Induction of Nonselective Permeability of the Inner Membrane in Deenergized Mitochondria
V. N. Dedov, O. V. Demin, V. Ya. Chernyak, and B. V. Chernyak*
Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-3181; E-mail: chernyak@pcman.genebee.msu.su* To whom correspondence should be addressed.
Received November 5, 1998; Revision received December 7, 1998
Induction of the nonselective cyclosporin-sensitive pore in the inner mitochondrial membrane under conditions of complete dissipation of ion gradients and transmembrane potential was studied. This approach allows the kinetics of Ca2+-dependent pore opening and the preceding processes of induction to be studied separately. The effects of mitochondrial heterogeneity were also minimized. We found that the kinetics of pore opening can be described by a minimal two-step scheme where only the rate constant at the first step depends on Ca2+ concentration. Oxidation of pyridine nucleotides in the matrix caused a slow transition in the pore complex and decreased the apparent dissociation constant of the Ca2+-binding site from >1 mM to ~30 µM. N-Ethylmaleimide (but not disulfide-reducing agents) prevented and slowly reverted the pore induction process. Data suggesting allosteric modulation of the pore by pyridine nucleotides are presented.
KEY WORDS: mitochondria, cyclosporin-sensitive pore, pyridine nucleotides
Abbreviations: DQ) duroquinone; HEDTA) N'-(2-hydroxyethyl)ethylenediamine-N,N,N'-triacetate; NEM) N-ethylmaleimide; CCCP) carbonyl cyanide m-chlorophenylhydrazone; FCCP) carbonyl cyanide p-trifluoromethoxyphenylhydrazone; PEG2000) polyethylene glycol with molecular mass ~2000 daltons; CsA) cyclosporin A.
Increase in the nonselective permeability («permeability
transition») of the inner mitochondrial membrane was long
attributed to nonspecific damage caused by lipid oxidation and
lipolysis. Later, however, convincing evidence for a specific mechanism
of the permeability transition was presented. It was shown that during
the transition a proteinaceous aqueous pore with 3-nm effective
diameter and cut-off molecular weight of 1500 daltons was opened in the
membrane. The probability of pore opening is increased by a decrease in
membrane potential, by an increase in Ca2+ concentration in
the matrix, by the oxidation of various mitochondrial components, and
also by agents of different nature («pore inducers») with
unknown mechanism of action. The pore opening is prevented by decrease
of pH, by increase of ADP and Mg2+ concentration, by the
immunosuppressive oligopeptide cyclosporin A (CsA), and by other less
specific inhibitors. In some cases the inhibitors can induce closure of
the pore.
The molecular structure of the pore is not known. It probably consists of cyclophilin (CsA-binding matrix protein), adenine nucleotide translocase (from the inner membrane), and a voltage-dependent anion channel (VDAC, from the outer mitochondrial membrane). This means that the pore may be localized in contact sites between the two mitochondrial membranes. An extensive review on the mitochondrial permeability transition pore (PTP) was recently published [1].
The mechanism of PTP formation has attracted more interest after indications of its role in important cellular reactions, Ca2+ signaling [2], and programmed cell death (apoptosis) [3] were found. Studies of this phenomenon are difficult because of the great number of factors affecting the pore opening and their complicated interplay. The «traditional approach» includes the loading of mitochondria Ca2+, addition of pore inducer or inhibitors, and monitoring permeability by the efflux of K+, Ca2+ or Mg2+ from the matrix, by the decrease of the membrane potential, or by swelling of mitochondria caused by entry of osmotically active substances through the pore. This method does discriminate between direct effects on the pore and secondary effects caused by changes in membrane potential, matrix pH, etc. Analysis of data is also hampered by pore-inducing effect of matrix components (especially Ca2+) which are released through open pores. These difficulties can be bypassed in studies of Ca2+-dependent pore opening in deenergized mitochondria, where all ion gradients and transmembrane potentials are dissipated by ionophores. Pore inducers strongly increase the efficacy of Ca2+ under these conditions [4, 5]. Using this approach, the kinetics of Ca2+-dependent pore opening and preceding processes induced by oxidation of pyridine nucleotides in the matrix were studied separately in the present work.
MATERIALS AND METHODS
Rat liver mitochondria were isolated as described [6]. The isolation medium additionally contained 0.25 mM EGTA, which was omitted from the last washing. The assay medium contained 200 mM sucrose, 20 mM Tris-Mops, pH 7.4, 20 µM EGTA, 2 µM rotenone, 1 µM A23187, and 5 µM CCCP. The mitochondrial protein concentration was 0.6 mg/ml. In experiments where shrinkage was measured (Fig. 5), the medium contained 40 mM sucrose, 20 mM Tris-Mops, 20 µM EGTA, 2 µM rotenone, 5 mM succinate, and 1 mM KPi, pH 7.4. The mitochondrial protein concentration was 1 mg/ml. Total Ca2+ concentration in the media was measured by atomic absorption and free Ca2+ concentration in the presence of EGTA or HEDTA (Fig. 5) was calculated using standard software [7]. All measurements were done at 25°C with constant stirring.
Light scattering was measured as the optical density of a mitochondrial suspension at 540 nm in spectrophotometers Cary-219 (Varian, USA) or DW-2000 (SLM Aminco, USA) (Fig. 5). In control experiments intact and completely swollen mitochondria were mixed in different proportions and their optical density was measured. The value was proportional to the fraction of intact mitochondria in the full range of optical densities used in the experiments on swelling.
Kinetics of NAD(P)H oxidation were measured at 340 nm (versus 370 nm) in DW-2000 spectrophotometer. The initial content of NADH and NADPH determined after alkaline extraction as described [8] was 1.6 ± 0.2 and 1.8 ± 0.4 nmoles/mg protein (5 independent experiments).
RESULTS
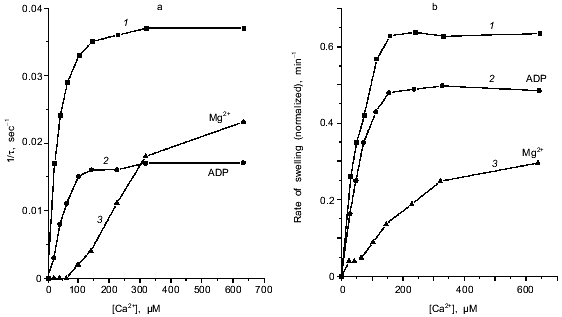
Induction of the mitochondrial pore by oxidation of pyridine nucleotides. The data of Fig. 1 show the induction of the pore in deenergized mitochondria. In the absence of inducers no significant swelling was observed after addition of up to 1-2 mM Ca2+. Incubation with duroquinone (5 µM) caused rapid swelling triggered by 24 µM Ca2+ (free) (40 µM CaCl2 added) in the major fraction of mitochondria (Fig. 1a). The maximal rate of swelling was observed at 0.1 mM Ca2+ (Fig. 2). The effect was prevented by cyclosporin A (Fig. 1a) and inhibited by ADP and Mg2+ (Fig. 2). These data confirm that Ca2+-induced swelling of deenergized mitochondria is due to opening of the mitochondrial pore in the inner membrane. The kinetics of this process was studied in detail (see below).
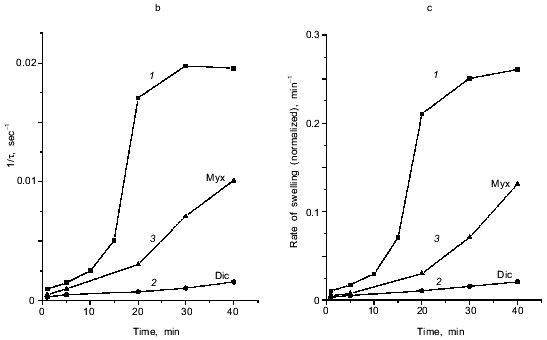
Fig. 1. Ca2+-dependent swelling of deenergized mitochondria preincubated with duroquinone (DQ) (a). Dependence of the lag-phase tau (b) and the rate of swelling gamma (c) on time of preincubation. Mitochondria were preincubated with 5 µM duroquinone for 10 (trace 6 in (a)) or 20 min. Swelling was triggered by 40 µM CaCl2 (24 µM free Ca2+). Cyclosporin A (CsA, 1 µM ), 25 µM NEM (NEM), 1 µM dicumarol (Dic), or 5 µM myxothiazol (Myx) were added 1 min before duroquinone. The graphical method for determination of the lag-phase and rate of swelling is shown.
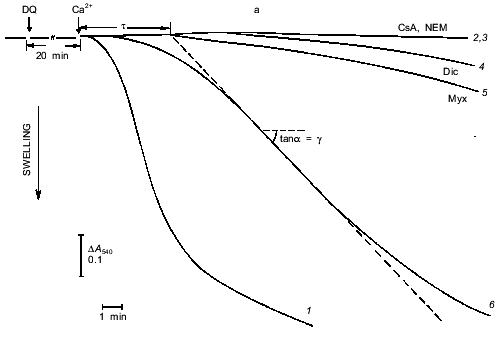
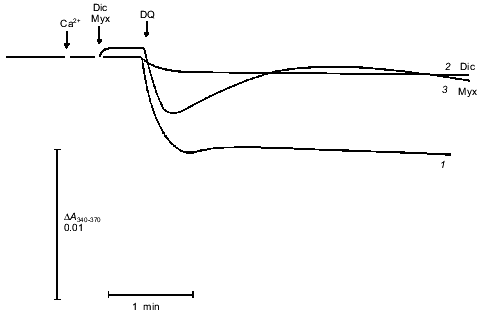
The approach used allowed study of the effects of pore inducers in intact mitochondria since no pore was opened before addition of Ca2+. Under these conditions the effect of duroquinone correlated with the oxidation of pyridine nucleotides in the matrix. Duroquinone (5 µM) induced rapid oxidation (Fig. 3), and more than 90% of both NADH and NADPH were oxidized within 5 min. Duroquinone did not oxidize NAD(P)H so strongly in the presence of substrates of dehydrogenases (glutamate/malate mixture or 2-hydroxybutyrate). Incubation for 15-20 min did not induce pore opening in this case (data not shown). Dicumarol (an inhibitor of D,T-diaphorase that catalyzes he oxidation of NAD(P)H by duroquinone) and myxothiazol (an inhibitor of the bc1-complex of the respiratory chain which blocks reoxidation of reduced duroquinone) prevented oxidation of NADH and NADPH in the matrix (Fig. 3). Both inhibitors block redox cycling of the quinone, but in the first case quinone is completely oxidized and in the second case quinone is completely reduced. Dicumarol as well as myxothiazol prevented the pore induction by duroquinone (Fig. 1), so the redox state of the quinone was not significant for this effect. The possible reduction to semiquinone catalyzed by NADPH-cytochrome P450 reductase and coupled generation of superoxide- and hydroxyl-radicals also are not important for the effect of duroquinone but are probably significant when the pore is induced by menadione [9], benzoquinone [10], or dibromothymoquinone [11].Fig. 2. Dependence of kinetic parameters of swelling on concentration of free Ca2+. Mitochondria were preincubated with 5 µM duroquinone for 20 min. ADP (0.1 mM, traces 2) and 0.25 mM MgSO4 (traces 3) were added 1 min before addition of duroquinone. The lag-phase (a) and the rate of swelling (b) were determined as in Fig. 1a.
The pore in deenergized mitochondria was also induced by oxidation of NAD(P)H by dehydrogenases of the Krebs cycle (in the presence of acetoacetate or oxaloacetate) or by NADH oxidase in respiratory chain (in the absence of rotenone). In the latter case glutamate/malate or 2-hydroxybutyrate in combination with rotenone caused reduction of NAD(P)H and rapid (2-3 min) decrease in the Ca2+-sensitivity, i.e., reversal of the pore induction (data not shown). These data indicate that possible hydrolysis of oxidized nucleotides and coupled ADP-ribosylation of membrane proteins do not determine the pore induction (as was postulated in [12]) under the conditions of our experiments.Fig. 3. Kinetics of NAD(P)H oxidation by duroquinone in mitochondria. The measuring procedure is described in «Materials and Methods». Cyclosporin A (1 µM) was added in all assays to prevent swelling of mitochondria and possible optical artifacts. The concentration of free Ca2+ was 24 µM (40 µM CaCl2 added). Additions: 5 µM duroquinone (DQ, traces 1-3), 1 mM dicumarol (Dic, trace 2), 5 µM myxothiazol (Myx, trace 3).
Thus, pore induction in the experiments presented here was determined by the oxidation of NADH and/or NADPH. This conclusion is in a perfect agreement with earlier results of Lehninger and co-workers [13] who found in 1978 that oxidation of pyridine nucleotides caused Ca2+ release from mitochondria.
Inhibition of pore induction by N-ethylmaleimide. As shown in Fig. 3, maximal oxidation of NAD(P)H was reached after 2-3 min. On the other hand, pore induction takes much longer. The rate of Ca2+-dependent pore opening (measured at fixed Ca2+ concentration) reached its maximal value only after 20-30 min (Fig. 1, b and c). We suggest that slow rearrangement (or formation) of the pore complex may occur after rapid oxidation of NAD(P)H. This process is blocked by N-ethylmaleimide (NEM) which does not affect NAD(P)H oxidation (data not shown) but inhibits the pore opening (Fig. 1a). NEM did not affect the pore in some other experimental models (e.g., uncoupler-induced pore opening in Ca2+-loaded mitochondria [14]), so it did not interfere with the mechanism of permeability.
NEM did not affect the rate of swelling if it was added to mitochondria treated with duroquinone (for 20 min) 1-3 min before addition of Ca2+. When the time between additions of NEM and Ca2+ was increased, inhibition of pore opening was observed. The maximal inhibition was reached after 20-30 min (Fig. 4). NEM decreased the rate and maximal amplitude of swelling. These data indicate that NEM causes slow reversal of pore induction when pore induction is a result of NAD(P)H oxidation.
The effect of NEM points to a possible role of oxidation of membrane dithiols in the pore induction caused by NAD(P)H oxidation. However, dithiothreitol and 2-mercaptoethanol, active disulfide-reducing agents, and the specific SH-reagent monobromobimane (MBM) did not prevent the effect of duroquinone and other NAD(P)H oxidizing agents (data not shown). It was shown earlier that oxidation or cross-linking (caused by hydroperoxides or arsenite, for example) caused pore induction in deenergized mitochondria, but their effect was prevented by MBM [5]. Perhaps NAD(P)H oxidation oxidizes specific dithiol(s) unavailable for dithiothreitol and MBM. An alternative mechanism suggests allosteric regulation of the pore by pyridine nucleotides.Fig. 4. Dependence of the lag-phase (a) and the rate of swelling (b) on time of incubation with NEM. Reversal of the pore induction by NEM. Mitochondria were preincubated with 5 µM duroquinone for 20 min and then 25 µM NEM was added. Swelling was triggered by 40 µM CaCl2 (24 µM Ca2+ free) at different times after NEM addition.
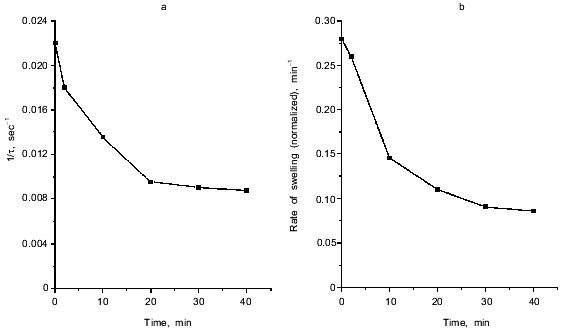
Kinetics of Ca2+-dependent opening of the mitochondrial pore. The swelling of mitochondria triggered by Ca2+ has a long lag phase (Fig. 1). The dependence of lag phase and maximal rate of swelling on concentration of Ca2+ is shown in Fig. 2. For both parameters the half-maximal effect is reached at 20-30 µM Ca2+ and the maximal effect at 100-150 µM Ca2+. The pore inhibitors ADP and Mg2+ decrease the rate of swelling and increase the lag phase simultaneously. These parameters are also changed in parallel at different times of incubation with duroquinone (Fig. 1, b and c). Similar mitochondrial pore opening properties were observed earlier under various experimental conditions and various methods of detection (for review, see [1]). In the majority of the works the lag phase was attributed either to slow action of the inducers (oxidative agents, for example) or to secondary redistribution of Ca2+ released from the mitochondria with open pores and taken up by energized organelles to promote pore opening there. Both possibilities were excluded in our experiments. First, the lag phase was observed after a long incubation with duroquinone and did not change with further increase of incubation time (Fig. 1b). Second, Ca2+ redistribution was excluded in the presence of uncoupler (CCCP) and Ca2+ ionophore (A23187).
A possible reason for the lag phase is based on the heterogeneity of the mitochondrial population, where pore opening has different probability in different fractions. The main sources for heterogeneity, membrane potential and ion gradients, were minimized in our experiments. More complete dissipation of membrane potential (including Donnan potential) was obtained in the medium with high K+ concentration in the presence of the K+ ionophore valinomycin, and the lag phase during pore opening persisted [5].
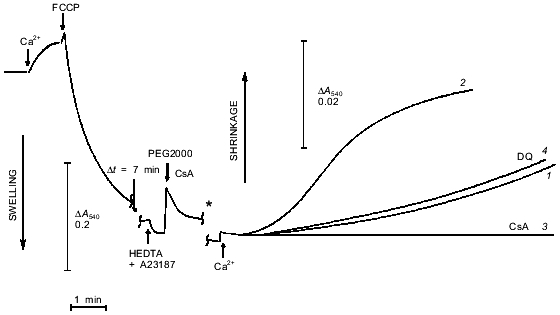
Another approach to studies of pore opening is based on measurement of mitochondrial shrinkage in medium with a non-permeable polymer of high molecular weight [15]. In our experiments (Fig. 5), mitochondria were loaded with Ca2+, and the pore was completely opened with uncoupler. This caused maximal swelling. Then the Ca2+-chelator HEDTA and the non-permeable polymer PEG2000 were added. No shrinkage was observed because in the absence of free Ca2+, the pore closed [1] and concentration of polymer was low (4 mM). Subsequent addition of Ca2+ caused a slow increase in light scattering that is attributed to opening of a pore permeable for sucrose. Shrinkage was completely prevented by cyclosporin A. This process had a pronounce lag phase that was shortened by increasing the Ca2+ concentration (Fig. 5). During these experiments mitochondria were swollen and had an open pore for a long time (10 min), so soluble matrix components with molecular mass < 1500 daltons were released. The release of ADP, Mg2+, and probably some other pore inhibitors resulted in a significant increase in the Ca2+ sensitivity of the pore. We think that the role of mitochondrial heterogeneity in the kinetics of pore opening is minimal under these conditions.
Incubation of mitochondria with duroquinone (and oxidation of NAD(P)H by other mechanisms) did not affect the kinetics of shrinkage caused by repeated pore opening (Fig. 5, trace 4). Dithiol cross-linking agents strongly accelerated pore opening in similar experiments [5]. These data confirm the conclusion that NAD(P)H oxidation does not cause covalent modification but rather modulates the pore allosterically.Fig. 5. Kinetics of osmotic mitochondrial shrinkage caused by pore opening in the presence of PEG2000. Mitochondria were incubated in hypotonic medium (see «Materials and Methods»). Maximal swelling was induced by subsequent additions of CaCl2 (500 µM) and FCCP (0.4 µM). HEDTA (5 mM), 1 µM A23187, and 4 mM PEG2000 were added after 10 min. Mitochondrial shrinkage was triggered by addition of 3.1 mM CaCl2 (free Ca2+ 1 µM) (1, 3, 4) or 4 mM CaCl2 (free Ca2+ 4 µM) (2). Shrinkage corresponds to increase in optical density of the mitochondrial suspension (a change in scale is marked by the asterisk). Cyclosporin A (CsA, 1 µM) was added simultaneously with PEG2000 (3). Duroquinone (DQ, 5 µM) was added 20 min before first addition of Ca2+ (4).
The kinetics of pore opening is analyzed below assuming that
mitochondrial suspension is homogenous. The following basic
propositions are used in analysis of the data: 1) pore opening and
mitochondrial swelling is an «all-or-nothing» phenomena
without intermediate states [1]; 2) the optical
density of the mitochondrial suspension is proportional to the fraction
of swollen mitochondria (see «Materials and Methods»). A
model containing only two steps satisfactorily describes the kinetics
of the Ca2+-triggered swelling (Fig. 1a):
where A represents intact mitochondria, B represents an intermediate state with modified but still closed pore, and C represents swollen mitochondria with opened pores. The concentration of swollen mitochondria depends on time after Ca2+ addition as:
where k1 and k2 are the rate constants of the steps and Cm is the total concentration of mitochondria (proportional to the maximal amplitude of swelling). The maximal rate of the process normalized to the amplitude of swelling (gamma. and the lag phase (tau. determined as shown in Fig. 1a are described as:
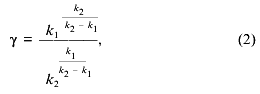
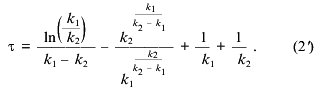
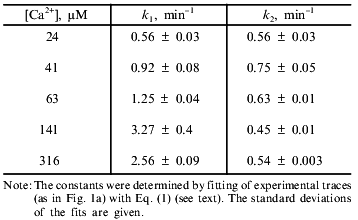
Equations (2) and (2´) allowed calculation of the values of the constants from the measurements of lag phase and maximal rate, but more reliable results were obtained by fitting of the experimental traces (e.g., the traces in Fig. 1a) with Eq. (1). The values of k1 and k2 determined according to this procedure at different concentrations of Ca2+ are shown in the table. The first constant was found to increase with Ca2+ concentration and to reach a plateau at 140 µM Ca2+. The second constant did not depend on Ca2+ concentration in the range 20-300 µM. We suggest that the Ca2+ dependence of k1 reflects the effective dissociation constant of a Ca2+-binding site which determines the pore opening. The second step is probably a spontaneous, Ca2+-independent process.
Kinetic constants of Ca2+-dependent mitochondrial swelling
DISCUSSION
The approach developed in this study allowed separate investigation of the kinetics of Ca2+-dependent pore opening and «induction» preceding opening. The Ca2+-dependent process was perfectly fitted by a two-step scheme, and heterogeneity of mitochondria did not interfere with the kinetics. Analysis of the pore opening at different Ca2+ concentrations showed that only the rate constant of the first step is Ca2+ dependent; the second step is Ca2+ independent. Oxidation of pyridine nucleotides causes slow transformation of the pore and decreases the effective dissociation constant of the Ca2+-binding site from >1 mM to ~30 µM.
With our method secondary effects coupled to release of matrix components through the pore are excluded, and this allowed the study of the reversal of the «induction» process. The data indicate that the effect of NAD(P)H oxidation does not depend on oxidation of SH-groups or other kinds of covalent modification of mitochondrial proteins, but rather is allosteric. These data are in agreement with the observation that NADH, when it appears in the matrix, can inhibit pore opening [16]. Oxidation of NAD(P)H triggers a very slow process of pore transformation which is blocked by NEM but is insensitive to disulfide-reducing agents. Reduction of NAD(P)H causes rapid reversal of this process. A similar reversal is induced by NEM, and it is much slower. We suggest that the NEM-sensitive residue becomes unavailable after NAD(P)H oxidation. It is probably localized in a putative NAD(P)+-binding site and is shielded by the nucleotide.
It is interesting that various ion channels contain nucleotide-binding sites and are regulated by nucleotides. ATP-dependent and voltage-sensitive (alpha.beta, Kv) K+ channels belong to this group. In the first case regulation by nucleotides is the most distinctive feature of the family. Sequence analysis of voltage-dependent channels have shows that a fragment of beta-subunit is strongly homologous with the NAD(P)H-binding domain of a large family of NAD(P)H-dependent dehydrogenases [17, 18]. This fragment is conserved in Kv channels from mammals to drosophila, so it is likely to be functionally significant. A similar domain has been identified in another protein involved in K+ transport (TrkA) in Escherichia coli [19]. Modulation of channel activity by pyridine nucleotides was described for a «Ca2+-nonselective cation channel» (CaNS+), but no difference between reduced and oxidized forms of nucleotides was observed [20]. Physiological modulation of the channel activity by the redox state of pyridine nucleotides was postulated for the voltage-dependent anion channel (VDAC) from the outer mitochondrial membrane. The permeability of this channel for ADP and other compounds is decreased by NADH but not by NAD+ or NADP(H) [21]. These examples suggest that the mitochondrial pore is evolutionary related to a large family of channels regulated by nucleotides.
The authors are indebted to Prof. V. P. Skulachev and B. N. Kholodenko for helpful discussions. The work was supported by grants from the Russian Foundation for Basic Research (Nos. 96-04-49330 and 98-04-48868).
REFERENCES
1.Zoratti, M., and Szabo, I. (1995) Biochim.
Biophys. Acta, 1241, 139-176.
2.Ichas, F., Jouaville, L. S., and Mazat, J.-P.
(1997) Cell, 89, 1145-1153.
3.Kroemer, G., Petit, P. X., Zamzami, N., Vayssiere,
J.-L., and Mignotte, B. (1995) FASEB J., 9,
1277-1287.
4.Chernyak, B. V., Dedov, V. N., and Chernyak, V. Ya.
(1995) FEBS Lett., 365, 75-78.
5.Chernyak, B. V., and Bernardi, P. (1996) Eur. J.
Biochem., 238, 623-630.
6.Johnson, D., and Lardi, H. (1967) Meth.
Enzymol., 10, 90-96.
7.Fabiato, A. (1988) Meth. Enzymol.,
157, 378-417.
8.Beatrice, M. C., Siers, D. L., and Pfeiffer, D. R.
(1984) J. Biol. Chem., 259, 1279-1287.
9.Rizzuto, R., Pitton, G., and Azzone, G. F. (1987)
Eur. J. Biochem., 162, 239-249.
10.Moore, G. A., Weis, M., Orrenius, S., and
O'Brien, P. J. (1988) Arch. Biochem. Biophys.,
267, 539-550.
11.Harris, E. J., and Baum, H. (1980) Biochem.
J., 186, 725-732.
12.Lehninger, A. L., Vercesi, A. E., and Bababunmi,
E. A. (1978) Proc. Natl. Acad. Sci. USA, 75,
1690-1694.
13.Richter, C., and Frei, B. (1988) Free Rad.
Biol. Med., 4, 365-375.
14.Petronilli, V., Costantini, P., Scorrano, L.,
Cola, C., Colonna, R., Passamonti, S., and Bernardi, P. (1994) J.
Biol. Chem., 269, 16638-16642.
15.Hunter, D. R., and Haworth, R. A. (1979) Arch.
Biochem. Biophys., 195, 453-459.
16.Haworth, R. A., and Hunter, D. R. (1979) Arch.
Biochem. Biophys., 195, 460-467.
17.McCormack, T., and McCormack, K. (1994)
Cell, 79, 1133-1135.
18.Milkman, R. (1994) Proc. Natl. Acad. Sci.
USA, 91, 3510-3514.
19.Shlosser, A., Hamann, A., Bossemeyer, D.,
Schneider, E., and Bakker, E. P. (1993) Mol. Microbiol.,
9, 533-543.
20.Reale, V., Hales, S. N., and Ashford, M. L.
(1994) J. Membr. Biol., 142, 299-307.
21.Lee, A.-C., Zizi, M., and Colombini, M. (1994)
J. Biol. Chem., 269, 30974-30980.