Translation Termination and Yeast Prions (Introductory Remarks of the Guest Editor of This Special Issue)
L. L. Kisselev
Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, Moscow, 117984 Russia; fax: (095) 135-1405; E-mail: kissel@imb.ac.ru
Received July 10, 1999
Protein biosynthesis is the final step in the transfer of genetic information in the cell. In turn, its last step is the release of a nascent polypeptide from the ribosome. Therefore, termination of translation may be considered (if we do not take into account protein post-translational modification and folding) as a final step of the transition from genotype to phenotype through the classic DNA--RNA--protein pathway. In a narrow sense, termination of translation is the hydrolytic cleavage of peptidyl-tRNA into free tRNA and completed polypeptide chain carrying all the information encoded in the corresponding mRNA and DNA. Then the completed protein molecule is released from the ribosome and the ribosome dissociates into its components (subunits, factors, mRNA, tRNA, etc.). After the synthesis is completed, the polypeptide chain is folded either cotranslationally or by an additional specialized mechanism, depending on the nature of the protein, organism, and other factors. This issue of Biochemistry (Moscow) highlights from various points of view the problem of translation termination, excluding protein folding. Yeast termination factors with prion-like properties are also considered.
KEY WORDS: protein biosynthesis, translation termination, prokaryotes, eukaryotes, release factors, prions
There are two aspects to translation termination studies. The narrow one is restricted to the peptidyl-tRNA cleavage in the ribosome involving a termination codon, a termination factor, and the ribosomal peptidyl transferase center. A wider aspect considers also the events following the step of chemical hydrolysis. Termination and posttermination are often studied together, though, as one may see from the papers in this issue, these two processes are fundamentally different in mechanisms and in proteins involved. The effect of mRNA structure on termination and posttermination efficiency was recently discussed in detail (see [1-3]).
Termination of translation is interesting and attracts attention from various points of view. It remains unknown how the termination codon at the ribosomal A site interacts with the release factor and with the substrate of the reaction, peptidyl-tRNA. It is still not proved whether the release factor interacts directly with a stop codon, or a complex is first formed between ribosomal RNA region(s) and mRNA and is then recognized by the release factor. At present the hypothesis of direct recognition is preferred [4-8], however, in parallel new data obtained [9-11] pointing to the important role of rRNA in translation termination. Though about 30 years have passed since the basic discoveries around translation termination were made (see reviews [12, 13]), still neither a catalytic mechanism of the reaction nor a route of signal transduction from stop codon in mRNA toward the catalytic center has been established. Therefore, enzymological study of translation termination remains essential.
A nonsense mutation in mRNA has two possible consequences: this mRNA either undergoes degradation by the special complex mechanism recognizing mRNAs with a stop codon in a wrong position (see [14-16] for details), or, if this mechanism fails for any reason to degrade the defective mRNA, the truncated polypeptide chain is released.
Contrary to the mutations from one sense codon to another which are in most cases not reflected in the phenotype, a mutation of most sense codons in any of the stop codons may be fatal to the cell. It becomes more and more evident that a number of diseases including hereditary and oncological ones is caused by premature termination producing the C-truncated proteins. Nonsense mutations form a significant part of pathological mutations in general. Therefore, it is a medically important problem to "cure" these mutations to recover normal size of the proteins and thus their functions.
A traditional treatment is replacing the damaged gene with the normal one--gene therapy. However, if one considers that there are hundreds of diseases caused by protein truncation by nonsense mutations, it becomes evident that development of special gene-replacing therapy for every gene will take a lot of time and effort. In spite of the differences in the pathological genes, these diseases have similar mechanisms; therefore, it appears more reasonable to search for a universal mechanism for their repair.
It is well known that stop signals may be read as sense codons by suppressor tRNAs which recognize each of the three stop codons. Usually in a normal cell the concentration of suppressor tRNAs is very low, and they are unable to compete efficiently with the release factors. Therefore, most of the nonsense codons are read as termination signals, or this mRNA is degraded as mentioned above.
Both in vitro and in vivo there is direct competition between the release factor and suppressor tRNAs, because both occupy the ribosomal A site. This phenomenon has been known for a long time for prokaryotes and recently shown also for eukaryotes [17, 18]. The competition between the release factor and the suppressor tRNAs may form the basis for a new approach to "cure" nonsense mutations. There are two possible ways to achieve a shift toward the readthrough inhibiting termination: to increase the concentration of the suppressor tRNA, for example, by their hyperexpression in damaged cells, or to suppress the release factor activity, for example by specially generated inhibitors. These two strategies may be used concomitantly.
Any of these methods may be efficient enough for total or partial functional recovery of the damaged truncated protein. However, it is also evident that weakened termination and enhanced suppression should cause increase in the concentration of elongated proteins in which the amino acid sequence is encoded by mRNA part downstream from the stop codon toward the 3´-end of the mRNA. It is known that mRNAs vary in the length of the 3´-untranslated region (UTR), and the 3´-UTR may comprise a rather long mRNA fragment. Consequently, many proteins of excessive length should appear as a side effect of nonsense-codon therapy. How dangerous may this be for a cell? First, a significant part of these extra C-terminal fragments may be destroyed by proteolysis, because contrary to the conventional proteins they have no regular evolutionarily selected secondary and tertiary structure. Second, these «extra» polypeptides may not interfere with functional activity of the normal protein: most probably the extra part will form a spatially isolated domain. Third, termination is only weakened, not stopped, and obviously normal proteins will be produced along with the extended ones. The amount of normal protein may be sufficient to provide normal functioning, because usually cells produce proteins «in excess».
To summarize, the presence of many extended proteins may be much less harmful than the absence of an essential functional protein. This makes it reasonable to develop experimentally the suggested strategy to cure nonsense mutations. This method has two important advantages: a) a cellular gene with its own normal regulation system is preserved instead of introduction of a new gene and its regulation system; b) there is no need to develop a special method for each new disease, because the approach is unspecific toward the genes affected by nonsense mutations.
Obviously, problems which are now difficult to predict may arise on practical realization of this approach. For some genes and mRNAs the inhibition of termination factors will probably be more efficient, and for others it will be better to increase the concentration of suppressor tRNAs. Efficiency of the treatment should depend on the stop codon context: the variability of possible contexts is virtually unlimited. Inhibition of termination and/or enhancement of suppression will probably vary depending on the given situation.
A very interesting discovery in translation termination studies was the observation of the similarity between prions, proteins responsible for some incurable neurodegenerative diseases in humans and animals, and a protein involved in termination of translation in baker's yeast Saccharomyces cerevisiae. This will also be discussed in this issue.
Briefly, it was shown that in yeast there is a phenotype named [psi+] which is inherited cytoplasmically (for details see [19-21]). Most yeast strains have [psi-] phenotype. The origin of the [psi+] remains unknown: the consequence of long-term yeast cultivation under laboratory conditions is not excluded (Yu. Chernov, unpublished).
Though [psi+] and [psi-] phenotypes have been known for a long time, only recently they were related to a certain yeast protein, Sup35p [22]. It was suggested that one and the same protein Sup35p has two alternative conformations corresponding to the [psi+] and [psi-] phenotypes.
It is known that a similar phenomenon was shown earlier for the prion proteins: a normal cellular protein PrPC affected by unknown factors may transform into the pathogenic form PrPSc. The «prionic» ([psi+]) form of Sup35p causes no pathology, but rather provides some advantages for survival under uncomfortable conditions [23]. Quite unexpectedly and unpredictably the yeast protein Sup35p became a model prion, and great attention was drawn to it. Independently it was shown that Sup35p belongs to the class II of termination factors named eRF3 [24].
The yeast protein Sup35p possesses not only prion-like properties, but also biochemical activity of the eRF3 protein family, that is, eRF1- and ribosome-dependent GTPase activity (L. Frolova, personal communication). While no activity which could be easily measured is known for mammalian prions, Sup35p possesses such activity; therefore, Sup35p appears to be a convenient model to study prionic properties.
The problem of termination including its "prion" aspects is highlighted from various points of view in the papers of this issue. There is no need to summarize the achievements in this area within these introductory notes. However, it seems reasonable rather to stress unsolved problems and unclear points.
After determination of structure for the class I release factors (eRF1) [25] the situation became similar to prokaryotes, where the structure of RF1 and RF2 has been established much earlier (see reviews [26, 27]). It then became possible to study the RFs of this class from various species in parallel.
Though the principal scheme of translation termination should be the same in prokaryotes, archaebacteria, and eukaryotes, some important differences have already been found. It remains unknown whether a common ancestral gene exists for eukaryotic and prokaryotic class I RFs. Different points of view have been presented. On one hand, RF1/2 and eRF1 are rather different [25], though they have a common functionally important GGQ motif [28]. On the other hand, alignment of the sequences for several class I RFs of various origin suggested the existence of a common ancestor [29, 30]. Computer-assisted structural analysis of many class I RFs representing all living kingdoms have shown structural homology of the eRF1 C domain (about 160 amino acids) with some ribosomal proteins, while neither RF1, nor RF2 from prokaryotes have shown similar homology. Moreover, no homology was found between C-domains of eRF1 and RF1/2. These data (N. Oparina, L. Frolova, and L. Kisselev, unpublished) confirm that at least the C-domain of eRF1 has no common ancestor with prokaryotic RF1/2.
Another important difference is found in the interaction with class II release factors, RF3 and eRF3. eRF1 has a specific region in its C domain which allows formation of a stable complex with eRF3 [31-34]. For RF1/2 no similar region was found, and no complex formation was shown between RF1/2 and RF3 [30]. The third difference is in stop codon recognition. While in eukaryotes eRF1 recognizes all three stop codons, prokaryotic RF1 and RF2 recognize UAA plus either UAG (RF1) or UGA (RF2) (see reviews [35, 36]).
Even more serious differences are found on comparison of RF3 and eRF3 (table). First, eRF3 is encoded by an essential gene, while this is not true for RF3. Second, though both RF3 and eRF3 are GTP-binding proteins, their structural differences are obvious: RF3 is similar to EF-G (elongation factor, translocase), while eRF3 is similar to eEF1alpha (elongation factor, analog of prokaryotic EF-Tu). Third, the C domain of eRF3 contains an eRF1-binding site [32, 33], while no similar site was found in RF3. Fourth, RF3 is active as a GTPase in the presence of ribosomes and in the absence of RF1/2 [37-39], while eRF3 requires not only ribosomes, but also eRF1 for its GTPase activity [40, 41].
TABLE 1. Comparison of RF3 (prokaryotes) and
eRF3 (eukaryotes)
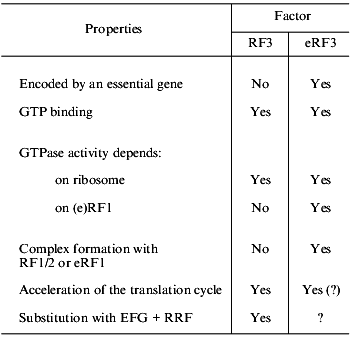
The functional role of RF3 is gradually becoming clear [37, 42-44]. It is already known that this factor is not involved in peptidyl-tRNA hydrolysis, but is essential for fast removal of RF1/2 from the ribosomes after termination. In other words, RF3 is involved in preparation of a ribosome for the next translation cycle and may be considered as a post-termination factor.
The situation with eRF3 remains rather unclear. Though it was shown that, similarly to RF3, it is not involved in peptidyl-tRNA hydrolysis and is rather a post-termination factor (see [45]), its more precise function has become the subject of discussion. It is suggested that it may be an analog of elongation factor eEF1alpha [30, 46], though there are arguments against this (L. Frolova, T. Merkulova, and L. Kisselev, in preparation), that it may be an analog of elongation factor eEF2 translocating from A to P ribosomal site not tRNA, but eEF1alpha [47], that it may have a function of proofreading [40], or that it may serve as a functional analog of two prokaryotic factors, RF3 + RRF [35], because putative eukaryotic RRF was shown to be a release factor from chloroplasts [48], and no cytoplasmic RRF has been found in eukaryotes so far.
A reconstructed system close to the natural one was created to study separately termination and post-termination stages in prokaryotes [49], but there are great difficulties in developing a similar system for eukaryotes, mainly because numerous proteins are involved in these processes.
Two periods are seen in the history of translation termination research during which all basic discoveries have been made. The first five years (1967-1971) resulted in the general scheme of the process with the essential role of stop codons and termination factors later assigned to class I [50], the post-termination stage remaining obscure. It is now evident that though these results were not essentially changed later, they were only the very first approximation.
The second five-year period (1994-1998) followed the first after a quarter-century break of no new knowledge about termination of translation. During the second period RF3, eRF1, and eRF3 were characterized structurally and functionally and a realistic system was developed to analyze termination in prokaryotes. Taken together, these results provided a new level of knowledge concerning the translation termination process.
The next five years at the transition to the third millennium should be a period of very extensive studies of prokaryotic and eukaryotic (and probably of archaebacterial) systems. These studies will employ site-directed mutagenesis, yeast two-hybrid and three-hybrid systems [51, 52], crystallization of the termination factors and their complexes with other proteins to establish their 3D structure, analysis of fast and stationary kinetics, and computer-assisted analysis of genes and proteins involved in termination and post-termination.
Of course, these introductory notes consider only a few aspects of the problem. The reader is invited to find the missing information in the papers of this issue; they are written by those who have made essential personal contributions to this area of biochemistry and molecular biology.
REFERENCES
1.Tate, W. P., and Mannering, S. A. (1996) Mol.
Microbiol., 21, 213-219.
2.Tate, W. P., Dalphin, M. E., Pel, H. Y., and
Mannering, S. A. (1996) Genet. Eng., 18, 157-182.
3.Pavlov, M. Y., Freistroffer, D. V., Dinchas, V.,
MacDougall, Y., Buckingham, R. H., and Ehrenberg, M. (1998) J. Mol.
Biol., 284, 579-590.
4.Tate, W., Greuer, B., and Brimacombe, R. (1990)
Nucleic Acids Res., 18, 6537-6544.
5.Brown, C. M., and Tate, W. P. (1994) J. Biol.
Chem., 269, 33164-33170.
6.Ito, K., Uno, M., and Nakamura, Y. (1998) Proc.
Natl. Acad. Sci. USA, 95, 8165-8169.
7.Poole, E. S., Brown, C. M., and Tate, W. P. (1995)
EMBO J., 14, 151-158.
8.Poole, E. S., Brimacombe, R., and Tate, W. P.
(1997) RNA, 3, 974-982.
9.Murgola, E. J. (1996) in Ribosomal RNA:
Structure, Evolution, Processing, and Function in Protein
Biosynthesis (Zimmermann, R. A., and Dahlberg, A. E., eds.) CRC
Press, Boca Raton, FL, pp. 357-369.
10.Arkov, A. L., Mankin, A., and Murgola, E. J.
(1998) J. Bacteriol., 180, 2744-2748.
11.Arkov, A. L., Freistroffer, D. V., Ehrenberg, M.,
and Murgola, E. J. (1998) EMBO J., 17, 1507-1514.
12.Caskey, C. T. (1977) in Molecular Mechanisms
of Protein Biosynthesis (Weissman, H., and Pestka, S., eds.)
Academic Press, N.-Y., pp. 1507-1514.
13.Caskey, C. T. (1980) Trends Biochem. Sci.,
5, 234-237.
14.Maquat, L. E. (1995) RNA, 1,
453-465.
15.Cui, Y., Gonzales, C. I., Kinzy, T. G., Dinman,
Y. D., and Peltz, S. W. (1999) RNA, 5, 794-804.
16.Hilleren, P., and Parker, R. (1999) RNA,
5, 711-719.
17.Drugeon, G., Jean-Jean, O., Frolova, L., Le Goff,
X., Philippe, M., Kisselev, L., and Haenni, A.-L. (1997) Nucleic
Acids Res., 25, 2254-2258.
18.Le Goff, X., Philippe, M., and Jean-Jean, O.
(1997) Mol. Cell Biol., 17, 3164-3172.
19.Lindquist, S. (1997) Cell, 89,
495-498.
20.Ter-Avanessyan, M. D., Paushkin, S. V.,
Kushnirov, V. V., and Kochneva-Pervukhova, M. V. (1998) Mol. Biol.
(Moscow), 32, 32-42.
21.Ter-Avanessyan, M., and Kushnirov, V. (1998)
Cell, 94, 13-16.
22.Wickner, R. (1994) Science, 264,
5676-5689.
23.Eaglestone, S. S., Cox, B. S., and Tuite, M.
(1999) EMBO J., 18, 1974-1981.
24.Zhouravleva, G., Frolova, L., Le Goff, X., Le
Guellec, R., Inge-Vechtomov, S., Kisselev, L., and Philippe, M. (1995)
EMBO J., 14, 4065-4072.
25.Frolova, L., Le Goff, X., Rasmussen, H. H.,
Cheperegin, S., Drugeon, G., Kress, M., Arman, I., Haenni, A.-L.,
Celis, J. E., Philippe, M., Justesen, J., and Kisselev, L. (1994)
Nature, 372, 701-703.
26.Craigen, W. Y., Lee, C. C., and Caskey, C. T.
(1990) Mol. Microbiol., 4, 861-865.
27.Tate, W., and Brow, C. M. (1992)
Biochemistry, 31, 2443-2450.
28.Frolova, L. Yu., Tsivkovskii, R., Sivolobova, G.,
Oparina, N., Serpinsky, O., Blinov, V., Tatkov, S., and Kisselev, L.
(1999) RNA, 5, 1014-1020.
29.Nakamura, Y., Ito, K., Matsumura, K., Kawazu, Y.,
and Ebihara, K. (1995) Biochem. Cell Biol., 73,
1113-1122.
30.Ito, K., Ebihara, K., Uno, M., and Nakamura, Y.
(1996) Proc. Natl. Acad. Sci. USA, 93, 5443-5448.
31.Ito, K., Ebihara, K., and Nakamura, Y. (1998)
RNA, 4, 958-972.
32.Merkulova, T. I., Frolova, L. Y., Lazar, M.,
Camonis, J., and Kisselev, L. (1999) FEBS Lett., 443,
41-47.
33.Ebihara, K., and Nakamura, Y. (1999) RNA,
5, 739-750.
34.Eurwilaichitr, L., Graves, F. M., Stansfield, I.,
and Tuite, M. F. (1999) Mol. Microbiol., 32, 485-496.
35.Buckingham, R. H., Grentzmann, G., and Kisselev,
L. (1997) Mol. Microbiol., 24, 449-456.
36.Nakamura, Y., and Ito, K. (1998) Genes to
Cells, 3,265-278.
37.Freistroffer, D. V., Pavlov, M. Yu., MacDougall,
J., Buckingham, R. H., and Ehrenberg, M. (1997) EMBO J.,
16, 4126-4133.
38.Pel, H. J., Moffat, J. G., Ito K., Nakamura, Y.,
and Tate, W. P. (1998) RNA, 4, 47-54.
39.Grentzmann, G., Kelly, P. J., Laalami, S., Shuda,
M., Firpo, M. A., Cenatiempo, Y., and Kaji, A. (1998) RNA,
4, 973-983.
40.Frolova, L., Le Goff, X., Zhouravleva, G.,
Davydova, E., Philippe, M., and Kisselev, L. (1996) RNA,
2, 334-341.
41.Frolova, L. Yu., Simonsen, J. L., Merkulova, T.
I., Litvinov, D. Y., Martensen, P. M., Rechinsky, V. O., Camonis, J.,
Kisselev, L. L., and Justesen, J. (1998) Eur. J. Biochem.,
256, 36-44.
42.Pavlov, M. Y., Freistroffer, D. V.,
Heurgue-Hamard, V., Buckingham, R. H., and Ehrenberg, M. (1997) J.
Mol. Biol., 273, 389-401.
43.Pavlov, M. Y., Freistroffer, D. V., MacDougall,
J., Buckingham, R. H., and Ehrenberg, M. (1997) EMBO J.,
16, 4133-4141.
44.Karimi, R., Pavlov, M. Y., Buckingham, R., and
Ehrenberg, M. (1999) Mol. Cell, 3, 601-609.
45.Kisselev, L., and Frolova, L. (1999)
Biochemistry (Moscow), 64, 8-16.
46.Nakamura, Y., Ito, K., and Isaksson, L. A. (1996)
Cell, 87, 147-150.
47.Nissen, P., Kjeldgaard, M., Thirup, S.,
Polekhina, G., Reshetnikova, L., Clark, B. F. C., and Nyborg, Y. (1995)
Science, 270, 1464-1472.
48.Rolland, N., Yanosi, L., Block, M. A., Shuda, M.,
Teyssier, E., Miege, C., Cheniclet, C., Carde, Y. P., Kaji, A., and
Joyard, Y. (1999) Proc. Natl. Acad. Sci. USA, 96,
5464-5469.
49.Pavlov, M. Y., and Ehrenberg, M. (1996) Arch.
Biochem. Biophys., 328, 9-16.
50.Tate, W. P., Poole, E. S., and Mannering, S. A.
(1996) Prog. Nucleic Acids Res. Mol. Biol., 52,
293-335.
51.SenGupta, D. Y., Zhang, B., Kraemer, B., Poshert,
P., Fields, S., and Wickens, M. (1996) Proc. Natl. Acad. Sci.
USA, 93, 8496-8501.
52.SenGupta, D. Y., Wickens, M., and Fields, S.
(1999) RNA, 5, 596-601.