REVIEW: Functions of Sphingosine in Cell Proliferation and Death
A. V. Alessenko
Institute of Biochemical Physics, Russian Academy of Sciences, ul. Kosygina 4, Moscow, 117977 Russia; fax: (095) 137-4101; E-mail: aless@center.chph.ras.ru
Submitted July 25, 1997
Interest in the biological functions of sphingosine, the metabolic product of sphingolipids, increased dramatically during the last few years. Sphingosine was found to be an exogenous inhibitor of protein kinase C and of many cell functions which depend on this enzyme including cell proliferation, differentiation, and programmed death. Sphingosine also activates some other protein kinases and regulates a variety of enzymes which are involved in the transmission of cell signals. Sphingosine mobilizes Ca2+ from intracellular stores and controls the specific Ca2+-channel. Sphingosine influences the synthesis of DNA and interacts with DNA in competition for the binding sites with histones, some enzymes, and transcriptional factors. Sphingosine is suggested to be a second messenger in the transmission of cell proliferation and apoptosis signals. The possible use of sphingosine in combined treatment of various diseases as a synergist of many drugs is considered.
KEY WORDS: sphingosine, cell proliferation, apoptosis, DNA synthesis, gene expression, nucleus, protein kinases, Ca2+ mobilization, signal transmission
Abbreviations: DAG) diacylglycerol; EGF) epidermal growth factor; ERK) extracellular signal-regulated kinase; JNK) c-Jun-N-terminal kinase; PI) phosphatidylinositol; PKC) protein kinase C; PMA) phorbol 12-myristate-13-acetate (phorbol ester); SAPK) stress-activated protein kinase; SPC) sphingosylphosphocholine; SPM) sphingomyelin; SPS) sphingosine; TNF) tumor necrosis factor.
A long-chain base sphingosine (SPS) (sphingenine) is one of the main
components of sphingolipid structure. The name sphingosine is related
to the word "sphinx"; it was proposed in 1884 by Thudichum
because of the enigmatic properties of SPS, and some mysteries of this
compound were disclosed only during the last decade.
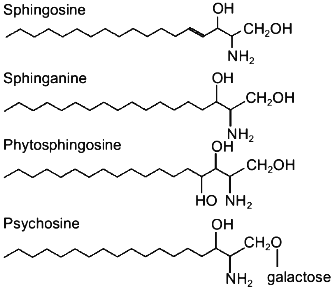
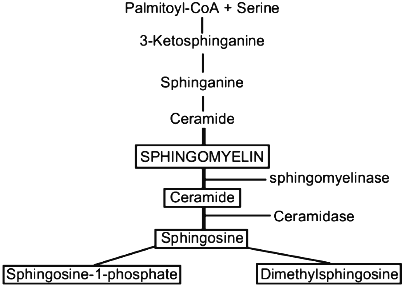
Sphingosine, sphinganine, phytosphingosine, and psychosine (their structures are presented in Fig. 1) are the most widely distributed natural sphingoid bases. It is now established that free sphingosine is a product of ceramide hydrolysis by ceramidase [1], but not of sphinganine oxidation by flavoprotein (Fig. 2).
Fig. 1. Structure of sphingoid bases.
Free SPS has been found in the liver of rats and mice [2, 3], HL-60 cells [4], neutrophils [5], and other cells. Sphingoid bases are found mainly in plasma membranes where they are generated from endogenous ceramide by ceramidase [6]. SPS in plasma membranes is produced in the presence of bivalent cations, at neutral pH, and with a dependence on temperature [6]. Sphingosine is produced in lysosomes at acidic pH, and this indicates that the cell contains two ceramidase isoforms (acidic and neutral) which digest ceramide to sphingosine. Free SPS was also found in cell nuclei of the liver of rats and mice [7-9]. Isolated plasma membranes contain 55 ng SPS per mg protein, i.e., half the SPS of liver cells [6]. The amount of SPS in nuclei of intact liver cells is not more than 0.45 nmole per mg protein [7]. It should be noted that the amount of SPS in the nucleus calculated per sphingomyelin (SPM) is significantly higher than in the whole cell. The generation of SPS is stimulated with an increase in the cell content of ceramide, either exogenous or resulting from treatment of membranes with sphingomyelinase which causes hydrolysis of the endogenous sphingomyelin [6]. Sphingomyelin is the only source of the active ceramide because other sphingolipids during enzymatic hydrolysis produce ceramides which fail to generate SPS. Quantitative analysis of free SPS was developed by Merrill et al. [2] and their method was later improved to decrease the loss of sphingoid bases during extractions and to increase the number of samples to be analyzed in clinical laboratories [10, 11].Fig. 2. Metabolism of sphingolipids.
In 1974 the activity of SPS in the cell was first shown by Italian scientists [12]; SPS was found to interact with DNA and influence its structure. Similarly to polyamines, the SPS molecule has a primary amino group which forms a ionic bond with the oxygen atom of the phosphate group of DNA molecules. This change in the structure of DNA via its interaction with SPS leads to alteration of the DNA substrate specificity. SPS inhibits DNA-primase in vitro and in vivo [13]. SPS, phytosphingosine, and N,N-dimethylsphingosine greatly inhibited purified DNA-primase and the growth of HL-60 leukemic cells due to their high cytotoxicity. Sphinganine and cis-sphingosine manifested a slight inhibiting effect and were not toxic for these cells; ceramides slightly inhibited both the enzyme activity and cell growth [13]. The interaction of DNA with SPS affected the ability of the DNA to be chemically modified, i.e., methylated [14]. The most interesting and worthy of attention is the reverse effects of SPM and SPS on heterologous methylation of DNA. Depending on the content, SPM either increased or decreased the extent of DNA methylation while SPS inhibited the methylation at all concentrations. Since SPS specifically interacts with DNA, it is suggested to bind to sequences which are the targets for DNA-methylases.
SPS was found to compete with H1 histone for the binding to DNA. Other lipids, such as phosphatidic acid and phosphatidylglycerol, firmly bound precisely with histones [15] and the treatment with some SPS derivatives, e.g., sphingosylphosphocholine (SPC), for 1-3 min sharply increased the DNA-binding activity of the AP-1 transcriptional factor and after 30 min this was accompanied by appearance of c-fos-transcripts [16]. The increase in the binding activity of AP-1 preceded the accumulation of c-fos mRNA; therefore, this derivative of SPS is suggested to cause post-translational modification. SPC induced the accumulation of c-Fos and c-Jun proteins, and this is likely to indicate its involvement in the regulation of expression of the early response genes. The content of SPS sharply increased in the liver nuclei during the cycloheximide-induced superexpression of nuclear oncogenes c-myc, c-fos, and c-jun, and this also seems to indicate the involvement of sphingoid bases in the regulation of gene expression [17]. SPS was also found to directly influence the accumulation level of c-myc-transcripts [18]. Sphinganine increased the gene expression of the retinoid acid-binding protein through an increase in the transcription rate [19]. To provide the increase in the transcription of this gene, an AP-1-binding site should be present in the promotor region whose activity is influenced by sphinganine similarly to SPS [16]. SPS influenced the activity of RNA-polymerases providing the free enzyme binding to the template [7].
SPS is of special interest because it influences the synthesis of DNA. Low concentrations (below 10 µM) of SPS stimulated the synthesis of DNA in Swiss- and A7r5-cells, but a higher content of SPS caused cell death [20, 21]. SPS and its numerous derivatives are extremely toxic for cells including tumor cells [22, 23]. SPS acts as a synergist with a number of antitumor drugs during combined treatment, and, consequently, it seems to be promising for pharmacology [24].
SPS was found to inhibit the activity of protein kinase C (PKC) [25] which is one of key enzymes in the regulation of cell proliferation, differentiation, and apoptosis, and this metabolite of sphingolipids is now believed to be a second messenger [26]. The PKC family is a set of isoenzymes which somewhat differ from the initially found enzyme isoform [27, 28]. Some isoforms are sensitive to the influence of ceramides and sphingomyelin [28, 29] but virtually all are inhibited by SPS and its derivatives. The extent of SPS-induced inhibition of the activity of PKC depends on the presence and quantitative ratios of phosphatidylserine, Ca2+, diacylglycerol, phorbol esters, lysophosphatidylcholine, and fatty acids [30]. Stearylamine, a structural analog of SPS, inhibited PKC similarly to SPS, whereas octylamine virtually failed to inhibit the enzyme activity [30]. Consequently, the effect of SPS is provided by the presence of both the primary amino group and the length of fatty acid residue. Similarly to SPS, sphinganine and other long-chain bases inhibited the activity of PKC [4]. SPS and sphinganine are now considered to be endogenous inhibitors of PKC [31]. An increase in the cell content of SPS in response to various treatments resulted due to enzymatic degradation of ceramide and not to synthesis de novo [5]. On the contrary, phorbol ester (PMA, 100 nM) decreased the amount of SPS nearly 1.5-fold with a simultaneous accumulation of ceramide. Arachidonic acid and ionophore A23187 decreased the cell content of SPS [5].
Numerous literature data and our own results show the cell nucleus to contain PKC, which seems to be a modified cytoplasmic form of the enzyme [32, 33]. The presence of SPS in the nucleus and changes in its content associated with activation of the cell nucleus suggest SPS to negatively regulate this enzyme which is located in nuclear structures. SPS is likely to control the replication and transcription of DNA through regulation of PKC activity because RNA-polymerases and topoisomerases II which are key enzymes in the activation of the cell genetic machinery, and histones and proteins of the nuclear matrix as well are substrates of the enzyme. Changes in the content of SPS were shown during both the synthesis of DNA and activation of RNA-polymerases in the nuclei of regenerating liver which is the most adequate model of these processes in vivo [7]. SPS inhibited the phosphorylation of nuclear proteins induced by retinoid acid, which is known to activate the nuclear form of PKC [34].
SPS is known to activate a number of protein kinases other than PKC. Thus, there is a variety of unidentified protein kinases which are sharply activated by SPS [35]. Among the identified protein kinases, special attention should be paid to casein kinase II [36], which is involved in the regulation of DNA synthesis and translocates into the nucleus during replication. SPS located in the nucleus is likely to control the activity of this enzyme. In some cases SPS is involved in the regulation of activity of mitogen-activated protein kinases [37] which transmit signals from receptors of the plasma membrane to transcriptional factors located in the nucleus. The cascade of these reactions is now divided into two subgroups, one of which is controlled by the protein kinase cascade transmitting mitogenic signals (ERK), and the other is related to the stress-activated protein kinase (SAPK) cascade which controls the inhibition of cell growth and inflammation reactions. On the rat glomerular mesogliar cells SPS was shown to activate only the ERK cascade and not influence that of SAPK [37]. These data demonstrate the opposite effects of SPS on inflammatory processes and cell proliferation due to distinguished activation of different kinase cascades [27]. A specific SPS- and dimethylsphingosine-dependent kinase has been found [38].
SPS activated the c-Jun-N-terminal kinase (JNK) and slightly influenced the extracellular signal-regulated kinase 2 (ERK-2); however, its phosphorylated derivative sphingosine-1-phosphate manifested the reverse influence on these enzymes in ASM-cells by stimulating the DNA synthesis [39, 40]. These works describe a complicated regulatory cycle which involves SPS and its derivatives. While SPS inhibited the cell growth through the activation of JNK, SPS-1-phosphate increased the synthesis of DNA induced by growth factors through the activation of ERK-2 [39]. Moreover, under certain conditions, SPS, similarly to ceramide, stimulated the generation of cAMP (a negative modulator of cell growth) while SPS-1-phosphate decreased the content of cAMP [39]. In A431 cells SPS reversibly activated protein kinase FA/GSK-3alpha which is involved in the transmission of thermal shock signal, and this suggests SPS to be a new regulatory agent in processes which depend on thermal shock [41].
Except kinase reactions, SPS controls the activity of many other enzymes in the cell in PKC-independent way. In particular, it induces GTP cyclohydrolase [42] and inhibits NADPH oxidase, preventing the translocation of one of the cytosol components of this enzyme (47-phox) to the membrane [43]. Similarly to ceramides with short acyl chains, SPS activates adenylate cyclase with the participation of PKC [44].
The treatment of isolated mitochondria with SPS resulted in a sharply increased generation of hydrogen peroxide while diacylglycerol (DAG) had no effect [45]. This seems to be especially important for understanding cytotoxicity of cytokines which are producers of sphingosine and active oxygen forms in the cell.
SPS is responsible for PKC-independent effects as follows: the inhibition of tissue growth factor [46], calmodulin-dependent kinase [47], and tyrosine kinase activity of the insulin receptor [48]; regulation of DAG kinase activity [49]; inhibition of phosphohydrolase of phosphatidic acid [50] and of cytidine triphosphate: phosphocholine cytidyl transferase [51]; activation of phospholipase D [52] and phospholipase Cdelta [53].
SPS influenced the phosphorylation of various receptor proteins [48, 54, 55], and the effects were multiple and ambiguous. Thus, the effects of SPS on phosphorylation of epidermal growth factor (EGF) receptor were quite opposite depending on the presence or the absence of phorbol ester [54]. In the absence of phorbol ester, SPS increased the affinity of receptors for EGF by increasing the number of EGF-binding sites on the cell surface. In the presence of phorbol ester, sphingosine inhibited the binding of 125I-labelled EGF to the highly specific sites. Such effects were not shown for ceramide. SPS inhibited the affinity properties of the muscarine cholinergic receptor regardless of the content of Ca2+ and PKC [56].
Recently the SPS-induced mobilization of Ca2+ from intracellular stores was shown in many studies [57-62]. SPS was shown to induce the dose-dependent release of Ca2+ from microsomal stores, although the maximal Ca2+ content was found in mitochondria [62]. SPS also inhibited binding of ryanodine to the sarcoplasmic reticulum and purified ryanodine receptor [61]. This effect was caused by an increase in the dissociation rate of bound ryanodine from membranes of the sarcoplasmic reticulum and a decrease in the rate of ryanodine binding to high-affinity sites. Bound ryanodine was released by sphingosine within some minutes [61]. However, ovocytes were found to have specific receptors for sphingosine, which was not similar to the ryanodine and inositol-1,4,5-triphosphate receptors, and this suggests that a new type of Ca2+ channel should exist which is controlled by SPS [63].
It should be noted that SPS stimulated the fast dose-dependent hydrolysis of phosphatidylinositol (PI) in fibroblasts accompanied by the accumulation of inositol-1,4,5-triphosphate and the subsequent accumulation of Ca2+; sphingosine-1-phosphate stimulated more rapid mobilization of Ca2+ but failed to affect the conversion of PI; ceramide was inert to both the metabolism of PI and the mobilization of Ca2+ [64].
The ability of SPS to influence a multiplicity of cell enzyme systems is described by the extreme diversity of its biological activity. SPS was found to influence cell growth and differentiation [4], platelet aggregation [30], inhibition of blood coagulation [46], stimulation of mitogenesis [20], and to show antitumor [26, 65] and antimicrobial activity [66]. SPS seems to be of special importance due to its ability to induce apoptosis [8, 18, 67-70].
Apoptosis, or programmed cell death, is a basis for various important biological processes, such as the positive and negative selection of T- and B-lymphocytes, the glucocorticoid-induced death of lymphocytes, the cell death caused by radiation, heating, or absence of specific growth factors. Apoptosis is of importance in protecting the organism under viral infections. The immune deficiency of HIV-infection is determined by disorders in the control of apoptosis.
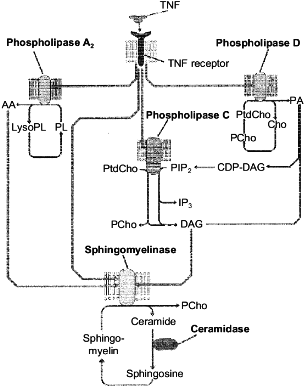
Among various models of cell death, apoptosis induced via receptors of tumor necrosis factor (TNF-R) and Fas-antigen (Fas/APO-1 or CD95) was mostly studied because of its high biological significance. Lipid components of the plasma membrane carrying the TNF- and Fas-receptors on the surface play an important role in the apoptosis signal transmission from the cell surface to the nucleus [71-73]. Phospholipids are a source of second messengers which are involved in the regulation of apoptotic enzymes, such as protein kinases, phosphatases, and proteases. The interaction of signal molecules with the receptor activates lipolytic enzymes (phospholipases A2, C, and D and sphingomyelinase) with subsequent accumulation of phospholipid hydrolysis products (arachidonic acid, diacylglycerol, ceramide, etc.). These products are immediately involved in the transmission of the signal induced by TNF-alpha and Fas L [69, 70, 73-77]. However, only products of the sphingomyelin cycle (ceramide and sphingosine) manifest a pronounced proapoptotic effect during direct contact with the cell [69, 70, 73, 75]. Products of enzymatic hydrolysis of other phospholipids were nontoxic [78, 79]. However, according to the literature data, arachidonic and phosphatidic acids and diacylglycerol activate the key enzyme of the sphingomyelin cycle, sphingomyelinase, and induce the accumulation of toxic products of this cycle in the cell [80-82], i.e., the lipid cascades which are induced during apoptosis are likely to activate the sphingomyelin cycle and its products are a toxic unit in the apoptosis signal transmission. This suggestion is presented in Fig. 3.
In spite of the rather strong evidence on the possible involvement of ceramide in the apoptotic signal transmission, the exact role of this compound as a mediator of the cell death is still unclear. Moreover, certain cells were found (L929) where the TNF-alpha failed to induce the generation of ceramide during the toxic signal passage [79]. It is also doubtful if the synthetic ceramides used in some studies to prove the induction of apoptosis by this sphingomyelin metabolite were adequate to the natural analog. The synthetic ceramides contain shortened fatty acid and are likely to be significantly more toxic for the cell than the natural compound. The treatment of neutrophils and cardiomyocytes with TNF-alpha and of HL-60 cells with phorbol ester was shown [18, 69, 74] to induce the accumulation of not only ceramide, but of SPS as well. We have also found that a single injection of TNF-alpha to mice resulted in the accumulation of ceramide and SPS in the liver cells and their nuclei [83].Fig. 3. Activation of signal phospholipases and enzymes of the sphingomyelin cycle induced by the binding of TNF-alpha to the TNF receptor. AA, arachidonic acid; Cho, choline; DAG, diacylglycerol; LysoPL, lysophospholipids; PA, phosphatidic acid; PL, phospholipids; PtdCho, phosphatidylcholine; PCho, phosphocholine; PIP2, phosphatidylinositol-4,5-diphosphate; IP3, inositol-1,4,5-triphosphate.
Six hours after the addition of SPS and N,N-dimethylsphingosine to tumor cells, apoptosis was observed in 90% of them which was manifested by the internucleosomal degradation of DNA and apoptosis-specific morphological changes (nuclear condensation, generation of apoptotic bodies, etc.) which were similar to changes induced in these cells by inhibitors of protein kinase C (staurosporine and H7) [74]. Ceramide which was added to the HL-60 cells during the differentiation stage easily converted to SPS, and this suggested an activation of ceramidase [18]. There are very interesting data on the synergic effects of SPS and sphinganine at sublethal concentrations (less than 750 nM) in increasing proapoptotic activity of ceramide (10 µM) [84]. Therefore, we think that the accumulation in the cell of both ceramide and sphingosine under the influence of apoptosis inducers is nonrandom. Other inhibitors of PKC, similarly to SPS, are synergists of ceramide in the effect on the cell, i.e., the inhibition of PKC by sphingoid bases and other inhibitors sharply increased the proapoptotic effect of ceramide [85], which is known to activate phosphatases. Apoptosis-resistant cells cannot simultaneously accumulate ceramide and SPS. Thus, the irradiation of radiation-resistant A431 cells failed to increase the content of ceramide, however, the level of SPS increased [86]. Ceramide induced apoptosis at higher concentration than SPS and is suggested to be partly utilized by the cell to produce sphingosine [18]. Precisely the combined influence of ceramide and sphingosine is likely to be the most efficient for apoptosis under various toxic influences on the cell.
In addition to PKC which is the main target of SPS, apoptosis seems to significantly depend on the dephosphorylation of the gene Rb (retinoblastoma) product regardless of PKC [87] and inhibition of DNA-primase [13]. The specific activation of the retinoblastoma protein correlates with the inhibition of cell growth and arrest at the G0/G1 stage of the cell cycle [87]. Recently it was found that SPS influences the expression of the bcl-2 gene which is responsible for the product preventing apoptosis. The induction of apoptosis in HL-60 cells by SPS and N,N-dimethylsphingosine was accompanied by a decrease in the expression of bcl-2 which was determined by an accumulation in the cells of the transcripts and the protein itself, while the expression of bcl-XL and bax mRNA was unchanged. Pharmacological inhibitors of PKC (H7 and staurosporine) failed to influence the expression of bcl-2 and induce the accumulation of SPS in the cells [88]. The authors conclude that SPS inhibited the expression of bcl-2 independently of PKC.
In spite of significant advances in studies on properties of SPS, the exact molecular mechanisms of its participation in the transmission of the apoptosis signals are not yet elucidated. To study in detail the involvement of SPS in the manifestation of TNF toxicity and apoptosis induction, we used mutant forms of human TNF-alpha which were obtained by site-directed mutagenesis [83]. They contained point (single- and double-site) and deletion mutations. The mutations changed the cytotoxic activity of the recombinant TNF-alpha. It was shown by the cytopathic testing on mouse L929 fibroblasts that the deletion-containing polypeptides were the most toxic, while those containing the double-site mutations lost toxicity and the single-site mutations decreased the toxic effects. The correlation was shown between the toxic effect of polypeptide and accumulation of SPS in mouse liver cells [2] after injection into animals of the mutant forms of TNF-alpha. In the same experiments no correlation was found between changes in the content of ceramide, sphingomyelin and in the sphingomyelinase activity and the toxicity of TNF-alpha, in spite of the accumulation of ceramide and activation of sphingomyelinase in the liver cells [83].
To test a possible involvement of SPS in the manifestations of TNF toxicity and it role in the apoptotic signal transmission, exogenous SPS and its phosphorylated analog sphingosine-1-phosphate were injected into mice at the dose of 20 µg per mouse. The single injection of SPS resulted in a reversible degradation of DNA in the liver and spleen to fragments of high molecular weight. In the thymus the fragmentation of DNA was still more pronounced. The combined injection to mice of SPS and its analog with TNF at doses of 20 µg per mouse resulted in a sharp increase in the apoptotic degradation of DNA in the spleen and thymus [70]. SPS was found to increase the toxic effects of various antitumor agents on the cell. It seems that in our case SPS increased the toxic effect of cytokine, and this seems to be of great interest for pharmacology and clinical use of TNF combined with SPS.
Fumonisin B1, an inhibitor of ceramide synthase which is a key enzyme of the sphingolipid biosynthesis de novo and the conversion of sphingosine into ceramide, was found to inhibit cell growth and induce their death due to apoptosis [89]. Fumonisin B1 caused the accumulation of sphinganine and sphingosine which are likely to be involved in the induction of apoptosis [89].
SPS is suggested to participate in apoptosis via interaction with DNA [12] and influencing the activity of enzymes responsible for replication and transcription [7]. By methods of fluorescent probes [7] and thermal denaturation [12], SPS was reliably shown to interact with DNA more efficiently than the other products of the sphingomyelin cycle. SPS also prevented frame-shift reading mutations induced by an intercalating antibiotic carminomycin [90]. SPS seems to similarly influence DNA-binding properties of a number of regulatory proteins, including transcription factors and topoisomerases which were spoken of earlier.
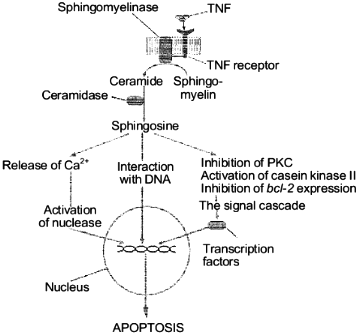
Thus, SPS properties suggest the following mechanism of its involvement in the apoptotic signal transmission on the level of the cytoplasm and cell nucleus. It seems that in the cytoplasm SPS regulates enzymes, such as phosphokinases and proteases, which are responsible for the apoptotic signal transmission and in the nucleus SPS influences the degradation of DNA by direct interaction with the latter. We present a scheme which describes the known pathways of the apoptotic signal transmission with the involvement of SPS (Fig. 4). The subsequent studies on SPS specific features should allow us to find targets of its influence during the induction in the cell of the cell death program.
Thus, it seems evident that SPS plays in the cell an extremely important role of an informational molecule in the transmission of extracellular signals of cell proliferation, differentiation, and death from the cell membrane surface into the nucleus.Fig. 4. Involvement of sphingosine in the TNF-alpha-induced apoptosis signal transmission.
This work was supported by the Russian Foundation for Basic Research (grant No. 95-04 12209a).
REFERENCES
1.Rother, J., van Echten, G., Schwarzmann, G., and
Sanhoff, K. (1992) Biochem. Biophys. Res. Commun., 189,
14-20.
2.Merrill, A. H., Jr., Wang, E., Mullins, R. E.,
Jamison, W. C., Nimcar, S., and Liotta, D. C. (1989) Analyt.
Biochem., 171, 373-381.
3.Rusakov, S. A., Filippova, G. N., Pushkareva, M.
Yu., Korobko, V. G., and Alessenko, A. V. (1993) Biochemistry
(Moscow), 58, 724-732 (Russ.).
4.Merrill, A. H., Jr., Sereni, A. M., Stevens, V. L.,
Hannun, Y. A., Bell, R. M., and Kinkade, J. M. (1986) J. Biol.
Chem., 261, 12610-12615.
5.Wilson, E., Wang, E., Mullins, R. E., Uhlinger, D.
J., Liotta, D. S., Lambeth, J. D., and Merrill, A. H., Jr. (1988) J.
Biol. Chem., 263, 9304-9309.
6.Slife, C. W., Wang, E., Hunter, R., Wang, S.,
Burgess, C., Liotta, D. C., and Merrill, A. H., Jr. (1989) J. Biol.
Chem., 264, 10371-10377.
7.Alessenko, A. V., Pantaz, E. A., Pushkareva, M.
Yu., Rusakov, S. A., and Filippova, G. N. (1993) Biochemistry
(Moscow), 58, 461-470 (Russ.).
8.Zhizhina, G. P., Korobko, V. G., and Alessenko, A.
V. (1994) Biochemistry (Moscow), 59, 1307-1313
(Russ.).
9.Alessenko, A., and Chatterjee, S. (1995) Mol.
Cell Biochem., 143, 169-174.
10.Van Veldhoven, P. P., Bishop, W. R., and Bell, R.
M. (1989) Analyt. Biochem., 183, 177-189.
11.Castegnaro, M., Garren, L., Gaucher, I., and
Wild, C. P. (1996) Nat. Toxins, 4, 284-290.
12.Manzoli, F. A., Muchmore, J. H., Bonora, B.,
Capitani, S., and Bartoli, S. (1974) Biochim. Biophys. Acta,
340, 1-15.
13.Tamiya-Koizumi, K., Murate, T., Suzuki, M.,
Simbulan, C. M., Nakagawa, M., Takemura, M., Furuta, K., Izuta, S., and
Yoshida, S. (1997) Biochem. Mol. Biol. Int., 41,
1179-1189.
14.Romanenko, E. B., Alessenko, A. V., and
Vanyushin, B. F. (1995) Biochem. Mol. Biol. Int., 35,
87-94.
15.Koiv, A., Palvimo, J., and Kinnunen, P. K. (1995)
Biochemistry, 34, 8018-8027.
16.Berger, A., Rozenthal, D., and Kinnunen, P. K.
(1995) Proc. Natl. Acad. Sci. USA, 92, 5885-5889.
17.Alessenko, A. V., Boikov, P. Ya., Drobot, L. B.,
Rusakov, S. A., and Filippova, G. N. (1994) Biochemistry
(Moscow), 59, 1076-1088 (Russ.).
18.Ohta, H., Sweeney, E. A., Masamune, A., Yatomi,
Y., Hakomori, S., and Igarashi, Y. (1995) Cancer Res.,
55, 691-697.
19.Wei, L. N., Lee, C. H., and Chang, L. (1995)
Mol. Cell. Endocrinol., 111, 207-211.
20.Zhang, H., Buckley, N. E., Gibson, K., and
Spiegel, S. (1990) J. Biol. Chem., 265, 76-81.
21.Jacobs, L. S., and Kester, M. (1993) Am. J.
Physiol., 265, 740-747.
22.Merrill, A. H., Jr. (1983) Biochim. Biophys.
Acta, 754, 284-291.
23.Merrill, A. H., Nimkar, S., Melandino, D.,
Hannun, Y. A., Loomis, C., Bell, R. M., Tayagi, S. R., Lambeth, J. D.,
Stevens, V. L., Hunter, R., and Liotta, D. C. (1989)
Biochemistry, 28, 3138-3145.
24.Sosnovski, J., Stetter-Neel, C., Cole, D.,
Durham, J. P., and Mawhinney, M. G. (1997) J. Urol.,
158, 269-274.
25.Hannun, Y. A., and Bell, R. M. (1987)
Science, 235, 670-674.
26.Merrill, A. H., and Stevens, V. L. (1989)
Biochim. Biophys. Acta, 1010, 131-139.
27.Nishizuka, Y. (1986) Science, 233,
305-312.
28.Dekker, L. V., and Parker, P. J. (1994) Trends
Biochem. Sci., 19, 73-77.
29.Mathias, S., Dressler, K. A., and Kolesnik, R. N.
(1991) Proc. Natl. Acad. Sci. USA, 88, 10009-10013.
30.Hannun, Y. A., Loomis, C. R., Merrill, A. H., and
Bell, R. M. (1986) J. Biol. Chem., 261, 12604-12609.
31.Smith, E. R., Jones, P. L., Boss, J. M., and
Merrill, A. H. (1997) J. Biol. Chem., 272,
5640-5646.
32.Leach, K. L., Powers, E. A., Ruff, V. A., Jaken,
S., and Kaufmann, S. (1989) J. Cell Biol., 109,
685-695.
33.Alessenko, A. V., Khan, W., Wetsel, C., and
Hannun, Y. (1992) Biochem. Biophys. Res. Commun., 182,
1333-1339.
34.Zorn, N. E., and Sauro, M. D. (1995) Int. J.
Immunopharmacol., 17, 303-311.
35.Pushkareva, M., Khan, W., Alessenko, A., Sahyoun,
N., and Hannun, Y. (1992) J. Biol. Chem., 267,
15246-15251.
36.McDonald, O. B., Hannun, Y. A., Reynolds, C. H.,
and Sahyoun, N. (1991) J. Biol. Chem., 266,
21773-21776.
37.Coroneus, E., Wang, Y., Panuska, J. R.,
Pempleton, D. J., and Kester, M. (1996) Biochem. J.,
316, 13-17.
38.Megidish, T., White, T., Takio, K., Titani, K.,
Igarashi, Y., and Hakomori, S. (1995) Biochem. Biophys. Res.
Commun., 216, 739-747.
39.Pyne, S., Chapman, J., Steele, L., and Pyne, N.
J. (1996) Eur. J. Biochem., 237, 819-826.
40.Pyne, S., and Pyne, N. J. (1996) Biochem.
J., 315, 917-923.
41.Yang, S. D., Chang, H. S., and Lee, S. C. (1996)
J. Cell Biochem., 60, 218-225.
42.Anastasiadis, P. Z., Kuhn, D. M., Blitz, J.,
Imerman, B. A., Louie, M. C., and Levine, R. A. (1996) Brain
Res., 713, 125-133.
43.Sasaki, J. I., Yamaguchi, M., Yamane, H.,
Okamura, N., and Ishibashi, S. (1996) J. Biochem., 120,
705-709.
44.Zhou, X. M. (1994) J. Neurochem.,
63, 1361-1370.
45.Garcia-Ruiz, C., Colell, A., Mari, M., Morales,
A., and Fernandes-Checa, J. C. (1997) J. Biol. Chem.,
272, 11369-11377.
46.Conkling, P. R., Patton, K. L., Hannun, Y. A.,
Greenberg, C. S., and Weinberg, J. B. (1989) J. Biol. Chem.,
264, 18440-18444.
47.Jefferson, A. B., and Schulman, H. (1988) J.
Biol. Chem., 263, 15241-15244.
48.Arnold, R. S., and Newton, A. C. (1991)
Biochemistry, 330, 7747-7754.
49.Sakane, I., Yamada, K., and Kanoh, H. (1989)
FEBS Lett., 255, 409-413.
50.Mulman, T. J., Siegel, M. J., Egan, R. W., and
Billah, M. M. (1991) J. Biol. Chem., 266, 2013-2016.
51.Sohal, P. S., and Cornel, R. B. (1990) J.
Biol. Chem., 265, 11746-11750.
52.Lavie, Y., and Liscovitch, M. (1990) J. Biol.
Chem., 265, 3868-3872.
53.Matecki, A., and Pawelczyk, T. (1997) Biochim.
Biophys. Acta, 1325, 287-296.
54.Davis, R. J., Girones, N., and Faucher, M. (1988)
J. Biol. Chem., 263, 5373-5379.
55.Faucher, M., Girones, N., Hannun, Y. A., Bell, R.
M., and Davis, R. J. (1988) J. Biol. Chem., 263,
5319-5327.
56.Fugita, Y., and Sugiya, H. (1992) Comp.
Biochem. Physiol. C. Comp. Pharmac. Toxicol., 1103,
269-272.
57.Wong, K., and Kwabyeung, L. (1993) Cell
Calcium, 14, 493-505.
58.Olivera, A., Zhang, H., Carlson, R. O., Mattie,
M. E., Schmidt, R. R., and Spiegel, S. (1994) J. Biol. Chem.,
269, 17924-17930.
59.Sabala, P., Wiktorek, M., Csarny, M., Chaban, V.,
and Baranska, J. (1996) Acta Neurobiol. Exp. (Warsz.),
56, 507-513.
60.Sakano, S., Takemura, H., Yamada, K., Imoto, K.,
Kaneko, M., and Ohshika, H. (1996) J. Biol. Chem., 271,
1148-1155.
61.Needleman, D. H., Aghdasi, B., Seryshev, A. B.,
Schroepfer, G. J., Jr., and Hamilton, S. L. (1997) Am. J.
Physiol., 272, C1465-C1474.
62.Huang, W. C., and Chueh, S. H. (1996) Brain
Res., 718, 151-158.
63.Kim, S., Lakhani, V., Costa, D. J., Sharara, A.
I., Fitz, J. G., Huang, L. W., Peters, K. G., and Kindman, L. A. (1995)
J. Biol. Chem., 270, 5266-5269.
64.Chao, C. P., Laulederkind, S. J. F., and Ballou,
L. R. (1994) J. Biol. Chem., 269, 5849-5856.
65.Hakomori, S.-I. (1992) J. Exp. Med.,
168, 211-222.
66.Bibel, D. J., Aly, R., and Shinefield, H. R.
(1995) Clin. Exp. Dermatol., 20, 395-400.
67.Nakamura, S., Kozutsumi, Y., Sun, Y., Miyake, Y.,
Fujita, T., and Kawasaki, T. (1996) J. Biol. Chem., 271,
1255-1257.
68.Krown, K. A., Page, M., Nguyen, C., Zechner, D.,
Gutierrez, V., Comstock, K. L., Gembotski, C. C., Quintana, P. J., and
Sabbadini, R. A. (1996) J. Clin. Invest., 98,
2854-2865.
69.Sweeney, E. A., Sakakura, C., Shirahama, T.,
Masamune, A., Ohta, H., Hakomori, S., and Igarashi, Y. (1996) Int.
J. Cancer, 66, 358-366.
70.Khrenov, A. V., Terent'ev, A. A., and Alessenko,
A. V. (1996) Eur. Cytol. Network, 7, 207.
71.Cifone, M. G., Roncaioli, P., de Maria, R.,
Camarada, G., Santoni, A., Ruberti, G., and Testi, R. (1995) EMBO
J., 71, 5859-5868.
72.De Valck, D., Beyaert, R., Van Roy, F., and
Fiers, W. (1993) Eur. J. Biochem., 212, 491-497.
73.Dressler, K. A., Mathias, S., and Kolesnik, R.
(1992) Science, 255, 1715-1718.
74.Ohta, H., Yatomi, Y., Sweeney, E. A., Hakomori,
S., and Igarashi, Y. (1994) FEBS Lett., 355,
267-270.
75.Obeid, L. M., Linardic, C. M., Koralak, L. A.,
and Hannun, Y. A. (1993) Science, 259, 1769-1771.
76.Schutze, S., Berkovic, D., Tomsing, O., Unger,
C., and Kronke, M. (1991) J. Exp. Med., 174,
975-988.
77.Schutze, S., Machleidt, T., and Kronke, M. (1994)
J. Leukocyte Biol., 56, 533-541.
78.Suffys, P., Beyaert, R., de Valck, D.,
Vanhaesebroeck, B., Van Roy, F., and Fiers, W. (1991) Eur. J.
Biochem., 195, 465-484.
79.Fiers, W. (1991) FEBS Lett., 285,
199-212.
80.Jayadev, S., Linardic, C. M., and Hannun, Y. A.
(1994) J. Biol. Chem., 269, 5757-5763.
81.Kolesnik, R. N. (1987) J. Biol. Chem.,
262, 16759-16762.
82.Liscovitch, M. (1992) Trends Biochem.
Sci., 17, 393-399.
83.Alessenko, A. V., Korobko, V. G., Khrenov, A. V.,
Rozhnova, U. A., Soloviev, A. S., and Shingarova, L. N. (1997)
Biochem. Mol. Biol. Int., 42, 143-154.
84.Jarvis, V. D., Fornari, F. A., Traylor, R. S.,
Martin, H. A., Kramer, L. B., Erukula, R. K., Bittman, R., and Grant,
S. (1996) J. Biol. Chem., 271, 8275-8284.
85.Chmura, S. J., Nodzenski, E., Crane, M. A.,
Virudachalam, S., Hallahan, D. E., Weichselbaum, R. R., and Quintans,
J. (1996) Adv. Exp. Med. Biol., 406, 39-55.
86.Balaban, N., Moni, J., Shannon, M., Dang, L.,
Murphy, E., and Goldkorn, T. (1996) Biochim. Biophys. Acta,
1314, 147-156.
87.Pushkareva, M., Chao, R., Bielawska, A., Merrill,
A. H., Jr., Crane, H. M., Lagu, B., Liotta, D., and Hannun, Y. A.
(1995) Biochemistry, 34, 1885-1892.
88.Sakakura, C., Sweeney, E. A., Shirahama, T.,
Hakomori, S., and Igarashi, Y. (1996) FEBS Lett., 379,
177-180.
89.Yoo, H. S., Norred, W. P., Showker, J., and
Riley, R. T. (1996) Toxicol. Appl. Pharmacol., 138,
211-218.
90.Soloviev, A. S., Korobko, V. G., Shingarova, L.
N., Vasil'eva, S. V., Makhova, E. V., and Alessenko, A. V. (1995)
Biochemistry (Moscow), 60, 1283-1291 (Russ.).