18:3omega6 | >41400±6000 | B | CP-55,940 | [32] |
| 18:3omega6 | 4600±300 | A | HU-243 | [39] |
| 18:4omega3 | >1000 | A | HU-243 | [39] |
| 16:0 | inactive | B | CP-55,940 | [32] |
| 16:0 | inactive | A | HU-243 | [39] |
| 16:0 | >100000 | A | CP-55,940 | [41] |
|
CB2 |
||||
| 20:4omega6 | 1600±400 | E | WIN-55,212-2 | [18] |
| 20:4omega6 | 1620±430 | F | CP-55,940 | [28] |
| 20:4omega6 | 1940±240 | F | CP-55,940 | [35] |
| 20:4omega6 | 85±13 | C | HU-243 | [20] |
| 20:4omega6 | 33±29 | G | WIN-55,212-2 | [23] |
| 20:3omega6 | 857±100 | F | CP-55,940 | [35] |
| 20:3omega9 | 1810±520 | F | CP-55,940 | [28] |
| 20:3omega9 | 1800±520 | F | CP-55,940 | [35] |
| 22:4omega6 | 9020±470 | F | CP-55,940 | [35] |
| 16:0 | 1.0±0.6 | G | WIN-55,212-2 | [23] |
Note: A, rat brain synaptosomal membranes; B, membranes of murine Ltk--(L) cells expressing human CB1; C, membranes of CHO-K1 cells expressing rat CB1 or human CB2; D, P2 membranes of rat forebrain; E, membranes of COS cells expressing human CB2; F, membranes of murine AtT-20 cells expressing human CB2; G, murine RBL-2H3 cells.
*Experiments performed in the presence of PMSF.
The CB2 peripheral cannabinoid receptor has good affinity for natural cannabinoids (for example, THC) and synthetic cannabimimetics (WIN-55,212-2 and CP-55,940) but low affinity to ANA [18] which displaced radiolabeled ligands from their complex with CB2 ~30-fold less efficiently than with CB1 (see the table). Synthetic cannabinoids significantly inhibited mouse anti-dinitrophenol monoclonal IgE antibody-stimulated secretion of [3H]serotonin by the rat basophilic leukemia RBL-2H3 cells which expressed CB2 [23]. ANA displaced [3H]WIN-55,212-2 from the membrane but did not block serotonin secretion. Addition of ANA (but not arachidonic acid and ethanolamine) together with WIN-55,212-2 and other active non-lipid cannabinoids weakened their effects. Hence, ANA is not an agonist but an antagonist of CB2. N-Palmitoylethanolamine (unlike ANA) exhibited high activity in [3H]WIN-55,212-2 displacement from the membrane and in inhibition of IgE-stimulated serotonin secretion. ANA weakened the effects of PEA and cannabinoids. The data suggested that PEA and possibly ethanolamides of other saturated fatty acids are endogenous ligands of CB2 [23]. However, other data indicate that PEA has low affinity for CB2 in COS-7 cells expressing the receptor [39]; this could be explained by the presence of several subtypes of CB2.
2-Arachidonylglycerol, a recently discovered endogenous cannabinoid, is another candidate for the natural ligand of CB2 [40, 41]; it also binds to CB1 [40] and exhibits specific cannabimimetic pharmacological characteristics [42]. In the near future, selective inhibitors of various types of CB receptors would enable definitive determination of the receptors that bind endogenous cannabinoids.
Physiological Effect of Ethanolamides of FA
Effects of ethanolamides of polyunsaturated FA. Physiological effects of ANA and similar compounds are similar to the effects of cannabinoids although less durable; this can be associated with their hydrolysis by amide hydrolases. Intraperitoneal administration of ANA in rats and mice results in catalepsy, analgesia, hypothermia, and loss of mobility [43, 44]. THC and ANA enhance the effects of muscimol (GABA receptor agonist) that induces catalepsy in rats [45]. More detailed investigation of physiological response of mice administered with ANA via various routes indicated that intravenous, intraperitoneal and intraspinal injections of ANA result in loss of sensitivity (by >80% maximally possible in intravenously and intraspinally injected animals), significant body temperature decrease (by 2-4°C), decreased motility, and catalepsy. Maximal effect on motility (decrease by ~85%) was detected immediately after intravenous and intraperitoneal administration, whereas the onset developed 10 min after intraspinal injection. The effects of ANA (1.3-18-fold) in all behavioral tests were weaker than that of THC. Intraspinal injection of ANA resulted in loss of sensitivity independently of nor-binaltorphimine, which blocks the similar effect of THC [46].
ANA and synthetic cannabimimetics inhibit ion channels coupled to the serotonin 5-HT3 receptor. The effect was not associated with changes in cAMP [47]. Also, ANA decreased the level of dopamine in rat striatum, lowered the activity of tyrosine hydroxylase, and changed the ratio of dopamine D1 and D2 receptors after intraperitoneal injection [48]. ANA also inhibited prolactin-luteinizing hormone and growth hormone secretion in gonadectomized rats [49].
Stimulation of neurons in various regions of the rat brain (with ionophores, depolarizing agents, and glutamate) induces the synthesis of N-acylethanolamines and accumulation of their precursors [50, 51]. Glutamate secretion by the neurons of rat hippocampus is inhibited by activation of the cannabinoid receptors coupled to a G-protein [52]. Thus, neuron functions can be regulated by N-acylethanolamines via ion permeability of the membrane, mediator secretion, and intracellular biochemical processes.
Considering the cardiovascular effects, it is important that ANA injection causes bradycardia and at first, induces rapid increase with subsequent prolonged lowering of blood pressure [53]. The hypertensive effect is evidently due to the direct action of ANA on the vessels and the hypotensive effect is mediated by ANA-induced inhibition of noradrenaline secretion by nerve endings in the heart and vessels because this was blocked by specific antagonist of CB1, SR141716A [54, 55]. ANA-dependent inhibition of noradrenaline secretion was also observed in rat atrium and vas deferens preparations [56].
ANA also affects reproductive function. ANA decreased the fertility of sea urchin spermatocytes by suppressing the acrosomal reaction which occurs during spermatozoa interaction with the ovarian cell [57]. Later on, it was shown that sea urchin eggs contain APE and a small amount of ethanolamides of palmitic, stearic, and arachidonic acids [58]. ANA and its precursors were detected in the rat testis and biosynthesis of these compounds was demonstrated in the same tissue [59].
Effects of ethanolamides of saturated fatty acids. The first detected effect of ethanolamides of saturated FA was the anti-inflammatory activity of PEA-containing lipid extracts of certain natural products [6, 60, 61]. At the end of the 1960s, it was shown that PEA increases nonspecific resistance to various bacterial toxins and traumatic shock [62, 63] and decreases the intensity of inflammatory and immune processes [64-66]. PEA enhanced the resistance of the organism to the toxic effects of anti-cancer drugs [67] and ethanol [68-70].
It has been shown that ethanolamides of saturated and monoenic FA influence the functioning of calcium channels by blocking calcium efflux from cells and organelles under hypoxia [71]. Such characteristics suggested that heart tissue can be protected from ischemic damage by ethanolamide derivatives of saturated FA; this was confirmed in guinea pigs. Oleoylethanolamine was the most potent protector of the heart muscle [72].
Biosynthesis
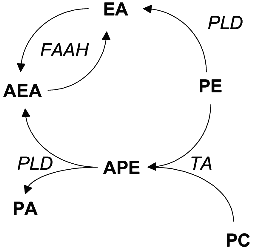
The first reports on the biosynthesis of ethanolamides of FA in mammalian tissues appeared long before their role as endogenous ligands of cannabinoid receptors was demonstrated. They are formed via two pathways: from free FA and ethanolamine and via hydrolysis of N-acylphosphatidylethanolamines (Fig. 2). By the beginning of the 1990s, most of the studies considered the amides of saturated and monoenic acids. Both synthetic pathways were confirmed for ethanolamides of most polyunsaturated FA only recently.
The study of phosphatidylethanolamine synthesis with [14C]ethanolamine revealed that rat liver microsomes can incorporate the label into a lipid identified as N-acylethanolamine [73]. The synthesis was detected for saturated and unsaturated FA with 12-24 carbon atom length. Amide derivatives were not formed if choline and various amino acids were used as amines; however, phenylamine, N-hydroxyphenylamine, and 1-amino-2-propanol were N-acylated [73]. Study of other biologically active amines as substrates of this enzyme system indicated that palmitic acid forms amides with histamine, tyramine, phenethylamine, and tryptamine but not with noradrenaline or serotonin [74]. The highest activity of N-acylethanolamine synthesis was detected in the homogenates of liver, brain, and kidney especially in the microsomes, although the synthesis was significant in the presence of mitochondria. Among saturated FA, the best substrates were myristic, palmitic, and stearic acids; and among unsaturated FA, these were oleic and linoleic acids [75-79] (Fig. 2).Fig. 2. Biosynthesis of acylethanolamines (AEA). EA, ethanolamine; PE, phosphatidylethanolamine; PC, phosphatidylcholine; PA, phosphatidic acid; APE, acylphosphatidylethanolamine; PLD, phospholipase D; FAAH, FA amide hydrolase; TA, trans-acylase.
The synthesis of ethanolamides of polyunsaturated FA is detected in homogenates of various mammalian tissues [80, 81]. Microsomes and cytosol of various rabbit tissues can synthesize ethanolamides of saturated and unsaturated FA [81]. ANA synthesis was the most active in brain microsomes and less active in liver, lung, and kidney microsomes. Among the cytosolic fractions, significant synthetic activity was detected only in the brain cytosol. Arachidonic acid was the best substrate (among all acids) for the membrane and cytosolic preparations from the brain. The selectivity of the enzyme system for this acid was over 50-fold versus palmitic acid. The synthesis is ATP- and CoA-independent was similar to the PEA synthesis by rat liver microsomes [75]. The data were confirmed by synthesis of ethanolamides of various FA in bovine brain membranes. The order of the kinetic characteristic of the substrates was as follows: arachidonic > dihomo-gamma-linolenic, 11,14-eicosadienoic > palmitic > adrenic, docosahexaenoic acids. Synthetic activity was the highest in hypothalamus, lower in thalamus, striatum, and cortex, and the lowest in the cerebellum and medulla [82]. The Michaelis constants for both substrates of the synthetase reaction were relatively high; their values in the enzyme systems from rabbit [81] and rat brain microsomes [59] were 130 and 135 mM for ethanolamine and 7 and 100 µM for arachidonic acid, respectively. Thus, ANA synthesis from arachidonic acid and ethanolamine requires significant local increase in these substances or in their release from precursors, for example by PLA2 and PLD.
An alternative pathway of FA ethanolamide biosynthesis by hydrolysis of the corresponding N-acylphosphatidylethanolamines was discovered in dog heart after myocardial infarction. The damaged myocardial regions accumulated N-acylethanolamines and their precursor APE; the fraction of radioactive ethanolamine in these lipids upon incubation with the heart tissue indicated that the former are synthesized from the latter [83-85] by a specific phospholipase. The enzyme catalyzing the APE hydrolysis was also detected in rat brain [86]. It is active over a wide pH range (from 4.5 to 8.5) and did not require Ca2+ or other divalent cations but was inhibited by Zn2+. Salts of bile acids and other ionic detergents inhibited the enzyme and Triton X-100 activated the reaction. The enzyme cleaved diacyl and plasmalogen forms of APE and lyso-APE but had no effect on phosphatidylcholine and phosphatidylethanolamine. Thus, the substrate specificity and inhibitor-activator profile indicate that this phospholipase is different from PLD previously detected in the brain and other mammalian tissues [87, 88]. Later on, the presence of enzyme reaction synthesizing ethanolamides of FA from exogenous APE was confirmed in rat [89] and canine brain [90].
In the studies performed in 1970s, the formation of ethanolamides of polyunsaturated FA was not detected and APE found in mammalian tissues primarily had saturated acids at the N-position, whereas ethanolamides of polyunsaturated FA longer than 18 atoms were not detected [84, 91, 92]. ANA synthesis from the corresponding APE was described for the first time in primary culture of rat brain neurons in 1994 [93]. The inhibitors of PLA2 (dimethyleicosadienoic and 3-(4-octadecylbenzoyl)acrylic acids) and PLC (neomycin) did not affect the ionomycin-stimulated ANA formation as well as the addition of arachidonic acid to intact neurons incubated with exogenous PLD, thus indicating that the free acid was not coupled to ethanolamine in the cells. On the other hand, addition of exogenous PLD (but not PLA2 and PLC) was essential for anandamide formation in the unstimulated cells and incubation of synthetic [3H]N-arachidonylphosphatidylethanolamine and other APE with the neuron homogenate also resulted in the synthesis of ANA and its analogs, thus confirming the PLD-dependent formation of ANA [93].
The mechanism of N-acylphosphatidylethanolamine synthesis involves enzymatic translocation of FA from sn-1-position of diacyl phospholipids; the amine group is not directly acylated by FA or acyl-CoA [3]. Trans-acylation is Ca2+-dependent and is catalyzed by a membrane enzyme of sarcoplasmic reticulum or mitochondria [3]. The donors of FA are phosphatidylcholine [94, 95], cardiolipin [96], and phosphatidylethanolamine; in the latter case, intermolecular and intramolecular acylation can occur [94]. This pathway results in the preferential formation of ethanolamides of saturated FA because unsaturated FA (arachidonic acid in particular) are rare in the sn-1-position although such phospholipids have been detected [41]. Thus, the ratio of synthesized N-acylethanolamines depends on the ratio of the corresponding FA esterified at the first position of phosphatidylcholine that is the trans-acylase substrate (Fig. 2).
Metabolism
In mammalian tissues, ethanolamides of FA decompose into fatty acid and ethanolamine. Amidases catalyzing this reaction were discovered in 1966 in rat liver microsomes [75]. Later it was demonstrated that the activity of amidase is the highest in the rat liver; the activity in the brain, testis, kidney, lung, and spleen was 5.5-10-fold lower and activity in the heart was over 60-fold lower than in the liver [86].
The rat liver enzyme was localized predominantly in microsomes but mitochondria also exhibited significant activity [79]. Hydrolysis occurred over a wide pH range (5-9) and activity increased at increasing pH. The enzyme was very specific towards the bases and FA included into N-acylethanolamines [79]. PEA-hydrolyzing amidase was detected also in canine brain microsomes [90].
The data on amidase activity in animal tissues considered only ethanolamides of saturated and monoenic FA. Among polyenic FA, ANA was the most studied amidase substrate. Its amidase activity was detected in brain cells and other rat tissues [80] and activity was the highest in the membrane fraction. The enzyme is not detected in the heart and muscle. Enzyme activity is maximal at pH 8-9 and at pH below 6 and above 10 it was significantly lower [97]. PMSF was a potent inhibitor of the enzyme (IC50 = 290 nM [98]). Its presence increases the activity of radioactively labeled cannabimimetic displacement by anandamide [99] and enhances the anandamide-dependent inhibition of electrically stimulated contraction of guinea pig ileum [100]. Trifluoromethyl ketones and ethyl alpha-keto esters of FA (especially arachidonyl trifluoromethyl ketone, IC50 = 1.9 µM [98], and ethyl-2-oxostearate) also efficiently inhibited ANA hydrolysis, whereas alpha-ketoethanolamides and ethanolamides were relatively weak inhibitors [101]. Recently, more active inhibitors of amidase were synthesized including lauryl sulfonyl fluoride (IC50 = 3 nM) [98] and methyl-arachidonyl-fluoro-phosphonate (IC50 = 2.5 nM) [102].
The amidase was specific to amides of FA, did not hydrolyze the peptide bond, and had no ceramidase activity [103]. Oleic acid amide (oleamide, sleep-inducing factor) was a substrate as well [104, 105]; currently, this enzyme is designated as a fatty acid amide hydrolase. Studies in various cells indicate that the substrate specificities of amidases are variable and depend on tissue type. For example, amidases of rat RBL-1 cells had higher activity towards palmitoylethanolamine (Km = 20 µM) versus anandamide (Km = 67 µM), whereas nerve cell amidases (N18TG2) had the opposite specificity (Km = 80 and 15 µM for PEA and ANA, respectively) [106]. It should be pointed out that the affinity of PEA is higher for peripheral (CB2) receptors and that of ANA is higher for CB1 brain receptors [23]. Thus, amidase activity is the universal inactivation mechanism of tissue-specific biologically active modulators, amidated derivatives of FA (Fig. 2).
Studies of hydrolase and synthetase activities of the enzymes from porcine brain indicate that both activities are attributable to the same protein [103]. Local increase in ethanolamine and arachidonic acid can provide the conditions required for the reversal of the amidase reaction and synthesis of ANA. However, the authors did not exclude a specific activation mechanism of the synthetase reaction of the isolated enzyme that lowers the Km values [103].
Similar to the corresponding FA, ethanolamides of polyunsaturated FA can form hydroxy derivatives under the action of lipoxygenases [107, 108]. Various types of lipoxygenases exhibit variable specificity towards ANA. For example, 12-LO of porcine leukocytes, 15-LO of rabbit reticulocytes, and soybean 15-LO (type I) catalyzed the formation of the corresponding 12- and 15-hydroxy derivatives. However, recombinant human platelet 12-LO had very weak activity and activity of porcine leukocyte 5-LO was undetectable [107]. The kinetic parameters of ethanolamides of arachidonic and eicosapentaenoic acids differed insignificantly in oxidation by soybean 15-LO (type I) versus the corresponding acids, thus indicating that they can be involved in enzymatic reactions of the arachidonic acid cascade [108].
Hydroxy derivatives of ANA had variable interaction with CB. The 15-hydroxy derivative of ANA was still able to inhibit the electrically stimulated contraction of murine vas deferens but 12-hydroxy derivative had low activity [107]. Thus, oxidized derivatives of ANA have their own spectrum of biological activities that is different from that of ANA. The presence of oxidized ethanolamides in tissues and their physiological role have yet to be determined.
PRIMARY AMIDES OF FATTY ACIDS
Simple amides of FA are also bioeffectors. For example, in chicken chorioallantoid membrane and rat cornea, it was shown that amide of 13-cys-docosenoic acid (erucamide) discovered in the bovine mesentery is an angiogenic factor [109]. Angiogenic activity was demonstrated by synthetic primary amides of 13-trans-docosenoic acid, 18:0, 20:0, 22:0, 20:4omega6 FA, and to the lesser extent of 16:0, 18:1omega9 FA [109] (Fig. 1). Interestingly, activities of the corresponding FA were sometimes identical or slightly lower that of their amide derivatives. Later, angiogenic activity of erucamide was demonstrated in regenerating skeletal muscle [110]. It was noted in both studies that erucamide had no proliferative activity in endothelium, muscle, and connective tissue.
Recently, the amide of cys-9,10-octadecenoic acid (oleamide) was isolated from the cerebrospinal fluid of sleep-deprived cats; it is the endogenous factor inducing sleep in mammals [8]. Structural and functional studies indicate that the trans-isomer of oleamide has similar but significantly weaker effect versus oleamide and change in the position of the double bond and increasing the length of the hydrocarbon chain cause dramatic lowering or complete loss of biological activity [8, 111]. Oleamide is synthesized by the brain cells from oleic acid and ammonium [112] and is degraded by FA amide hydrolase [113]. Apart from its effect on the central nervous system, oleamide modulates the function of the immune cells. For example, it inhibits lymphocyte proliferation [114] and exhibits synergism with ANA; the trans-isomer of oleamide and its saturated derivative were inactive. It is important that oleamide does not significantly bind to CB1 and CB2 [39], and its mechanism of action is yet to be discovered.
The PLA2 inhibitors as a mixture of simple amides of certain FA (14:3, 14:2, 18:3, 18:2) were isolated from the culture medium of actinomycetes (SC0043). Synthetic amides of oleic, arachidonic, and gamma-linolenic acids also inhibited PLA2 from porcine pancreas, human synovial fluid, and bee and snake venoms [115] and their activity was comparable to that of "natural" amides.
AMIDES OF FATTY ACIDS WITH BIOACTIVE AMINES
Studies of the lipid-soluble analogs of capsaicin (vanillinamide of 8-methyl-6-nonenic acid that is the main pungent principle in peppers of the Capsicum family) in mice indicated that they have anti-nociceptive and anti-inflammatory activities [116]. Vanillinamide of oleic acid (Fig. 1) was active after oral administration and its toxicity was lower versus other analogs. The effects can be associated with selective blockade of sensitive neurons by vanillinamide [116].
Amides of FA and biogenic amines (Fig. 1) have biological activity as well. Amides of palmitic, stearic, and linoleic acids and dopamine significantly decreased motility in mice and potentiated haloperidol-induced catalepsy in rats [117]. Dopamineamides and serotoninamides of arachidonic and eicosapentaenoic acids protect early sea urchin embryos from the cytotoxic effects of dopamine and serotonin antagonists and can be agonists of dopamine and serotonin receptors [118]. Amides of arachidonic acid with dopamine, histamine, and serotonin inhibit human platelet aggregation induced by adrenaline or arachidonic acid. The anti-aggregatory effect of these amides can be associated with their action on arachidonic acid-initiated biochemical processes in platelets [118].
Amides of oleic, linoleic, and linolenic acids and dopamine are inhibitors of 5-LO from RBL-1 cells. Linoleyldopamine had the highest activity, which was ~4-fold higher than that of the specific 5-LO inhibitor AA-861 [119]; serotoninamide of arachidonic acid was an irreversible inhibitor of 15-LO from soybean [120].
Amides and ethanolamides of saturated and unsaturated FA participate in bioregulation as endogenous bioeffector lipids; this was discovered and confirmed by recent studies. These relatively simple compounds can be highly efficient neuromodulators (ANA, oleamide) and regulators of peripheral organs and tissues (ANA, PEA, and erucamide). High biological activity, receptor-mediated mechanism (demonstrated for ethanolamides of FA) and the existence of systems synthesizing and degrading the amides allow them to be considered as the ancestors of a new class of bioregulators. This class apparently should include amides of FA and biologically active amines (serotonin, dopamine, etc.) the presence of which in intact cells is not yet demonstrated but whose high and specific biological activity indicates that they are plausible bioeffectors. Bioactive amides of FA are tightly associated with other lipid bioregulators. For example, formation of ANA and PEA during PLD-dependent hydrolysis of APE is associated with the generation of phosphatidic acid that is an important messenger of the cell regulation by itself (for reviews see [121]). Anandamide (and THC) initiate release of arachidonic acid via activation of cytoplasmic arachidonate-specific PLA2 that is associated with stimulation of eucosanoid synthesis [122]. On the other hand, FA amides can inhibit PLA2 by decreasing the content of polyenic FA and lysolecithin. ANA and other polyunsaturated ethanolamides can participate in oxidative metabolism similar to the corresponding free acids and potent inhibition of lipoxygenase by their amides with dopamine and serotonin is another link between oxylipins1 and bioactive amides of FA. Thus, bioactive amides of FA are organic components of the family of bioeffector lipids.
This work was supported in part by the Russian Foundation for Basic Research (grant No. 96-04-49191).
1 Oxylipins (prostaglandins, leukotrienes, etc.) are the oxidized metabolites of arachidonic and other polyenic FA formed by action of an enzyme at least on one stage of their generation.
REFERENCES
1.Hannun, Y. A., Obeid, L. M., and Dbaibo, G. S.
(1996) Handbook Lipid Res., 8, 177-204.
2.Muthing, J., Maurer, U., Sostaric, K., Neumann, U.,
Brandt, H., Duvar, S., Peter-Katalinic, J., and Weber-Schurholz, S.
(1994) J. Biochem., 115, 248-256.
3.Schmid, H. H. O., Schmid, P. C., and Natarajan, V.
(1990) Prog. Lipid Res., 29, 1-43.
4.Hantke, K., and Braun, V. (1973) Eur. J.
Biochem., 34, 284-296.
5.Towler, D., and Glaser, L. (1986)
Biochemistry, 25, 878-884.
6.Kuehl, F. A., Jr., Jacob, T. A., Ganley, O. H.,
Ormond, R. E., and Meisinger, M. A. P. (1957) J. Am. Chem. Soc.,
79, 5577-5578.
7.Devane, W. A., Hanus, L., Breuer, A., Pertwee, R.
G., Stevenson, L. A., Griffin, G., Gibson, D., Mandelbaum, A., Etinger,
A., and Mechoulam, R. (1992) Science, 258,
1946-1949.
8.Cravatt, B. F., Prospero-Garcia, O., Siuzdak, G.,
Gilula, N. B., Henriksen, J., Boger, D. L., and Lerner, R. A. (1995)
Science, 268, 1506-1509.
9.Devane, W. A., Dysarz, F. A., III, Johnson, M. R.,
Melvin, L. S., and Howlet, A. C. (1988) Mol. Pharmacol.,
34, 605-613.
10.Bramblett, R. D., Panu, A. M., Ballesteros, J.
A., and Reggio, H. (1995) Life Sci., 56, 1971-1982.
11.Houston, D. B., and Howlett, A. C. (1992) Mol.
Pharmacol., 43, 17-22.
12.Gerard, C. M., Mollereau, C., Vassart, G., and
Parmentier, M. (1991) Biochem. J., 279, 129-134.
13.Matsuda, L. A., Lolait, S. J., Brownstein, M. R.,
Young, A. C., and Bonner, T. I. (1990) Nature, 346,
561-564.
14.Rodríguez de Fonseca, F., Ramos, J. A.,
Bonnin, A., and Fernández-Ruiz, J. J. (1993)
Neuroreport., 4, 135-138.
15.Matsuda, L. A., Bonner, T. I., and Lolait, S. J.
(1993) J. Comp. Neurol., 327, 535-550.
16.Pacheco, M. A., Ward, S. J., and Childers, S. R.
(1993) Brain Res., 603, 102-110.
17.Bouaboula, M., Rinaldi, M., Carayon, C.,
Carillon, C., Delpech, B., Shire, D., Le Fur, G., and Casellas, P.
(1993) Eur. J. Biochem., 214, 173-180.
18.Munro, S., Thomas, K. L., and Abu-Shaar, M.
(1993) Nature, 365, 61-65.
19.Slipetz, D. M., O'Neill, G. P., Favreau, L.,
Dufresne, C., Gallant, M., Gareau, Y., Guay, D., Labelle, M., and
Metters, K. M. (1995) Mol. Pharmacol., 48, 352-361.
20.Bayewitch, M., Avidor-Reiss, T., Levy, R., Barg,
J., Mechoulam, R., and Vogel, Z. (1995) FEBS Lett., 375,
143-147.
21.Galiegue, S., Mary, S., Marchand, J., Dussossoy,
D., Carriere, D., Carayon, P., Bouaboula, M., Shire, D., Le Fur, G.,
and Casellas, P. (1995) Eur. J. Biochem., 232,
54-61.
22.Lynn, A. B., and Herkenham, M. (1994) J.
Pharmacol. Exp. Ther., 268, 1612-1623.
23.Facci, L., Dal Toso, R., Romanello, S., Buriani,
A., Skaper, S. D., and Leon, A. (1995) Proc. Natl. Acad. Sci.
USA, 92, 3376-3380.
24.Pertwee, R. G., Stevenson, L. A., Elrick, D. B.,
Mechoulam, R., and Corbett, A. D. (1992) Br. J. Pharmacol.,
105, 980-984.
25.Hanus, L., Gopher, A., Almog, S., and Mechoulam,
R. (1993) J. Med. Chem., 36, 3032-3034.
26.Pertwee, R., Griffin, G., Hanus, L., and
Mechoulam, R. (1994) Eur. J. Pharmacol., 259,
115-120.
27.Fride, E., Barg, J., Levy, R., Saya, D., Heldman,
E., Mechoulam, R., and Vogel, Z. (1995) J. Pharmacol. Exp.
Ther., 272, 699-707.
28.Priller, J., Briley, E. M., Mansouri, J., Devane,
W. A., Mackie, K., and Felder, C. C. (1995) Mol. Pharmacol.,
48, 288-292.
29.Makie, K., and Hille, B. (1992) Proc. Natl.
Acad. Sci. USA, 89, 3825-3829.
30.Caulfield, M. P., and Brown, D. A. (1992) Br.
J. Pharmacol., 106, 231-232.
31.Vogel, Z., Barg, J., Levy, R., Saya, D., Heldman,
E., and Mechoulam, R. (1993) J. Neurochem., 61,
352-355.
32.Felder, C. C., Briley, E. M., Axelrod, J.,
Simpson, J. T., Mackie, K., and Devan, W. A. (1993) Proc. Natl.
Acad. Sci. USA, 90, 7656-7660.
33.Makie, K., Devan, W. A., and Hille, B. (1993)
Mol. Pharmacol., 44, 498-503.
34.Mackie, K., Lai, Y., Westenbroek, R., and
Mitchell, R. (1995) J. Neurosci., 15,
6552-6561.
35.Felder, C. C., Joyce, K. E., Briley, E. M.,
Mansouri, J., Mackie, K., Blond, O., Lai, Y., Ma, A. L., and Mitchell,
R. L. (1995) Mol. Pharmacol., 48, 443-450.
36.Childers, S. R., Sexton, T., and Roy, M. B.
(1994) Biochem. Pharmacol., 47, 711-715.
37.Pinto, J. C., Potié, F., Rice, K. C.,
Boring, D., Johnson, M. R., Evans, D. M., Wilken, G. H., Cantrell, C.
H., and Howlett, A. C. (1994) Mol. Pharmacol., 46,
516-522.
38.Adams, I. B., Ryan, W., Singer, M., Thomas, B.
F., Compton, D. R., Razdan, R. K., and Martin, B. R. (1995) J.
Parmacol. Exp. Ther., 273, 1172-1181.
39.Sheskin, T., Hanus, L., Slager, J., Vogel, Z.,
and Mechoulam, R. (1997) J. Med. Chem., 40, 659-667.
40.Mechoulam, R., Ben-Shabat, S., Hanus, L.,
Ligumsky, M., Kaminski, N. E., Schatz, A. R., Gopher, A., Almog, S.,
Martin, B. R., Compton, D. R., Pertwee, R. G., Griffin, G., Bayewitch,
M., Barg, J., and Vogel, Z. (1995) Biochem. Pharmacol.,
50, 83-90.
41.Sugiura, T., Kondo, S., Sukagawa, A., Nakane, S.,
Shinoda, A., Itoh, K., Yamashita, A., and Waku, K. (1995) Biochem.
Biophys. Res. Commun., 215, 89-97.
42.Mechoulam, R., Ben Shabat, S., Hanus, L., Fride,
E., Vogel, Z., Bayewitch, M., and Sulcova, A. E. (1996) J. Lipid
Mediators Cell Signal., 14, 45-49.
43.Fride, E., and Mechoulam, R. (1993) Eur. J.
Pharmacol., 231, 313-314.
44.Crawley, J. N., Corwin, R. L., Robinson, J. K.,
Felder, C. C., Devane, W. A., and Axelrod, J. (1993) Pharmacol.
Biochem. Behav., 46, 967-972.
45.Wickens, A. P., and Pertwee, R. G. (1993) Eur.
J. Pharmacol., 250, 205-208.
46.Smith, P. B., Compton, D. R., Welch, S. P.,
Razdan, R. K., Mechoulam, R., and Martin, B. R. (1994) J. Pharmacol.
Exp. Ther., 270, 219-227.
47.Fan, P. (1995) J. Neurophysiol.,
73, 907-910.
48.Romero, J., García, L., Cebeira, M.,
Zadrozny, D., Fernández-Ruiz, J. J., and Ramos, J. A. (1995)
Life Sci., 56, 2033-2040.
49.Wenger, T., Tóth, B. E., and Martin, B. R.
(1995) Life Sci., 56, 2057-2063.
50.Cadas, H., Gaillet, S., Beltramo, M., Venance,
L., and Piomelli, D. (1996) J. Neurosci., 16,
3934-3942.
51.Hansen, H. S., Lauritzen, L., Strand, A. M.,
Moesgaard, B., and Frandsen, A. (1995) Biochim. Biophys. Acta,
1258, 303-308.
52.Shen, M., Piser, T. M., Seybold, V. S., and
Thayer, S. A. (1996) J. Neurosci., 16, 4322-4334.
53.Stein, E. A., Fuller, S. A., Edgemond, W. S., and
Campbell, W. B. (1996) Br. J. Pharmacol., 119,
107-114.
54.Varga, K., Lake, K., Martin, B. R., and Kunos, G.
(1995) Eur. J. Pharmacol., 278, 279-283.
55.Varga, K., Lake, K. D., Huangfu, D., Guyenet, P.
G., and Kunos, G. (1996) Hypertension, 28, 682-686.
56.Ishac, E. J., Jiang, L., Lake, K. D., Varga, K.,
Abood, M. E., and Kunos, G. (1996) Br. J. Pharmacol.,
118, 2023-2028.
57.Schuel, H., Goldstein, E., Mechoulam, R.,
Zimmerman, A. M., and Zimmerman, S. (1994) Proc. Natl. Acad. Sci.
USA, 91, 7678-7682.
58.Bisogno, T., Ventriglia, M., Milone, A., Mosca,
M., Cimino, G., and Di Marzo, V. (1997) Biochim. Biophys. Acta,
1345, 338-348.
59.Sugiura, T., Kondo, S., Sukagawa, A., Tonegawa,
T., Nakane, S., Yamashita, A., and Waku, K. (1996) Biochem. Biophys.
Res. Commun., 218, 113-117.
60.Couburn, A. F., Gracham, C. E., and Hahinger, J.
(1954) J. Exp. Med., 100, 425-435.
61.Long, D. A., and Martin, A. J. P. (1956)
Lancet, 1, 464-466.
62.Raskova, H., and Masek, K. (1967)
Therapie, 22, 1241-1246.
63.Raskova, H., Masek, K., and Linet, O. (1972)
Toxicon, 10, 485-490.
64.Perlik, F., Elis, J., and Raskova, H. (1971)
Acta Physiol. Hung., 39, 123-124.
65.Perlik, F., Elis, J., and Raskova, H. (1971)
Acta Physiol. Hung., 39, 395-400.
66.Masek, K., Perlik, F., Klima, J., and Kahlich, R.
(1974) Eur. J. Clin. Pharmacol., 7, 415-419.
67.Svec, P., Bederova, E., and Svec, J. (1975)
Neoplasma, 22, 625-630.
68.Krsiak, M., Dvorak, Z., Raskova, H., and Masek,
K. (1971) Activ. Nerv. Super., 13, 208-209.
69.Sechserova, M., and Elis, J. (1972) Activ.
Nerv. Super., 14, 188.
70.Krisak, M., Sechserova, M., Perlik, F., and Elis,
J. (1972) Activ. Nerv. Super., 14, 187.
71.Epps, D. E., Palmer, J. W., Schmid, H. H. O., and
Pfeiffer, D. R. (1982) J. Biol. Chem., 257,
1383-1391.
72.Epps, D. E., Grupp, I. L., Grupp, G., and
Schwartz, A. (1983) IRCS-Bioch., 11, 899-900.
73.Colodzin, M., Bachur, N. R., Weissbach, H., and
Udenfriend, S. (1963) Biochem. Biophys. Res. Commun.,
10, 165-170.
74.Bachur, N. R., Weissbach, H., and Udenfriend, S.
(1963) Fed. Proc., 22, 1520.
75.Bachur, N. R., and Udenfriend, S. (1996) J.
Biol. Chem., 241, 1308-1313.
76.Bachur, N. R., Masek, K., Melmon, K. L., and
Undenfriend, S. (1965) J. Biol. Chem., 240,
1019-1024.
77.Stoffel, W., Kriger, E., and Melzner, I. (1980)
Hoppe-Seyler’s Z. Physiol., 361, 773-779.
78.Morell, P., and Radin, N. S. (1970) J. Biol.
Chem., 245, 342-350.
79.Schmid, P. C., Zuzarte-Augustin, M. L., and
Schmid, H. H. O. (1985) J. Biol. Chem., 260,
14145-14149.
80.Deutsch, D. G., and Chin, S. A. (1993)
Biochem. Pharmacol., 46, 791-796.
81.Kruszhka, K. K., and Gross, R. W. (1994) J.
Biol. Chem., 269, 14345-14348.
82.Devane, W. A., and Axelrod, J. (1994) Proc.
Natl. Acad. Sci. USA, 91, 6698-6701.
83.Epps, D. E., Schmid, P. C., Natarajan, V., and
Schmid, H. H. O. (1979) Biochem. Biophys. Res. Commun.,
90, 628-633.
84.Epps, D. E., Natarajan, V., Schmid, P. C., and
Schmid, H. H. O. (1980) Biochim. Biophys. Acta, 618,
420-430.
85.Natarajan, V., Reddy, P. V., Schmid, P. C., and
Schmid, H. H. O. (1981) Biochim. Biophys. Acta, 664,
445-448.
86.Schmid, P. C., Reddy, P. V., Natarajan, V., and
Schmid, H. H. O. (1983) J. Biol. Chem., 258,
9302-9306.
87.Saito, M., and Kanfer, J. N. (1975) Arch.
Biochem. Biophys., 169, 318-323.
88.Chalifour, R. J., and Kanfer, J. N. (1980)
Biochem. Biophys. Res. Commun., 96, 742-747.
89.Natarajan, V., Schmid, P. C., and Schmid, H. H.
O. (1986) Biochim. Biophis. Acta, 878, 32-41.
90.Natarajan, V., Schmid, P. C., Reddy, P. V., and
Schmid, H. H. O. (1984) J. Neurochem., 42,
1613-1619.
91.Matsumoto, M., and Miwa, M. (1973) Biochim.
Biophys. Acta, 296, 350-364.
92.Gray, G. M. (1976) Biochim. Biophys. Acta,
431, 1-8.
93.Di Marzo, V., Fontana, A., Cadas, H., Schinelli,
S., Cimino, G., Schwartz, J.-C., and Piomelli, D. (1994) Nature,
372, 686-691.
94.Reddy, P. V., Natarajan, V., Schmid, P. C., and
Schmid, H. H. O. (1983) Biochim. Biophys. Acta, 750,
472-480.
95.Sugiura, T., Kondo, S., Sukagawa, A., Tonegawa,
T., Nakane, S., Yamashita, A., Ishima, Y., and Waku, K. (1996) Eur.
J. Biochem., 240, 53-62.
96.Reddy, P. V., Schmid, P. C., Natarajan, V., and
Schmid, H. H. O. (1983) Biochim. Biophys. Acta, 751,
241-246.
97.Omeir, R. L., Chin, S., Hong, Y., Ahern, D. G.,
and Deutsch, D. G. (1995) Life Sci., 56, 1999-2005.
98.Deutsch, D. G., Lin, S., Hill, W. A. G., Morse,
K. L., Salehani, D., Arreaza, G., Omeir, R. L., and Makriyannis, A.
(1997) Biochem. Biophys. Res. Commun., 231, 217-221.
99.Childers, S. R., Sexton, T., and Roy, M. B.
(1994) Biochem. Pharmacol., 47, 711-715.
100.Pertwee, R. G., Fernando, S. R., Griffin, G.,
Abadji, V., and Makriyannis, A. (1995) Eur. J. Pharmacol.,
272, 73-78.
101.Koutek, B., Prestwich, G. D., Howlett, A. C.,
Chin, S. A., Salehani, D., Akhavan, N., and Deutsch, D. G. (1994) J.
Biol. Chem., 269, 22937-22940.
102.Deutsch, D. G., Omeir, R., Arreaza, G.,
Salehani, D., Prestwich, G. D., Huang, Z., and Howlett, A. (1997)
Biochem. Pharmacol., 53, 255-260.
103.Ueda, N., Kurahashi, Y., Yamamoto, S., and
Tokunaga, T. (1995) J. Biol. Chem., 270,
23823-23827.
104.Maurelli, S., Bisogno, T., De Petrocellis, L.,
Di Luccia, A., Marino, G., and Di Marzo, V. (1995) FEBS Lett.,
377, 82-86.
105.Cravatt, B. F., Giang, D. K., Mayfield, S. P.,
Boger, D. L., Lerner, R. A., and Gilula, N. B. (1996) Nature,
384, 83-87.
106.Bisogno, T., Maurelli, S., Melck, D., De
Petrocellis, L., and Di Marzo, V. (1997) J. Biol. Chem.,
272, 3315-3323.
107.Ueda, N., Yamamoto, K., Yamamoto, S., Tokunaga,
T., Shirakawa, E., Shinkai, H., Ogawa, M., Sato, T., Kudo, I., Inoue,
K., et al. (1995) Biochim. Biophys. Acta, 1254,
127-134.
108.Bobrov, M. Yu., Archakov, A. V., Kogteva, G.
S., Fomina-Ageeva, E. V., Zinchenko, G. N., Gretskaya, N. M., Kuklev,
D. V., and Bezuglov, V. V. (1996) Bioorg. Khim., 22,
875-877.
109.Wakamatsu, K., Masaki, T., Itoh, F., Kondo, I.,
and Sudo, K. (1990) Biochem. Biophys. Res. Commun., 168,
423-429.
110.Mitchell, C. A., Davies, M. J., Grounds, M. D.,
McGeachie, J. K., Crawford, G. J., Hong, Y., and Chirila, T. (1996)
J. Biomat. Appl., 10, 230-249.
111.Cravatt, B. F., Lerner, R. A., and Boger, D. L.
(1996) J. Am. Chem. Soc., 118, 580-590.
112.Sugiura, T., Kondo, S., Kodaka, T., Tonegawa,
T., Nakane, S., Yamashita, A., Ishima, Y., and Waku, K. (1996)
Biochem. Mol. Biol. Int., 40, 931-938.
113.Maurelli, S., Bisogno, T., De Petrocellis, L.,
Di Luccia, A., Marino, G., and Di Marzo, V. (1995) FEBS Lett.,
377, 82-86.
114.Langstein, J., Hofstädter, F., and
Schwarz, H. (1996) Res. Immunol., 147, 389-396.
115.Jain, M. K., Ghomashchi, F., Yu, B.-Z.,
Bayburt, T., Murphy, D., Houck, D., Brownell, J., Reid, J. C.,
Solowiej, J. E., Wong, S.-M., Mocek, U., Jarrell, R., Sasser, M., and
Gelb, M. H. (1992) J. Med. Chem., 35, 3584-3586.
116.Janusz, J. M., Buckwalter, B. L., Young, P. A.,
LaHann, T. R., Farmer, R. W., Kasting, G. B., Loomans, M. E.,
Kerckaert, G. A., Maddin, C. S., Berman, E. F., Bohne, R. L., Cupps, T.
L., and Milstein, J. R. (1993) J. Med. Chem., 36,
2595-2604.
117.Kolocouris, N., Foscolos, G. B., Fytas, G.,
Sakellariou, A., Raftopoulos, C., Papadopoulou-Daifoti, Z., and
Vamvakides, A. (1991) Ann. Pharm. France, 49,
99-110.
118.Bezuglov, V. V., Manevich, E. M., Archakov, A.
V., Bobrov, M. Yu., Kuklev, D. V., Petrukhina, G. N., Makarov, V. A.,
and Buznikov, G. A. (1997) Bioorg. Khim., 23,
215-224.
119.Tseng, C. F., Iwakami, S., Mikajiri, A.,
Shibuya, M., Hanaoka, F., Ebizuka, Y., Padmawinata, K., and Sankawa, U.
(1992) Chem. Pharm. Bull., 40, 396-400.
120.Bezuglov, V. V., Archakov, A. V., Bobrov, M.
Yu., Kogteva, G. S., and Manevich, E. M. (1996) Bioorg. Khim.,
22, 878-880.
121.English, D., Cui, Y., and Siddiqui, R. A.
(1996) Chem. Phys. Lipids, 80, 117-132.
122.Wartmann, M., Campbell, D., Subramanian, A.,
Burstein, S. H., and Davis, R. J. (1995) FEBS Lett.,
359, 133-136.