Isolation and Primary Characterization of an Amidase from Rhodococcus rhodochrous
E. K. Kotlova*, G. G. Chestukhina, O. B. Astaurova, T. E. Leonova, A. S. Yanenko, and V. G. Debabov
Institute of Genetics and Selection of Industrial Microorganisms, 1-yi Dorozhnyi Proezd 1, Moscow, 113545 Russia; fax: (095) 315-0501; E-mail: lab11@vnigen.msk.su* To whom correspondence should be addressed.
Received November 11, 1998
Amidase (EC 3.5.1.4) was purified to homogeneity from Rhodococcus rhodochrous M8 using isopropanol fractionation and exchange chromatography on Mono Q. The isolated amidase consists of four identical subunits with molecular weight 42 ± 2 kD. The activity of the enzyme is maximal at 55-60°C and within the pH range 5-8. The amidase from R. rhodochrous M8 is highly sensitive to such sulfhydryl reagents as Hg2+ and Cu2+. Chelators (EDTA and o-phenanthroline) and serine proteinase inhibitors (PMSF and DIFP) did not inhibit the activity of the enzyme. The enzyme exhibits hydrolytic and acyl transferase activity and does not possess urease activity. Aliphatic amides (acetamide and propionamide) were the best substrates for the amidase from R. rhodochrous M8, whereas bulky aromatic amides were poor substrates of this enzyme. The properties of the isolated enzyme are similar to those found in the corresponding amidase from Arthrobacter sp. J-1 and an amidase with wide substrate specificity from Brevibacterium sp. R312.
KEY WORDS: Rhodococcus rhodochrous, amidase, purification, properties, substrate specificity
Abbreviations: DIFP) diisopropyl fluorophosphate; DTT) dithiothreitol; PMSF) phenylmethylsulfonyl fluoride.
Acylamide amidohydrolases (amidases, EC 3.5.1.4) catalyze hydrolysis of
carboxylic acid amides to free carboxylic acids and free ammonium [1]. These enzymes are involved in nitrogen metabolism
in cells and are widely distributed in nature: they have been found in
procaryotic and eucaryotic cells. These enzymes are often found in
bacteria related to the group of nocardia-like actinomycetes (nocardia,
rhodococci, arthrobacteria). Amidases isolated from various sources [2-5] are characterized by distinct
substrate specificities. Some of them catalyze hydrolysis of amides of
aliphatic acids [2, 6], others
cleave amides of aromatic acids [4, 7], and still others hydrolyze amides of alpha-
or omega-amino acids [5, 8]; some amidases are stereoselective [4, 7]. In spite of significant
progress in expanding our knowledge of amidases, the spatial
organization of these proteins remains unknown and systematic
comparative analysis of structural--functional organization of amidases
is far from complete.
Among bacterial enzymes, amidases from Brevibacterium sp. R312,
Rhodococcus rhodochrous J1, and Pseudomonas aeruginosa
are the best studied enzymes of this type. Native enzymes consist of a
few identical subunits and are obviously related to the class of
sulfhydryl enzymes [2, 5]. They
exhibit two types of activity--amidohydrolase (1) and amidotransferase
(2) [6, 9]:
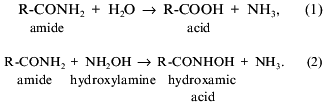
Based on data on sulfhydryl-group inhibition and kinetic analysis, amidase was suggested to catalyze by the “ping-pong” mechanism [10]. However, this mechanism is now strongly criticized [11].
The amidase of R. rhodochrous which we are studying is involved in nitrile metabolism in strain R. rhodochrous M8, which is employed as a biocatalyst in the industrial production of acrylamide. This microorganism was isolated from soil by scientists of our institute. It grows utilizing nitrile via the coupled action of nitrile hydratase (EC 4.2.1.84), which converts nitriles into corresponding amides, and the amidase hydrolyzing amides to free acids. In contrast to the well-studied nitrile hydratase from this strain, the spatial organization and properties of the amidase remain unknown.
In the present report we describe a method for isolation and purification of amidase from a mutant of R. rhodochrous M8, and we also compare some physicochemical properties of amidases from various sources.
MATERIALS AND METHODS
PMSF, DTT, and DIFP were purchased from Sigma (USA).
Isolation of amidase from R. rhodochrous. Biomass of the mutant strain R. rhodochrous M50 obtained from the wild strain R. rhodochrous M8 [12] was used as a starting material for the purification of the enzyme. The biomass was grown on MB medium containing (g/liter): KH2PO4, 0.5; K2HPO4, 0.5; MgSO4·7H2O, 0.5; FeSO4·7H2O, 0.005; pH 7.2. Glucose (20 g/liter) and urea (5 g/liter) were used as the sources of carbon and nitrogen, respectively. The cultivation was carried out in flasks (medium volume 150 ml) for 72 h at 30°C with constant shaking. The biomass was collected by centrifugation.
All stages of purification were carried out at 4°C.
Biomass (80 g) was suspended in 360 ml of 50 mM sodium phosphate buffer containing 1 mM MgCl2, 0.5 mM EDTA, and 1 mM DTT; cells were separated by centrifugation at 2,500 rpm for 30 min. The pellet was resuspended in 360 ml of the same buffer and disintegrated using an extrusion cell disintegrator (KB AIKhF, Russia) at 1,500 atm. The suspension was then centrifuged at 5,000 rpm for 30 min and the clear supernatant was used in the next stage.
For removal of contaminating proteins, 340 ml of the supernatant was mixed with 146 ml of isopropanol (final concentration 30%), stirred for 1 h using a magnetic stirrer, and left for 10-16 h for precipitate formation; the precipitate was separated by centrifugation as described above. The procedure was repeated at 40% and then at 50% isopropanol concentration. The precipitate obtained with 50% isopropanol contained the amidase; it was dissolved in 30 mM Tris-HCl buffer, pH 7.4.
Ion-exchange chromatography was carried out using a chromatograph for fast protein chromatography (Pharmacia, Sweden). An aliquot of the resulting solution (11 ml) was filtered through a lavsan filter (0.22 µm), and the filtrate was then applied onto an anion-exchange Mono Q HR 5/5 column equilibrated with 30 mM Tris-HCl buffer, pH 7.4. Chromatography was carried out using a gradient of NaCl concentrations (from 0 to 1 M during 30 min) at the flow rate 0.4 ml/min. Fractions containing amidase activity were collected and analyzed by PAGE.
The electrophoresis was carried out in a 10% polyacrylamide gel in the presence of 0.1% SDS at 50 mA and 150 V using the Laemmli method [13]. The sample (10-50 µl, 1 mg/ml) obtained by dissolution of the protein in 8 M urea containing 10 mM sodium phosphate buffer, pH 7.5, 1% SDS, and 1% mercaptoethanol was placed on the gel. Protein bands were stained with 0.25% solution of Coomassie Blue R-250, and the excess dye was washed out using 7% acetic acid at 100°C.
Transferase activity of the amidase was determined by formation of acetylhydroxamic acid from hydroxylamine and acetamide with subsequent photometric measurement of the colored complex of acetylhydroxamic acid with FeCl3 [14]. One unit of activity corresponds to the amount of the enzyme that converts 1 µmole of substrate at 37°C during 1 min.
Hydrolytic activity was determined by measuring ammonia liberation by Nessler's method [15] or using indophenol [16].
Homogeneity and molecular mass of amidase subunits were determined by electrophoresis in 10% polyacrylamide gels in the presence of 0.1% SDS [13]. Bovine serum albumin (BSA, 66 kD), ovalbumin (45 kD), glyceraldehyde-3-phosphate dehydrogenase (36 kD), carbonic anhydrase (29 kD), trypsinogen (24 kD), and soybean trypsin inhibitor (20.1 kD) were used as molecular mass markers.
The subunit composition of native amidase was evaluated by gel-filtration on a Superose 12 column using a moderate pressure chromatograph (Pharmacia). The flow rate was 0.4 ml/min. BSA (66 kD), ferritin (480 kD), catalase (240 kD), and aldolase (147 kD) were used as the standards.
The amino acid composition was determined using a Biotronik LC 5001 amino acid analyzer (Germany).
For investigation of enzymatic properties, the amidase (1 mg/ml) was dissolved in 0.05 M Tris-HCl buffer, pH 7.4.
The influence of metal ions and inhibitors on transferase and/or hydrolytic activities was investigated after preincubation of the enzyme with 5 mM reagent solutions at 22°C for 10 min. The amidase was preincubated with DIFP, PMSF, or o-phenanthroline (1 mM in each case) at 22°C for 1 h. For determination of the enzyme activity, an aliquot of preincubated enzyme solution was diluted 5-fold with reaction mixture so that the final concentration of the tested reagent was 1 mM (in the case of o-phenanthroline, it was added to the reaction mixture to final concentration of 1 mM). Aqueous 0.01 M solution of o-phenanthroline containing 10% DMFA, 0.5 M PMSF in absolute dioxane, and 3% DIFP in isopropanol were the stock solutions.
The dependence of amidase hydrolytic activity on pH was determined using the indophenol method after preincubation with the required pH for 1 h. Phosphate--citrate (pH 2.2-8.0) and Tris-HCl buffers (pH 7.6-8.8) were used in the studies of pH dependence.
The temperature dependence of the hydrolytic activity of the amidase was determined at pH 7.4 using the indophenol method after preincubation at the required temperature for 20 min.
RESULTS AND DISCUSSION
Many microorganisms utilize organic nitriles as sources of nitrogen and carbon. Microbial enzymes metabolizing nitrile-containing compounds are now used for industrial production of acrylamide and nicotinamide [17, 18]. The use of stereoselective amidases for a large-scale production of optically pure organic acids from their racemic derivatives seems very promising.
Strain R. rhodochrous M8, widely used in the industrial production of acrylamide, produces nitrile hydratase and amidase. In earlier studies [12, 19], we characterized some properties of the nitrile hydratase. In the present study we isolated and purified the coupled enzyme, amidase, and investigated its properties.
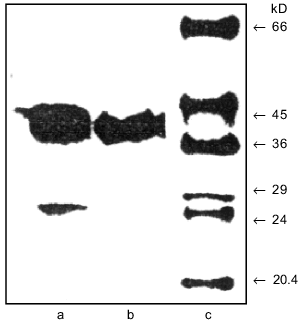
Isolation and purification of the amidase. Biomass of the mutant R. rhodochrous M8 (named M50) was used as the starting material for amidase isolation. In contrast to the wild strain, this mutant constitutively synthesizes amidase and does not synthesize nitrile hydratase. The purification procedure was monitored by measuring transferase activity as it is less sensitive to the composition of the reaction mixture and, in particular, to the presence of metal ions that influence the colorimetric determination of ammonium ions. Table 1 lists the purification stages and Fig. 1 shows the electrophoregram illustrating purification of the amidase. Fractionation of the cell homogenate with isopropanol resulted in ~20-fold purification and high yield of the amidase (Fig. 1a).
Table 1. Amidase isolation from 80 g of
R. rhodochrous M8 cells
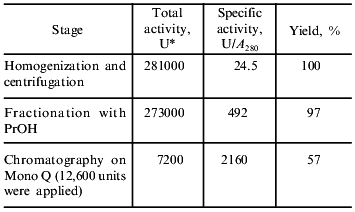
*Activity was determined by the transferase reaction. One unit
corresponds to the amount of enzyme which converts 1 µmole of
substrate per min at 37°C.
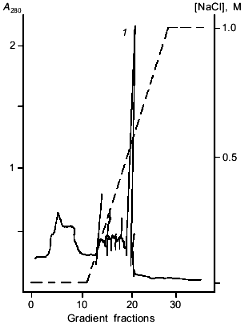
Using fast protein chromatography on Mono Q (Fig. 2), the preparation containing homogenous amidase was isolated from the amidase-enriched fraction (Fig. 1b). Specific transferase activity of the resulting enzyme was 2160 µmoles/A280 per min; this corresponds to 88-fold purification.Fig. 1. SDS-PAGE in 10% polyacrylamide gel demonstrating amidase purification: a) amidase enriched preparation after isopropanol fractionation; b) enzyme preparation after chromatography on a Mono Q column; c) standard proteins. BSA (66 kD), ovalbumin (45 kD), glyceraldehyde-3-phosphate dehydrogenase (36 kD), carbonic anhydrase (29 kD), trypsinogen (24 kD), and soybean trypsin inhibitor (20.1 kD) were used as standards.
Subunit composition. Almost all amidases studied have quaternary structure and consist of an even number of identical subunits. However, the size of subunits and their number vary over a wide range. For example, an enanthioselective 2-aryl-propionamide-specific amidase from Brevibacterium sp. R312 is a homodimer (2 x 54.671 kD) [4], whereas the amidase from R. erythropolis MP50 with similar specificity consists of eight subunits with molecular mass of 61 kD [7].Fig. 2. Chromatography of the amidase-enriched preparation on a Mono Q column. The amidase-containing peak is designated by number 1.
Polyacrylamide gel electrophoresis revealed that the amidase from R. rhodochrous M8 consists of identical subunits with molecular mass of 42 ± 2 kD. Gel-filtration on a Superose 12 column revealed that the molecular mass of the native protein is 150 ± 20 kD; this corresponds to four subunits in the native enzyme molecule. A heteromeric molecule (4 x 43 kD) of an amidase with wide specificity from Brevibacterium sp. R312 [6] has similar sizes. Similar subunit sizes were previously reported for aliphatic amidases from Arthrobacter sp. J1 (8 x 39.0 kD) [2] and P. aeruginosa (6 x 38.4 kD) [5, 20]. The amidase from R. rhodochrous M8 is shorter by 150 amino acid residues than the amidase from another studied strain, R. rhodochrous J1 [3]. Both strains can utilize nitriles using nitrile hydratases and amidases. In contrast to the amidases, the nitrile hydratases are almost identical in the two strains [21].
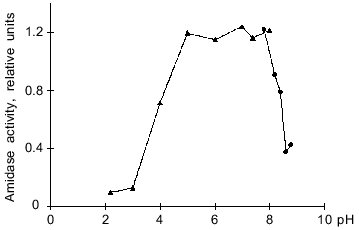
Optimal pH for enzyme activity. The optimal pH was determined by assaying hydrolytic activity in the pH range 2.2-8.8. The amidase was highly active within a rather wide pH range, from 5 to 8 (Fig. 3). Beyond these pH limits the enzyme activity is sharply decreased. For example, at pH 3.7 and 8.5 the enzyme exhibits 50% of its activity. The behavior of pH-dependence curve and the pH optimum are the same as for the amidase from Arthrobacter sp. J-1 [2].
Temperature optimum. The temperature optimum for the amidase activity lies within the range 55-60°C (Fig. 4). At 37°C the enzyme exhibits only 34% of its maximal activity. In spite of various subunit composition and specificity, studied amidases are most active at relatively high temperatures, about 55°C [2, 5-7, 22]. The only exception is a peptide amidase from Stenotrophomonas maltophilia [8] possessing maximal activity at 39-45°C. However, this enzyme also differs from the group of considered hydrolases in other characteristics.Fig. 3. Dependence of the hydrolytic activity of the amidase on pH. Citrate--phosphate (pH 2.2-8.0) and Tris-HCl buffers (pH 7.6-8.8) were used in these experiments. The amidase activity is expressed in relative units that correspond to optical density of the solution at lambda = 590 nm.
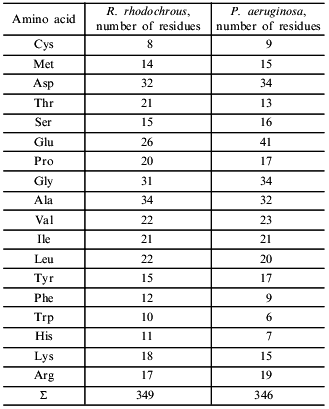
Amino acid composition. The amino acid composition of the amidase is characterized by a predominance of acidic residues over basic residues (Table 2). A similar ratio was found for the amidase from P. aeruginosa [20], which contains about 1.5-fold more acidic residues. The content of hydrophobic amino acid residues forming the protein core was also similar.Fig. 4. Temperature dependence of the hydrolytic activity of the amidase at pH 7.4. Amidase activity is expressed in relative units that correspond to optical density of thee solution at lambda = 590 nm.
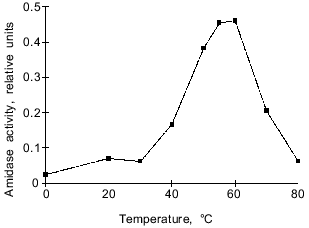
Table 2. Amino acid composition of amidases
from R. rhodochrous M8 and P. aeruginosa [20]
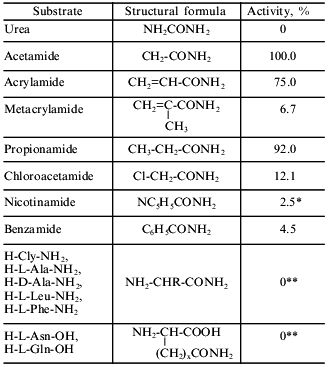
Substrate specificity. The substrate specificity of the amidase from R. rhodochrous M8 was determined by hydrolysis of amides of organic acids. The ability of the enzyme to hydrolyze amides and omega-amides of alpha-amino acids was also tested. Table 3 shows that amides of aliphatic organic acids (acetamide and propionamide) were the best substrates for the amidase from R. rhodochrous M8. The presence of a double bond near the hydrolyzing group markedly decreased enzyme activity (the rate of acrylamide hydrolysis was 75%), whereas the presence of a methyl radical at the alpha-carbon atom in the case of metacrylamide decreased hydrolysis of the amide bond by one order of magnitude (to 6.7%). The enzyme hydrolyzes bulky aromatic amides (at low rates), and it does not possess urease activity. The amidase from R. rhodochrous M8 does not cleave amides of alpha-amino acids of either L- and D-configuration. omega-Amides of alpha-amino acids are not amidase substrates as well. Even the relatively small amide of aminoacetic acid is not a substrate for this enzyme, in contrast to acetamide. Apparently the presence of a protonated NH2 group (in the working pH range, ~7.5) at the Calpha-carbon atom prevents substrate binding to enzyme and/or the proceeding of the catalytic cycle.
Table 3. Substrate specificity of the
amidase from R. rhodochrous M8. The activity was determined by
the indophenol reaction
*Activity was determined by the reaction with Nessler's reagent.
**Determined using an amino acid analyzer.
Comparison of the present results with data from the literature places the amidase from R. rhodochrous M8 in the group of aliphatic amidases. Amidases with similar specificities were isolated from Arthrobacter sp. J-1 [2], Aspergillus nidulans [5], Pseudomonas sp. [22], and Brevibacterium sp. R312 [5, 23]. For example, an amidase with “wide substrate specificity” from Brevibacterium sp. R312, like the enzyme described here, hydrolyzes benzamide one order of magnitude slower than acetamide. However, for the enzyme from Brevibacterium sp. R312 propionamide is the best substrate. In contrast to the amidase from R. rhodochrous M8, the amidase with “wide substrate specificity” hydrolyzes D-alpha-aminopropionamide (D-Ala-NH2) and beta-aminopropionamide. However, Maestracci et al. [5] suggest that the latter may be due to the presence of traces of membrane bound L-alpha-aminoamidase in the enzyme preparation.
Influence of metal ions and inhibitors on the activity of the amidase (Table 4). Heavy metal ions (characteristic sulfhydryl reagents) and Fe3+ inhibit the enzyme completely. Chelators (EDTA and o-phenanthroline) do not influence the enzyme activity. The latter fact obviously suggests that this is not a metal-dependent enzyme. Serine proteinase inhibitors (PMSF and DIFP) also do not inhibit the enzyme. The presence of DTT causes a ~1.5-fold increase in the amidase activity. These data suggest an important role of a sulfhydryl group in the mechanism of action of the enzyme. The presence of a sulfhydryl group essential for catalysis was noted in most studies, irrespectively of enzyme source (amidases from Corynebacterium sp. C5 [20] and S. maltophilia [8] were exceptions). The amidases are generally thought to be sulfhydryl-containing enzymes. However, recent data [11] on the active site of an amidase from R. rhodochrous J1 questions this viewpoint. Kobayashi et al. [11] studied the influence of substitutions Cys203 --> Ala, Asp191 --> Glu, Asp191 --> Asn, and Ser195 --> Ala on the enzyme activity; they also revealed some homology of primary structures of amidases and aspartyl proteinases. These data suggest that amidases might belong to a class of aspartyl enzymes; however, this suggestion requires further confirmation.
Table 4. Influence of metal ions and
inhibitors on the activity of the amidase
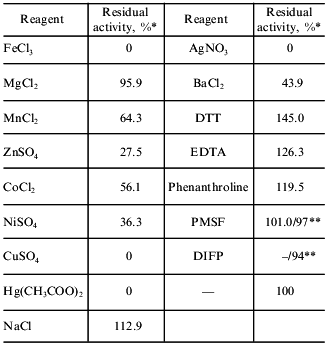
*Activity was determined by the transferase reaction.
**Numbers in the denominator indicate the residual hydrolase activity
determined by the indophenol method.
Our study provides a good basis for further detailed study of the amidase from R. rhodochrous M8. We have obtained enough of the enzyme for crystallization and analysis of its spatial structure.
We thank L. P. Revina and I. A. Zalunin for their advice and skillful help.
REFERENCES
1.Short Chemical Encyclopedia (1961) Vol. 1,
Sovetskaya Entsiklopediya, Moscow, p. 165.
2.Asano, Y., Tashibana, M., Tani, Y., and Yamada, H.
(1982) Agric. Biol. Chem., 46,
1175-1181.
3.Kobayashi, M., Komeda, H., Nagasawa, T., Nishiyama,
M., Horinouchi, S., Beppu, T., Yamada, H., and Shimizu, S. (1993)
Eur. J. Biochem., 217, 327-336.
4.Mayaux, J.-F., Cerbelaud, E., Soubrier, F.,
Faucher, D., and Petre, D. (1990) J. Bacteriol., 172,
6764-6773.
5.Maestracci, M., Bui, K., Thiery, A., Arnaud, A.,
and Galzy, P. (1988) Adv. Biochem.
Eng./Biotechnol., 36, 67-115.
6.Thiery, A., Maestracci, M., Arnaud, A., Galzy, P.,
and Nicolas, M. (1986) J. Basic Microbiol.,
26, 299-311.
7.Hirrlinger, B., Stolz, A., and Knackmuss, H.-J.
(1996) J. Bacteriol., 178, 3501-3507.
8.Stelkes-Ritter, U., Wyzgol, K., and Kula, M.-R.
(1995) Appl. Microbiol. Biotechnol., 44,
393-398.
9.Thiery, A., Maestracci, M., Arnaud, A., Galzy, P.,
and Nicolas, M. (1986) J. Gen. Microbiol.,
132, 2205-2208.
10.Maestracci, M., Thiery, A., Arnaud, A., and
Galzy, P. (1986) Agric. Biol. Chem., 50,
2237-2241.
11.Kobayashi, M., Fujiwara, Y., Goda, M., Komeda,
H., and Shimizu, S. (1991) Proc. Natl. Acad. Sci.
USA, 94, 11986-11991.
12.Yanenko, A. S., Astaurova, O. B., Gerasimova, T.
V., Polyakova, I. N., Pogorelova, T. E., and Paukov, V. N. (1995)
Proc. Ninth Symp. on the Actinomycetes, pp.139-144.
13.Laemmli, U. K. (1970) Nature, 227,
680-685.
14.Brammar, W. J., and Clarke, P. H. (1964) J.
Gen. Microbiol., 37, 307-319.
15.Gerhardt, F., et al. (eds.) (1984) Methods of
General Bacteriology [Russian translation] Vol. 2, Mir, Moscow, pp.
355-356.
16.Weatherburn, M. W. (1967) Anal. Chem.,
39, 971-974.
17.Kobayashi, M., Komeda, H., Nagasawa, T., Yamada,
H., and Shimizu, S. (1993) Biosci. Biotech. Biochem., 57,
1949-1950.
18.Nagasawa, T., and Yamada, H. (1991) in
Biocatalysis (Abramovich, D. A., ed.) Van Nostrand Reinhold, New
York, pp. 277-318.
19.Sinolitskii, M. K., Poltavskaya, S. V.,
Rogatcheva, S. M., Sevryugina, I. V., and Voronin, S. P. (1997)
Prikl. Biokhim. Mikrobiol., 33, 383-387.
20.Ambler, R., Auffret, A., and Clarke, P. (1987)
FEBS Lett., 215, 285-290.
21.Veiko, V. P., Yanenko, A. S., Alekseeva, M. G.,
Sintin, A. A., Gul'ko, L. B., Ratmanova, K. I., Ovcharova, I. V.,
Andreeva, L. B., Astaurova, O. B., Polyakova, I. N., Paukov, V. N.,
Voronin, S. P., and Debabov, V. G. (1995) Biotekhnologiya,
5/6, 3-5.
22.Tani, Y., Kurihara, M., and Nishise, H. (1989)
Agric. Biol. Chem., 53, 3151-3158.
23.Soubrier, F., Levy-Schil, S., Mayaux, J.-F.,
Petre, D., Arnaud, A., and Crouzet, J. (1992) Gene, 116,
99-104.