REVIEW: Influence of Sphingolipids on T Lymphocyte Activation
E. A. Martinova
Institute of Nutrition, Russian Academy of Medical Sciences, Ustinsky proezd 2/14, Moscow, 109240 Russia; fax: (095) 298-1872
Submitted June 30, 1997
Sphingolipid metabolism in immune cells results in formation of a second lipid messenger, e.g., ceramide, sphingosine, ceramide-1-phosphate, and sphingosine-1-phosphate. They are involved in a common signaling which controls the main stages of the lymphocyte development, differentiation, activation, and proliferation in response to a mitogenic and antigenic stimuli, and to initiate programmed cell death. Both, the sphingomyelin cycle products and inhibitor of ceramide synthase--fumonisin B1--have been shown to affect the CD3, CD4, CD8, CD45, and other T lymphocyte surface antigen expression, to disrupt lymphocyte subpopulation balance, to inhibit DNA synthesis in normal lymphocytes, and to suppress an immune response to T-dependent antigens in vivo. The common targets of the TCR/CD3-derived and sphingolipid-mediated signaling pathways may provide the sphingolipid effect on immune cells.
KEY WORDS: sphingosine, ceramide, fumonisin B1, T lymphocyte, TCR/CD3 complex, activation, signal transduction
Abbreviations: Ag) antigen; Ab) antibody; B7-1, B7-2) ligands for CD28 receptor; [Ca2+]i) intracellular calcium; CAPK) ceramide-activated protein kinase; CAPP) ceramide-activated protein phosphatase; CD) cluster designation; CD2, CD3, CD4, CD8, CD28, CD44, CD45) surface T lymphocyte receptors; CD25) interleukin-2 receptor alpha-chain; CD95/Fas/Apo-1) apoptosis receptor; cdk) cyclin-dependent kinase; Cer) ceramide; C2-Cer) N-acetyl sphingosine; Cer-1-P) ceramide-1-phosphate; DAG) diacylglycerol; DN) double negative thymocyte; DP) double positive thymocyte; ERK) extracellular receptor kinase; Fas-L) ligand for Fas-receptor; GPI) glycosylphosphatidylinositol; GSL) glycosphingolipids; IFN-gamma) gamma-interferon; IL) interleukin; JNK) NH2-terminal nuclear kinase; PI3-kinase) phosphatidylinositol-3 kinase; mAb) monoclonal antibodies; MAPK) mitogen-activated protein kinase; MHC) major histocompatibility complex; NK) natural killer cell; PIP2) phosphatidylinositol-4,5-bis-phosphate; PI) phosphatidylinositol; PKC) protein kinase C; PLC) phospholipase C; PTK) protein tyrosine kinase; PTP) protein tyrosine phosphatase; R) receptor; Rb) product of retinoblastoma gene; SAPK) stress-activated protein kinase; SH2) src homology 2; SM) sphingomyelin; SMase) sphingomyelinase; Sph) sphingosine; Sph-1-P) sphingosine-1-phosphate; Spha) sphinganine (dihydrosphingosine); Spha-1-P) sphinganine-1-phosphate; TCR) antigen-specific T lymphocyte receptor; TGF-beta) transforming growth factor beta; Th1) T helper of 1 subset; Th2) T helper of 2 subset; TNF-alpha) tumor necrosis factor alpha; TNF-beta) tumor necrosis factor beta.
SPHINGOLIPID METABOLISM AS A CONSTITUENT PART OF T LYMPHOCYTE
SIGNALING
Sphingolipid metabolism in immune cells exerts a control over their development, differentiation, activation, and apoptosis. The distinguishing features of the sphingolipid turnover in each subpopulation of immune cells are stemming from a wide variety of lymphocyte receptors, their ligands, and regulative cytokines in lymphoid tissues and organs. Sphingolipid turnover in T lymphocytes is accomplished by de novo synthesis and is activated by mitogens, foreign antigens, ligands for cell surface glycoproteins along with a cell--cell interaction and a contact with the extracellular matrix [1, 2]. The free sphingoid bases into cells may be up-regulated also by changing culture conditions in vitro or by lipid composition of very-low-density lipoproteins in vivo [3-5].
SM hydrolysis under plasma membrane SMase activation has been found to initiate a so-called "sphingomyelin cycle" resulting in a generation of second lipid messengers Cer, Sph, Sph-1-P, Cer-1-P, and other bio-active molecules in signal transduction [6-8]. The sphingomyelin cycle is associated with signaling via an antigen-specific receptor of a T cell to motivate the influence of sphingolipids on both the lymphocyte effector function and activation-induced apoptosis [9].
T lymphocyte receptor complex includes the alphabetazetazeta-chains of TCR, gammaepsilon/epsilondelta- or gammaepsilon/zetaeta-chains of CD3, and glycoprotein CD4 (or CD8) that is responsible for recognition of beta2-chain of MHC II class molecule (or alpha3-chain of MHC I class molecule for CD8). CD4 phenotype is inherent in T helper subpopulation which coordinate the immune response to T-dependent antigens. Glycoprotein CD8 has been shown to be primarily expressed on cytotoxic and suppressor T lymphocytes. Immune recognition suggests a physical association between the complex including processed Ag peptide and two MHC molecules with an active center of the TCR/CD3 complex. Unlike growth factor receptors, TCR/CD3 complex does not have an intrinsic kinase activity that assumes the close interaction with the membrane-associated PTKs p72syk, p56lck, p70ZAP, p59fyn, etc. [10]. PTK p59fyn is coupled with TCR zeta-chain. PTK p70ZAP is coupled with both CD8 molecule and TCR zeta-chain and is stabilized by PTK p56lck. These PTKs phosphorylate the cytoplasmic domains of the gammaepsilondelta of CD3, zetazeta of TCR, and CD4 (CD8) as well as the adapter proteins Shc, Vav, GRB-2, and other SH2-domain containing proteins, namely, c-Src, PI3-kinase, rasGAP, and PTP 1D (Syp) [10-12]. A kinase activity of the PTK p72syk was shown to be maximal for carboxy-terminal SH2-domain PLC-gamma-1 [13]. Most PTKs catalyze inter- and intramolecular autophosphorylation to exhibit an optimal activity, and to create new sites for SH2-domains on additional substrates [13]. In T lymphocytes, the non-receptor PTKs may be phosphorylated independently of their catalytic domains [14]. Cross-binding TCR/CD3 and CD4 causes the phosphorylation of TCR zetazeta-chains and the adapter protein Shc. Phosphorylated tyrosine of Shc may recognize the SH2-domain Grb2 protein followed by an activation of the Ras-signaling pathway via a Shc/Grb-2/mSos complex [15]. Anti-TCR mAbs activate T lymphocyte receptor without CD4 ligation that leads to the phosphorylation of the adapter proteins Crk and p116, increases stability in the p116/Crk/C3G complex, and provides additional signal towards the Ras-pathway [16].
Different isoforms of a transmembrane PTP CD45 were found to be expressed on T and B lymphocytes. CD45 phosphorylates the tyrosine of the cytoplasmic domain TCR under antigenic stimulation, and couples the TCR/CD3-derived signals with the intracellular signaling pathways [17]. The PTKs p56lck and p59fyn have been found to be the usual substrates for PTP CD45 [18]. The absence of CD45 in the mutant T cell lines attenuates the coupling between TCR/CD3 signal pathway and Ca2+-channels in a cell [18]. The impaired expression of CD45 up-regulates a threshold sensitivity of TCR/CD3 complex to affect both the positive and the negative selection of lymphocytes [19] and to activate Fas-dependent apoptosis [20]. A mycotoxin fumonisin B1 produced by the fungi Fusarium moniliforme and F. proliferatum has been found to inhibit a ceramide synthase followed by predominant accumulation of Spha and less Sph [1]. Both fumonisin B1 and Sph have been shown to increase CD45 expression in mouse lymphocytes within several minutes and to prolong its activation at the primary immune response to T-dependent antigens [21].
Lymphocyte activation leads to a high-rate transfer of lipids between plasma membrane bilayers (flip-flop) [22] that disrupts the phospholipid asymmetry keeping by APT-dependent aminophospholipid translocase [23]. The second lipid messenger PIP2 is a substrate for PI-PLC, PLC-gamma, and PLC-beta. PIP2 catalyses the generation of DAG and regulates the G-protein-dependent activation of PLD. Molecular types of DAG are determined by the phospholipid composition of the plasma membrane [24]. DAG activates the PKC alpha-, beta-, and gamma-isoforms and increases [Ca2+]i. Sphingolipids have been shown to modulate in the DAG generation, PKC activity, and PKC-dependent activation of PLD, and to create common targets between the signaling pathways into the cell [25].
The Sph-1-P-dependent signal pathway is derived rather from SM hydrolysis initiated by a membrane-associated sphingosine kinase than from de novo synthesis [2, 26]. Sph-1-P has been shown to exhibit the opposite effects in relation to Sph and Cer. In transformed T cell lines, Sph-1-P was found: 1) to activate G-protein-coupling sphingolipid receptors; 2) to increase [Ca2+]i; 3) to activate PLD; 4) to inhibit IP3-kinase; 5) to cancel an apoptotic effect of Cer and Sph; and 6) to up-regulate DNA synthesis via activation of a sphingosine kinase and PKC [27]. In the fibroblasts Swiss 3T3, Cer activates the Sph-1-P hydrolase and acts as an antagonist for Sph-1-P in DNA synthesis. C2-Cer prevents Ca2+-dependent PKC-alpha translocation and suppresses the Ag-induced activation of PLD [28, 29]. PKC-lambda/iota and PKC-zeta are the second messengers in Cer-initiated transactivation of kappaB-dependent promoter [30].
Prolonged activation of PKC and PLD are required for the production of some cytokines, and it may be regulated by Cer and Sph [28]. A competition between DAG and Cer in the PKC active sites is specified by both the DAG and Cer molecular types and by the time of DAG and Cer exposure. An extended DAG exposure in parallel with a decline in Cer concentration keeps the PKC activity and increases cell proliferation [24]. In vitro, serum factor deprivation causes SM hydrolysis, Cer formation, and cell arrest in the G0/G1-phase of the cell cycle that initiates an apoptosis in a dose-dependent manner. DAG may keep the Cer activity to initiate an apoptosis but not to arrest the cell growth in G0/G1-phase [31].
In T cell line Jurkat, Sph forms its own signal pathway that inhibits the CD3-derived signaling. Sph exerts the greatest effect on DAG kinase in comparison with Sph-1-P. Cer was not effective in these cells [31]. C8-Cer-1-P stimulates the cell growth via DNA synthesis and G0/G1-phase transmitting but does not involve PLD and MAPKs p42 and p44. C2-Cer makes a major contribution to metabolism of the exogenous C8-Cer-1-P in C8-Cer but does not take part in the formation of Sph and Sph-1-P [32].
MAPK kinase signaling provides the information from the plasma membrane receptors to the nuclear transcription factors. Lymphocyte activation initiates two MAPK kinase pathways: ERK cascade may serve as mitogen- and growth factor-transmitting pathway, and SAPK cascade provides signaling primarily from the pro-inflammatory cytokines [14, 33]. The fast agonist-dependent metabolism of sphingolipids and glycosphingolipids may regulate the lymphocyte proliferation via MAPK kinase pathways. C2-Cer activates SAPK/JNK kinases and suppresses the Sph-1-P-initiated activity of ERK kinases to prevent cell proliferation [34-36]. Kinases Erk-1 and Erk-2 have been found to regulate the activity of nuclear transcription factors, in particular AP-1, and to phosphorylate c-jun in its activation domain. MAPK kinase cascade also activates factor TCF/Elk1 increasing the c-fos oncogene transcription [37]. The Sph-1-P signal pathway includes ERK kinases Erk-2, Raf, MKK1, and MKK2. Sph-1-P has been shown to inhibit the Cer-initiated SAPK/JNK kinase cascade [38] and to provide mitogen signaling through the activation of AP-1 and c-fos [39]. PKC serves as a common messenger for signal transduction pathways to activate both p72raf1 and p42erk2b kinases as opposed to p21ras which serves for p72raf1 only. The possible cross-talk between the TCR/CD3-derived signals and the sphingomyelin cycle product signaling is presented in Fig. 1.
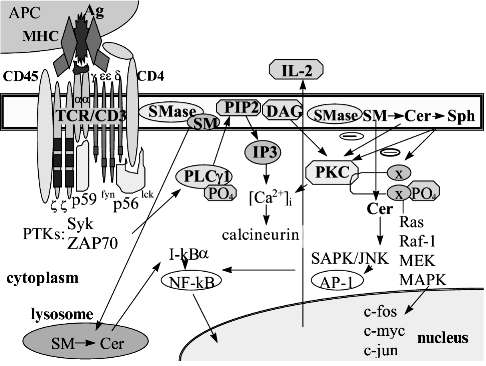
T cell activation via TCR/CD3 complex causes phosphorylation of kinases Erk1, Erk2, Erk3 and SAPK/JNK1,2; this defines the association between the cyclins and cyclin-dependent kinases regulating the cell cycle [14]. A pair cyclin E/cdk2 dictates the entering into G1-phase during differentiation as well as transmitting from G1 to S-phase during proliferation. The pair cyclin D1/cdk5 is believed to prevent differentiation of transformed lymphocytes. Cyclin D1 over-expression leads to B lymphocyte malignization [40, 41]. The kinases cdk4 and cdk6 up-regulate the G1/S-phase transmission. The phosphorylation of cdk1 and cdk2 results in the accumulation of cells in S-phase [42]. Sph was shown to arrest the cells in G1-phase via an unstable complex cyclin E/cdk2. The endogenous Cer accumulation, particularly under inhibition of GlcCer synthesis, has been found to decrease the p34cdc2 and cdk2 kinase activity [43]. A pair cyclin E/cdk2 regulates Rb-protein phosphorylation. Sph has been shown to arrest cell growth in G0/G1-phase that is due to Rb-protein activation followed by the loss of E2F-DNA-binding activity [44]. E2F ligation prevents E2F-dependent gene expression that is associated with G1/S-phase transmission. Cell arrest in G0/G1-phase may disrupt the DNA synthesis program and cause apoptosis [45].Fig. 1. Cross-talk between signaling pathways initiated through T lymphocyte receptor complex and sphingomyelin cycle products. The active center of T lymphocyte receptor (TCR/CD3) recognizes simultaneously both the processed antigenic peptide (Ag) and two molecules of major histocompatibility complex (MHC) expressed on the antigen presenting cell (APC). Co-receptor CD4 serves for the MHC II class molecule recognition. Receptor-associated protein tyrosine kinases (PTK) p56lck, p59fyn, src, Syk, and ZAP-70 are phosphorylated followed by an activation of phospholipase Cgamma1 (PLCgamma1) and its translocation to the plasma membrane. Lipid second messengers phosphatidylinositol-4,5-bisphosphate (PIP2) and diacylglycerol (DAG) are included into protein kinase C (PKC) signaling followed by the initiation of MAPK kinases Ras, Raf-1, MEK, etc. The activation of the nuclear transcription factors AP-1, NF-ATp, etc. up-regulates the transcription of c-fos, c-myc, and c-jun oncogenes. So-called "early" genes and interleukin-2 (IL-2) gene activation results in IL-2 synthesis. PKC and MAPK kinases Raf and Erk and the nuclear transcription factors AP-1, NF-ATp, NF-kappaB are shared by both TCR/CD3- and sphingolipid-derived signaling. cAMP also may serve as a common target to block the TCR/CD3 signaling.
The free Sph as well as Spha accumulation in lymphocytes was shown to inhibit DNA synthesis and to disrupt lymphocyte proliferation in response to mitogens and T-dependent antigens [21, 46, 47]. Sph has been found to inhibit IL-8-induced and integrin-dependent lymphocyte migration. Sph also prevents both the MHC-restricted and non-restricted cell adhesion, and it inhibits cytotoxic lymphocyte proliferation in response to IL-2 and in mixed lymphocyte culture [48]. Sph, Sph-1-P, and Cer have been shown to exhibit a mitogenic effect in Swiss 3T3 and NIH 3T3 fibroblasts [1, 49]. C2-Cer and Sph decrease the DNA synthesis and T lymphocyte proliferation in response to IL-2 and phorbol esters in contrast to C8-Cer [5, 38, 49]. In normal T lymphocytes and T helper cell clones, the selective activation of cAMP-dependent kinase I type was found to block the signals transducing through TCR/CD3 complex. Cer and Sph increase cAMP activity [50-52], but Sph-1-P inhibits both the adenylate cyclase activity and forskolin-stimulated cAMP accumulation [50, 53]. In transformed Swiss 3T3 fibroblasts, cAMP accumulation may activate the MAPK kinase cascade, G1/S-phase transmission and cell differentiation [1, 54]. Sphingolipid balance defines the proliferation of murine cytotoxic lymphocytes [55]. The mitogenic or suppressive effect of sphingolipids on lymphocyte proliferation is dependent on the cell cycle and additional signal pathways in the cell.
Lymphocyte activation results in additional GlcCer-derived signal transduction pathways. GalCer may promote transfer of GTP on Ras and activate MAPK kinases p44, MEK, and Raf [56]. The immune effect of gangliosides was shown in vitro when GM3 decreased CD4 molecule expression and inhibited Ag-dependent lymphocyte proliferation in response to allogenic stimuli [57]. In contrast to synthetic C2-Cer, endogenous GM3-derived Cer has been shown to exhibit anti-proliferation capacities but not to potentiate apoptosis [57]. GalCer and GlcCer up-regulate NK cell activity. Both, lysophosphatidic acid and lysophosphatidylserine may activate T cell lines via GTP-coupling receptors that leads to increasing [Ca2+]i. Sphingosylphosphorylcholine inhibits T lymphocyte proliferation and IL-2 production [58].
Biosynthesis of GPI-anchored glycoproteins is associated with GlcCer biosynthesis and may be prevented by the inhibitors of ceramide synthase, particularly by fumonisin B1 [59]. In the plasma membrane of resting T cells, both, GPI-anchored glycoproteins and sphingolipids are contained in so-called "GPI-domains" in which SM may physically interact with the membrane domain of receptors, for example, with Thy1,2 antigen. In the GPI-domains, associated src-kinases Lck and Fyn have the optimal conformation required to transmit signals from GPI-anchored receptors [60, 61].
SPHINGOLIPID REGULATION OF T LYMPHOCYTE DIFFERENTIATION
Thymus is a major site for development and selection of most T lymphocytes. The bone marrow-derived blast progenitors pass through a thymus from the medula to the cortex [62, 63]. They interact with epithelium- and dendritic cell-derived factors (IL-1, -2, -7, IFN-gamma, TNF-alpha, TGF-beta, stem cell factor and its receptor c-kit, etc.) which are responsible for thymocyte functional activity in situ and regulate the negative selection [63-66]. The growth factor recognition by thymocytes achieves their development that is characterized, particularly, by transient expression of cell surface glycoproteins--CD-antigens CD2, CD3, CD4, CD5, CD8, CD25, CD44, etc. Blast progenitor development should be ended at the stage CD4+- or CD8+-thymocytes to recognize either foreign or self antigens ( pro-T: CD2+CD3-CD4-CD8-; DN: CD4-CD8-, about 3% of all thymus cells; DP: CD4+CD8+, about 80% of immature thymocytes; and about 15% of thymocytes expressed the mature phenotype, either CD2+CD3+CD4+ or CD2+CD3+CD8+) [62, 67]. Within DN-stage, CD44 Ag expression is down-regulated but CD25 expression is up-regulated. The next transformation into DP-stage requires the synthesis of CD3, TCR-beta, and per-TCR-alpha glycoproteins and their expression on the cell surface [68].
Thymic selection is inherent in DP-stage for CD4+CD8+ thymocytes. Cells bearing the MHC . class molecule are believed to be differentiated into CD8+-lymphocytes, in contrast to cells bearing MHC .. class molecule which should be differentiated into CD4+-cells [62]. The positive selection requires the suppression of programmed cell death unlike the clonal deletion which requires the activation of this program [62, 69]. Thymocyte recognition of self antigen as well as imperfect TCR/CD3 complex may cause apoptosis followed by elimination of this cell by macrophages [63, 70]. Growth factor deprivation, glucocorticoid over-expression, stress, etc. may initiate apoptosis in thymocytes without macromolecule biosynthesis de novo [71].
From 1 to 5% of mature CD4+- and CD8+-thymocytes stream out of the thymus in the peripheral blood, spleen, regional lymph nodes, and non-lymphoid tissues [72]. Extra-thymic sites for T lymphocyte development were found in the intestinal epithelium, epidermis, and testicular epithelium. These T cells express TCR-gammadelta with low affinity. Their differentiation is coupled with unusual PTKs and does not require PTK p56lck [73-75].
Intensive sphingolipid metabolism is unique for the thymus. In mouse thymus, neutral SMase activity per SM quantity is 1.5-fold higher compared with liver [9]. Exogenous Sph, C2-Cer, and fumonisin B1 have been shown to regulate CD3, CD4, and CD8 molecule expression on thymocytes in early stages of the immune response in a dose-dependent manner. In vivo, Sph down-regulates CD3 and CD4 molecule expression but up-regulates the CD8 molecule. In vitro, Sph action was less significant [76]. In vitro, C2-Cer down-regulates CD3, CD4, and CD8 but is less effective compared with Sph [76]. Fumonisin B1 disrupts CD3 and CD4 expression in the same manner as Sph, but its action on CD8 was different from Sph; this is believed to be dependent primarily on Spha accumulation [76]. The common effect of sphingolipids on mouse thymus may be summarized as negative influence on both the thymocyte maturation and cell subpopulation balance.
Rat thymus cell differentiation has been found to be connected with gangliosides GD1c(NeuGc,NeuGc) and GD1b(NeuGc,NeuGc), whose expression is elevated during activation of most CD4+ cells and a small population of CD8+ thymocytes [77].
Consecutive transient glycoprotein expression on the plasma membrane provides the means for all stages of the immune response. Gangliosides, galactocerebrosides, and sulfatides have a common negative influence on CD4 expression [57, 78]. C2-Cer decreases CD4+- and CD8+-lymphocyte number in the peripheral blood [76]. Endogenous Cer depletion in parallel with SMase activation after fumonisin B1 exposure in vivo leads to impaired CD3-glycoprotein expression [9]. CD8 expression was affected more significantly than CD3 on mouse spleen cells after Sph exposure in vivo. In vitro, Sph was found to be most effective for CD8. Under these conditions, C2-Cer down-regulated the CD4+ lymphocytes more effectively than Sph. Fumonisin B1 significantly affects CD4+ and CD8+ lymphocyte subpopulations [76]. Long-chain (sphingoid) bases may interact with glycoproteins of the T cell receptor complex to affect the T cell activation at the early stages of the immune response.
In vitro, exogenous bacterial SMase causes fast and significant modification in CD3, CD4, CD8, and CD25 expression and, in the absence of specific antigen, leads to apoptosis in a dose-dependent manner [79]. On the basis of data that endogenous Cer accumulation under bacterial SMase exposure did not affect the normal lymphocyte proliferation, some different subsets of Cer are believed to be in human T lymphocytes [48].
Under specific Ag recognition, co-receptor CD4 up-regulates the avidity of the complex TCR/Ag/MHC class II molecule and enhances the proliferate response. Differentiation of CD4+-lymphocytes into Th1 or Th2 subsets is following by contact with an antigen presenting cell. This process is genetically restricted and controlled by the density of lymphocyte surface receptors CD4, CD28, MEL-14, and others [80, 81]. After activation, a small subpopulation of CD4+-lymphocytes begins to express the activation-induced phenotype which is intrinsic for memory cells (CD69high, CD45RBlow, CD44high, L-selectin, etc.) [82]. Fumonisin B1 has been found to regulate cell memory formation in response to T-dependent antigens [47].
At the late stages of activation and differentiation, T lymphocytes have been shown to express some sphingomyelin cycle-coupled receptors, namely, TNF-alpha-R55; TNF-beta-R; CD95/Fas/APO-1; IL-1-beta-R; IFN-gamma-R; etc. [83-87]. TNF-alpha-derived signal transduction includes SAPK/JNK kinases and activates the nuclear transcription factor NF-kappaB which is required for T lymphocyte proliferation and IL-2 synthesis [88]. In the normal T lymphocytes, only TNF-alpha provides the signal for NF-kappaB activation but both exogenous Cer or SMase do not. Cer accumulation under acid SMase action is required but not sufficient for NF-kappaB activation [89].
T lymphocyte activation results in the synthesis of IL-2 which takes part in all stages of the immune response and up-regulates the release of other cytokines such as TNF-alpha and IFN-gamma [27, 45, 90]. CD4+Th1-lymphocytes produce IL-2, IL-12, IFN-gamma, TNF-alpha, and TNF-beta. Three last are associated with sphingomyelin cycle products [91]. CD4+Th2 cells produce the interleukins IL-3, -5, -6, -4, -10. The last two interleukins down-regulate the CD4+Th1 subset and its proliferation [92]. TNF-alpha up-regulates IFN-gamma and IL-10 synthesis but down-regulates IL-5 production; this is also favorable for the differentiation of "naive" CD4+ T cells into the Th1 subset. Both, IFN-gamma and IL-4 may affect the T lymphocyte response to IL-12 (a unique regulatory cytokine in the immune network) via their influence on IL-12 receptor affinity [93]. IL-4 is required not only for Th2 development but also for IgG1 synthesis by mitogen-activated B lymphocytes as well as for CD40 antigen expression on activated B cells to block programmed cell death. Sph inhibits DNA synthesis in CD4+Th2-lymphocytes but increases IL-4-R expression and IL-4 synthesis; this results in cell growth arrest in the G1/S-phase and disrupts the balance between Th1/Th2 subsets. Fumonisin B1 also elevates the IL-4-R expression on mouse lymphocytes and simultaneously decreases IL-2-R+-lymphocyte number and antigen density on the cell surface [46, 79]. C2-Cer has been shown to elevate both the IL-6 gene expression and IL-6 release [94]. IL-6 supports "naive" CD4+-cell differentiation into Th2-lymphocytes [95]. IL-6 may be released also by antigen presenting cells and is shared by both so-called "natural" and cellular immune responses. The CD4+ T lymphocyte development during the primary immune response is presented in Fig. 2.
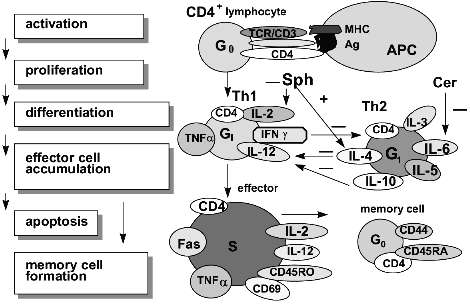
T lymphocyte activation via the TCR/CD3 complex requires the additional signal coupled with the membrane-associated type of acid SMase. This signal is provided by ligation of T cell receptor CD28 and its ligand B7 to initiate the signaling pathway including src-kinases Lck and Fyn, tec-kinase EMT, and then PLC, PI3-kinase, JNK/SAPK cascade followed by the activation of c-jun and NF-kappaB [80, 96, 97]. This prolongs IL-2 production by CD4+ cells, but CD8+ lymphocyte express higher cytolytic activity and undergo apoptosis [81]. Ligand B7.1 activates CD28+CD4+Th1 cells but ligand B7.2 interacts with the CD28+CD4+Th2 subset [98]. The density of MHC .. class molecules on B7.2-bearing cells may direct CD4+-cells toward the Th1 or Th2 subset. This process may be regulated by both, Sph and Cer [98]. The sphingomyelin cycle initiation is a critical point in T lymphocyte activation. The absence of CD28 antigen on the T cell has been shown to prevent proliferation in response to mitogenic stimuli and to block a helper signal for B cells to produce specific antibodies [96]. Nonetheless, these T lymphocyte are allowed for receiving additional signal (which is connected with the sphingomyelin cycle too) under the recognition 4-1BB-receptor/4-1BB-ligand (both are the members of the TNF family) [99].Fig. 2. Stages of CD4+ T lymphocyte development during the primary immune response. The "naive" CD4+ lymphocytes recognize the specific antigen (Ag) in parallel with major histocompatibility complex molecules (MHC) expressed on the antigen presenting cell (APC). T lymphocyte activation causes either CD4+Th1 or CD4+Th2 subset formation. CD4+Th1 cells produce such cytokines as interleukins (IL-2, IL-12), tumor necrosis factors (TNF-alpha, TNF-beta), interferon (IFN-gamma), etc. CD4+Th2 cells produce the interleukins (IL-3, -4, -5, -6, -10) and other cytokines. IL-4 and IL-10 suppress CD4+Th1 cell formation. In contrast, INF-gamma inhibits generation of CD4+Th2 cells. Ceramide (Cer) may modulate T subset development via IL-6 synthesis. Sphingosine (Sph) and fumonisin B1 have been shown to elevate both IL-4 synthesis and IL-4 receptor expression and to decrease IL-2 receptor expression. At the end of the immune response to specific Ag, most lymphocytes undergo apoptosis, and a small part of lymphocytes begins to express the memory cell phenotype.
At early stages of the immune response, specific progenitors undergo proliferation in a special micro-environment of lymphoid tissue followed by differentiation into effector lymphocytes and migration to the blood and non-lymphoid tissues. The latter is strongly dependent on both the specific antigen distribution in the non-lymphoid organs and the density of MHC molecules in situ. Sphingolipids may regulate the migration of activated lymphocytes toward tissues via modification in the expression of molecule adhesion and MHC as well as in the factor of cell migration [85, 100].
The last stages of the immune response are characterized by the expression of TNF-alpha-R, TNF-beta-R, Fas-R, etc. The specific recognition of these receptor results in part in endogenous Cer accumulation and in the activation of programmed cell death--apoptosis [94]. Apoptosis is an active gene-regulated process. The morphological picture includes chromatin condensation, cytoskeleton reorganization, loss of contact with the extracellular matrix, DNA fragmentation and cytoplasm reorganization, and, finally, phagocytosis of the isolated cell compartments without contact of their enzymes with the nearest microenvironment in contrast to necrosis. There are several biochemical types of the apoptosis in immune cells that are dictated by their participation in both, the Ag-dependent recognition and "natural" immunity. For example, the biological significance of Fas-dependent apoptosis is to prevent both unregulated cytokine production and effector cell accumulation. Fas-dependent elimination of CD4+- or CD8+-lymphocytes at the end of the immune response has been found to be initiated by Fas-receptor/Fas-ligand recognition. This is a Cer-dependent Ras-signaling pathway including JNK/SAPK cascade, protease Yama/CPP32, etc. The significant step is the transient synthesis of active O2· forms [101-103]. Fas-dependent apoptosis may be up-regulated during G1A/G1B-phase transmission but may be blocked under the stabilization of cell culture in either G1A- or S-phase of the cell cycle [104].
Fas-R/Fas-L recognition is involved in the immune network and can be promoted by IFN-gamma [84]. In thymus, Fas-dependent apoptosis may cause the almost complete depletion of the cortical thymocytes. Cytotoxic lymphocyte effector function is by way of a Fas-dependent mechanism [105]. Fas-R expression on intact lymphocytes causes them to undergo apoptosis [106]. In contrast to peripheral blood lymphocytes, most lymphocytes from human intestine lamina propria are Fas-R+ and CD45RO+. A small part of lymphocytes from this tissue has been shown to express Fas-L and to be sensitive to Fas-dependent apoptosis initiated by acid SMase [73, 74]. Sph, Spha, and fumonisin B1 increase Fas-R internalization during the first 2 h after T lymphocyte activation that results in apoptosis (E. A. Martinova and A. H. Merrill, submitted for publication).
Sphingolipid metabolism in the T lymphocyte regulates the immune response via lipid second messenger interaction with TCR/CD3-derived signaling. The universal role of sphingolipids in T cells might be explained by the sphingomyelin cycle-coupled receptor expression (CD28, TNF-alpha-R55, TNF-beta-R, Fas-R, etc.) during the main stages of cell activation, proliferation, differentiation, and death. Sphingolipids have been shown to modulate PTP CD45 activity which may regulate the TCR/CD3 signaling, differentiation, and memory cell formation. Furthermore, sphingolipid cross-talking with the cAMP system in lymphocytes (that suppresses the TCR/CD3 signaling in normal cells and up-regulates this in transformed cell lines) may explain both the mitogenic and inhibitory effect of sphingolipids on immune cells. Sphingolipid-derived signaling is associated with the MAPK kinase cascades and with cyclins/cyclin-dependent kinases pairs to regulate the cell cycle phases. Nuclear transcription factor activation is under sphingolipid control in immune cells. Short-term accumulation of sphingolipids in immune cells has been shown to disrupt both the T cell surface antigen expression and subpopulation balance, to suppress the proliferation in response to mitogens and T-dependent antigens as well as to IL-2, and to inhibit both lymphocyte migration and their contacts with extracellular matrix.
The universal negative influence of sphingolipids on normal T lymphocyte activation may be considered as signals of two-way communication to prevent effector cell accumulation and to end the immune response to foreign antigens. The well known inhibitors and activators of sphingolipid metabolism may be used as possible modulators under immune tolerance, anti-cancer immunity, and immune deficiency.
REFERENCES
1.Merrill, A. H., and Schroeder, J. J. (1993)
Annu. Rev. Nutr., 13, 539-559.
2.Merrill, A. H., Lingrell, S., Wang, E.,
Nikolova-Karakashian, M., Vales, T. R., and Vance, D. E. (1995) J.
Biol. Chem., 270, 13834-13841.
3.Lavie, Y., Blusztajn, J. K., and Liscovitch, M.
(1994) Biochim. Biophys. Acta, 1220, 323-328.
4.Inooka, S., and Toyokuni, T. (1996) Biochem.
Biophys. Res. Commun., 218, 872-876.
5.Hauser, J. M., Buehrer, B. M., and Bell, R. M.
(1994) J. Biol. Chem., 269, 6803-6839.
6.Hannun, Y. (1994) J. Biol. Chem.,
269, 3125-3128.
7.Jungner, M., Ohvo, H., and Slotte, J. P. (1997)
Biochim. Biophys. Acta, 1344, 230-240.
8.Ballou, L. R., Laulederkind, S. J., Rosloniec, E.
F., and Raghow, R. (1996) Biochim. Biophys. Acta, 1301,
273-287.
9.Martinova, E. A., Solov’ev, A. S., Khrenov, A.
V., Zabotina, T. N., and Alesenko, A. V. (1995) Biochemistry
(Moscow), 60, 461-465 (Russ.).
10.Furlong, M. T., Mahrenholz, A. M., Kim, K.-H.,
Ashendel, C. L., Harrison, M. L., and Geahlen, R. L. (1997) Biochim.
Biophys. Acta, 1355, 177-190.
11.Katsav, S., Sutherland, M., Packham, G., Yi, T.,
and Weiss, A. (1994) J. Biol. Chem., 269,
32579-32585.
12.Pawson, T. (1995) Nature, 373,
573-580.
13.Claesson-Welsh, L. (1994) J. Biol. Chem.,
269, 32023-32026.
14.Pinna, L. A., and Ruzzene, M. (1996) Biochim.
Biophys. Acta, 1314, 191-225.
15.Ravichandran, K. S., Lee, K. K., Songyang, Z.,
Cantley, L. C., Burn, P., and Burakoff, S. J. (1993) Science,
262, 902-904.
16.Sawasdikosol, S., Ravichandran, K. S., Lee, K.
K., Chang, J. H., and Burakoff, S. J. (1995) J. Biol. Chem.,
270, 2893-2898.
17.Robson, S. C., Siegel, J. B., and Kirsch, R. E.
(1996) Immunol. Cell Biol., 74, 65-71.
18.Shiroo, M., Goff, L., Biffen, M., Shivnan, E.,
and Alexander, D. R. (1992) EMBO J., 11, 4887-4892.
19.Shivnan, E., Clayton, L., Alldridge, L., Keating,
K. E., Gullberg, M., and Alexander, D. R. (1996) J. Immunol.,
157, 101-109.
20.Latinis, K. M., and Koretzky, G. A. (1996)
Blood, 87, 871-875.
21.Martinova, E. A. (1996) Adv. Exp. Med.
Biol., 391, 331-342.
22.Williamson, P., Verhoven, B., and Schlegel, R. A.
(1995) in Lipid Diversity in Membrane and Cellular Functions, La
Londes les Maures, France, Abst. 12.
23.Zwaal, R. F. A., and Schroit, A. J. (1997)
Blood, 89, 1121-1132.
24.Laurenz, J. C., Gunn, J. M., Jolly, C. A., and
Chapkin, R. S. (1996) Biochim. Biophys. Acta, 1299,
146-154.
25.Steed, P. M., Nagar, S., and Wennogle, L. P.
(1996) Biochemistry, 35, 5229-5237.
26.Van Veldhoven, P. P., and Mannaerts, G. P. (1991)
J. Biol. Chem., 266, 12502-12507.
27.Goodemote, K. A., Mattie, M. E., Berger, A., and
Spiegel, S. (1995) J. Biol. Chem., 270, 10272-10277.
28.Jones, M. J., and Murray, A. W. (1995) J.
Biol. Chem., 270, 5007-5013.
29.Lauritzen, L., and Hansen, H. S. (1995)
Biochem. Biophys. Res. Commun., 217, 747-754.
30.Blobe, G. C., Stribling, D. S., Fabbro, D.,
Stabel, S., and Hannun, Y. A. (1996) J. Biol. Chem.,
271, 15823-15830.
31.Jayadev, S., Liu, B., Bielawska, A. E., Lee, J.
Y., Nazaire, F., Pushkareva, M. Yu., Obeid, L. M., and Hannun, Y. A.
(1995) J. Biol. Chem., 270, 2047-2052.
32.Hannum, Y. A., and Linardic, C. M. (1993)
Biochim. Biophys. Acta, 1154, 223-236.
33.Alessi, D. R. (1997) FEBS Lett.,
402, 121-123.
34.Coroneos, E., Martinez, M., McKenna, S., and
Kester, M. (1995) J. Biol. Chem., 270, 23305-23309.
35.Cuvillier, O., Pirianov, G., Kleuser, B., Vanek,
P. G., Coso, O. A., Gutkind, S., and Spiegel, S. (1996) Nature,
381, 800-803.
36.Coroneos, E., Wang, Y., Panuska, J. R.,
Templeton, D. J., and Kester, M. (1996) Biochem. J.,
316, 13-17.
37.Goldstone, S. D., and Hunt, N. H. (1997)
Biochim. Biophys. Acta, 1355, 353-360.
38.Wu, J., Spiegel, S., and Sturgill, T. W. (1995)
J. Biol. Chem., 270, 11484-11488.
39.Su, Y., Rosenthal, D., Smulson, M., and Spiegel,
S. (1994) J. Biol. Chem., 269, 16512-16517.
40.Zhan-rong, L., Hromcha, K., and Bloch, A. (1997)
Biochim. Biophys. Acta, 1356, 149-159.
41.Uchimaru, K., Taniguchi, T., Yoshikawa, M.,
Asano, S., Arnold, A., Fujita, T., and Motokura, T. (1997)
Blood, 89, 965-974.
42.Wang, Q., Worland, P. J., Clark, J. L., Carlson,
B. A., and Sausville, E. A. (1995) Cell. Growth Differ.,
6, 927-936.
43.Obeyesekere, M. N., Herbert, J. R., and
Zimmerman, S. O. (1995) Oncogene, 11, 1199-1205.
44.Dbaibo, G. S., Wolff, R. A., Obeid, L. M., and
Hannun, Y. A. (1995) Biochem. J., 310, 453-459.
45.Berry, D. E., Lu, Y., Schmidt, B., Fallon, P. G.,
O'Connell, C., Hu, S. X., Xu, H. J., and Blanck, G. (1996)
Oncogene, 12, 1809-1819.
46.Martinova, E. A., Ivanchenko, O. B., Morozov, I.
A., Dobrovolskaya, M. A., Bakkes, P., Gelderblom, W. C. A., and
Marasas, W. F. O. (1998) J. Immunol., in press.
47.Martinova, E. A., and Merrill, A. H. (1995)
Mycopathologia, 130, 163-170.
48.Borchardt, R. A., Lee, W. T., Kalen, A., Buckley,
R. H., Peters, C., Schiff, S., and Bell, R. M. (1994) Biochim.
Biophys. Acta, 1212, 327-336.
49.Merrill, A. H., Wang, E., Gilchrist, D. G., and
Riley, R. T. (1993) Adv. Lipid Res., 26, 215-234.
50.Pyne, S., and Pyne, N. J. (1996) Biochem.
J., 315, 917-923.
51.Tamir, A., Granot, Y., and Isakov, N. (1996)
J. Immunol., 157, 1514-1522.
52.Szamel, M., Ebel, U., Uciechowski, P., Kaever,
V., and Resch, K. (1997) Biochim. Biophys. Acta, 1356,
237-248.
53.Van Koppen, C., Meyer, Z., Heringdorf, M., Laser,
K. T., Zhang, C., Jakobs, K. H., Bunemann, M., and Pott, L. (1996)
J. Biol. Chem., 271, 2082-2087.
54.Spiegel, S., Olivera, A., Zhang, H., Thompson, E.
W., Su, Y., and Berger, A. (1994) Breast Cancer Res. Treat.,
31, 337-348.
55.Yatomi, Y., Ruan, F., Megidish, T., Toyokuni, T.,
Hakomori, S., and Igarashi, Y. (1996) Biochemistry, 35,
626-633.
56.Abe, A., Radin, N. S., and Shayman, J. A. (1996)
Biochim. Biophys. Acta, 1299, 333-341.
57.Olshefski, R., Taylor, B., Heitger, A., Hasegawa,
A., and Ladisch, S. (1996) Eur. J. Immunol., 241,
47-55.
58.Xu, Y., Casey, G., and Mills, G. B. (1995) J.
Cell Physiol., 163, 441-450.
59.Zurzolo, C., van't Hof, W., van Meer, G., and
Rodrigues-Buolan, E. (1994) EMBO J., 13, 42-53.
60.Fujimoto, T. (1996) J. Histochem.
Cytochem., 44, 929-941.
61.Arni, S., Ilangumaran, S., van Echten-Deckert,
G., Sandhoff, K., Poincelet, M., Briol, A., Rungger-Brandle, E., and
Hoessli, D. C. (1996) Biochem. Biophys. Res. Commun.,
225, 801-807.
62.Weissman, I. L. (1996) J. Immunol.,
156, 2019-2025.
63.Volkmann, A., Zal, T., and Stockinger, B. (1997)
J. Immunol., 158, 693-706.
64.Gao, Y., Kinoshita, Y., Hato, F., Tominaga, K.,
and Tsuji, Y. (1996) Cell. Mol. Biol. Noisy. Grand, 42,
227-234.
65.Yasuda, Y., Nishijima, I., Watanabe, S., Arai,
K.-i., Zlotnik, A., and Moore, T. A. (1997) Blood, 89,
1349-1356.
66.Delgado, M., Garrido, E., Martinez, C., Leceta,
J., and Gomariz, R. P. (1996) Blood, 87, 5152-5161.
67.Tourigny, M. R., Mazel, S., Burtrum, D. B., and
Petrie, H. T. (1997) J. Exp. Med., 185, 1549-1556.
68.Shibata, S., Asano, T., Ogura, A., Hashimoto, N.,
Hayakawa, J., Naiki, M., and Doi, K. (1997) Immunol. Cell Biol.,
75, 154-160.
69.Nakayama, K., Nakayama, K.-i., Nagishi, I.,
Kuida, K., Louie, M. C., Kanagawa, O., Nakauchi, H., and Loh, D. Y.
(1994) Science, 263, 1131-1133.
70.Chen, J. J. (1993) Immunol. Today,
14, 126-130.
71.Gabai, V. L., Mosina, V. A., Makarova, I. M.,
Maliutina, I. V., Budagova, K. R., and Mosin, A. F. (1995)
Biochemistry (Moscow) 60, 1201-1208 (Russ.).
72.Gretz, J. E., Kaldjian, E. P., Anderson, A. O.,
and Shaw, S. (1996) J. Immunol., 157, 495-499.
73.Kirkham, P. A., Takamatsu, H. H., and Parkhouse,
R. M. (1997) Eur. J. Immunol., 27, 717-725.
74.Spinozzi, F., Nicoletti, I., Agea, E., Beli, S.,
Moraca, R., Migliorati, G., Riccardi, C., Grignani, F., and Bertotto,
A. (1995) Immunology, 86, 379-384.
75.Fujise, S., Matsuzaki, G., Kishihara, K., Kadena,
T., Molina, T., and Nomoto, K. (1996) J. Immunol., 157,
247-254.
76.Martinova, E. A. (1997) in Animal Cell
Technology (Carrondo, M. J. T., Griffiths, B., and Moreira, J. L.
P., eds.) Kluwer Academic Publishers, Dordrecht, pp. 625-632.
77.Nohara, K., Ozawa, H., Tai, T., Saji, H., and
Fujimaki, H. (1997) Biochim. Biophys. Acta, 1345,
207-214.
78.Tamma, S. L., Sundaram, S. K., Lev, M., and
Coico, R. F. (1996) Biochem. Biophys. Res. Commun., 220,
916-921.
79.Martinova, E. A. (1995) in Lipid Diversity in
Membrane and Cellular Functions, La Londes les Maures, France,
Abst. 20.
80.Noel, P. J., Boise, L. H., Green, J. M., and
Thompson, C. B. (1996) J. Immunol., 157, 636-642.
81.Deeths, M. J., and Mescher, M. F. (1997) Eur.
J. Immunol., 27, 598-608.
82.Muralidhar, C., Koch, S., Broome, H. E., and
Swain, S. L. (1996) J. Immunol., 157, 625-635.
83.Wu, M. X., Zhaohui, A., Hegen, M., Morimoto, C.,
and Schlossman, S. T. (1996) J. Immunol., 157,
707-713.
84.Maciejewski, J. P., Selleri, C., Anderson, S.,
and Young, N. S. (1995) Blood, 85, 3183-3188.
85.Masamune, A., Igarashi, Y., and Hakomori, S.
(1996) J. Biol. Chem., 271, 9368-9375.
86.Nagata, S. (1997) Cell, 88,
355-365.
87.Chen, J., Nikolova-Karakashian, M., Merrill, A.
H., and Morgan, E. T. (1995) J. Biol. Chem., 270,
25233-25238.
88.Cogswell, P. C., Mayo, M. W., and Baldwin, A. S.
(1997) J. Exp. Med., 185, 491-497.
89.Higuchi, M., Singh, S., Jaffrezou, J.-P., and
Aggarwal, B. B. (1996) J. Immunol., 156, 297-304.
90.Kelly, K., and Siebenlist, U. (1995) Curr.
Opin. Immunol., 7, 327-332.
91.Chen, M., Quintans, J., Fuks, Z., Thompson, C.,
Kufe, D. W., and Weichselbaum, R. R. (1995) Cancer Res.,
55, 991-994.
92.Ahmad, S., Choudhry, M. A., Shankar, R., and
Sayeed, M. M. (1997) FEBS Lett., 4021, 213-218.
93.Gollod, J. A., Kawasaki, H., and Ritz, J.
(1997) Eur. J. Immunol., 27, 647-652.
94.Laulederkind, S. J., Bielawska, A., Raghow, R.,
Hannun, Y. A., and Ballou, L. R. (1995) J. Exp. Med.,
182, 599-604.
95.Rincon, M., Anguita, J., Nakamura, T., Fikrig,
E., and Flavell, R. A. (1997) J. Exp. Med., 185,
461-470.
96.King, P. D., Sadra, A., Teng, J. M. C., Liu,
X.-R., Han, A., Selvakumar, A., August, A., and Dupont, B. (1997) J.
Immunol., 158, 580-590.
97.Boucher, L. M., Wiegmann, K., Futterer, A.,
Pfeffer, K., Machleidt, T., Schutze, S., Mak, T. W., and Kronke, M.
(1995) J. Exp. Med., 181, 2059-2068.
98.Wakita, H., Tokura, Y., Yagi, H., Nishimura, K.,
Furukawa, F., and Takigawa, M. (1994) Arch. Dermatol. Res.,
286, 350-354.
99.DeBenedette, M. A., Shahinian, A., Mak, T. W.,
and Watts, T. H. (1997) J. Immunol., 158, 551-559.
100.Hidari, K. P., Ichikawa, S., Fujita, T.,
Sakiyama, H., and Hirabayashi, Y. (1996) J. Biol. Chem.,
271, 14636-14641.
101.Sytwu, H.-K., Liblau, R. S., and McDevitt, H.
O. (1996) Immunity, 5, 17-23.
102.Gulbins, E., Bissonnette, R., and Mahboubi, A.
(1995) Immunity, 2, 341-351.
103.Um, H. D., Orenstein, J. M., and Wahl, S. M.
(1996) J. Immunol., 156, 3469-3477.
104.Komada, Y., Zhou, Y. W., Zhan, X. L., Xue, H.
L., Sakai, H., Tanaka, S., Sakatoku, H., and Sakurai, M. (1995)
Blood, 86, 3848-3860.
105.Heinkelein, M., Pilz, S., and Jassoy, C. (1996)
Clin. Exp. Immunol., 103, 8-14.
106.Laurence, J., Mitra, D., Steiner, M., Lynch, D.
H., Siegal, F. P., and Staiano-Coico, L. (1996) J. Clin.
Invest., 97, 672-680.