Mini-REVIEW: Ribosomal RNAs in Translation Termination: Facts and Hypotheses
A. L. Arkov and E. J. Murgola*
Department of Molecular Genetics (Box 11), The University of Texas M. D. Anderson Cancer Center, 1515 Holcombe Blvd., Houston, TX 77030, USA; fax: (713) 794-4295; E-mail: ejm@mdacc.tmc.edu* To whom correspondence should be addressed.
Received June 2, 1999
It is now well established that ribosomal RNAs (rRNAs) play an active role in every aspect of translation. This review focuses on recent evidence for the involvement of rRNAs from both subunits of the ribosome in translation termination. This evidence comprises data obtained with rRNA mutants both in vivo and in vitro. In particular, mutations in specific regions of rRNAs caused readthrough of nonsense codons in vivo. Consistent with their in vivo characteristics, the mutations decreased the productive association of the ribosome with release factor 2 (RF2) and the efficiency of catalysis of peptidyl-tRNA hydrolysis in the presence of RF2 in realistic in vitro termination systems. It is now evident that genetic selections for termination-defective mutants in vivo and their characterization in realistic in vitro termination assays will rapidly advance our understanding of the mechanism of termination.
KEY WORDS: termination, ribosomal RNA, release factor, helix 34, helix 44, GTPase center, peptidyl transferase, Escherichia coli
BASIC FEATURES OF TRANSLATION TERMINATION
When the A site of the ribosome encounters a stop codon, a cytoplasmic protein release factor (RF) binds to the ribosome and triggers hydrolysis of peptidyl-tRNA. In Escherichia coli, there are two codon-dependent RFs that are necessary for termination, RF1 and RF2. RF1 functions at UAA and UAG and RF2 at UAA and UGA. Until recently the role of ribosomal RNA in this process has been unclear. For recent reviews on translation termination and RFs see [1-6].
INVOLVEMENT OF SMALL-SUBUNIT rRNA IN TRANSLATION TERMINATION
Small-subunit rRNA was implicated in termination based on evidence obtained in vivo and in vitro. In particular, a mutation in E. coli 16S rRNA, C1054A, was isolated as a suppressor of UGA nonsense mutations [7, 8]. C1054 is a virtually universally conserved nucleotide [9, 10] of helix 34 (Fig. 1a). Furthermore, it was shown that C1054A did not suppress either UAA or UAG under the same conditions where UGA suppression was detected, that is on glucose minimal medium [7, 8]. Two other substitutions at position 1054 caused suppression of not only UGA but also UAG (C1054U), and UAA and UAG (C1054G) [8]. However, none of the mutations at position 1054 caused missense suppression of UGG or several other sense codons different from stop codons by just one nucleotide [8]. Besides, the mutations at position 1054 did not suppress two +1 frameshift mutants and one -1 frameshift mutant [8]. Therefore in vivo data pointed to a defect in termination caused by substitutions at position 1054 because the mutations caused readthrough of nonsense codons but were not seen to affect other misreading events under certain conditions [7, 8]. For the eukaryote Saccharomyces cerevisiae, mutations at the position of 18S rRNA that corresponds to position 1054 of E. coli 16S rRNA were shown to affect suppression of nonsense mutations, implicating this nucleotide in eukaryotic termination [13]. Consistent with the in vivo characteristics of mutations at position 1054 of 16S rRNA were the recent observations in a realistic in vitro termination assay [14, 15] that C1054A decreased the effective association rate constant, kcat/Km, for ribosome and RF2 [16] and the catalytic rate of peptidyl-tRNA hydrolysis after RF2 was bound to the ribosome [17]. On the other hand, C1054A caused a very small decrease in kcat/Km for RF1-dependent termination that was significantly smaller than that in the presence of RF2 [16]. Furthermore the catalytic rate of peptidyl-tRNA hydrolysis in the presence of RF1 was very close to normal [17]. These in vitro data provided an explanation for the UGA-specific suppression caused by C1054A in vivo, namely, mutant ribosomes read through UGA because they were defective in RF2-dependent termination, but readthrough of UAA and UAG was not seen due to the almost normal function of RF1 at these codons. Furthermore, other nucleotides of helix 34 are implicated in termination (Fig. 1a). In particular the mutation C1200U in helix 34 also caused readthrough of nonsense codons [18, 19], however, similar to C1054A, it did not suppress several missense mutations, including UGG, AAG, and GAA (F. T. Pagel and E. J. Murgola, unpublished data). In addition, changes of C1192 in the helix caused defects in the binding of both RF1 and RF2 to the ribosome in vitro [20]. Similar to the effect of C1054A on termination, defects in RF binding caused by the mutations C1192U and C1192G were larger for RF2 than for RF1.
There is evidence that another region of 16S rRNA, helix 44, is involved in termination. In particular for E. coli, it was shown that cloacin DF13 cleavage (between A1493 and G1494) of the part of the helix that contributes to the A site (Fig. 1a) inhibits formation of a complex between ribosomes, UAA, and RF1 and RF2 [21]. In addition, the aminoglycoside antibiotics hygromycin and neomycin, which interact with the A-site portion of the helix 44 [22], substantially inhibited termination driven by either RF1 or RF2 in vitro [20]. On the other hand, UAA, bound to the A site, was cross-linked to helix 44 and RF2 [23] demonstrating the close proximity of RF2 to this helix and suggesting a direct functional interaction between RF2 and helix 44 in the A site and/or the stop codon during termination. While the importance of the A-site portion of the helix 44 for termination is well substantiated, some recent data indicate involvement of other sites in that helix in the termination process (S. Q. Zhao, F. T. Pagel, and E. J. Murgola, unpublished data).Fig. 1. rRNA regions implicated in translation termination. a) Top: the secondary structure of 16S rRNA (small subunit) with helix 34 and the base of helix 44, the major part of which is involved in the A site [11], indicated in bold. Bottom left: detail of helix 34, with nucleotides implicated in termination encircled. Bottom right: detail of the base of helix 44 with the site of cloacin DF13 cleavage indicated (arrow). b) Top: The secondary structure of 23S rRNA (large subunit) with the GTPase center indicated in bold. The general location of the RNA component of peptidyl transferase in domains IV and V [12] is indicated (PT). Location of the two-nucleotide deletion that apparently compensates for termination defects caused by the mutation G1093A in the GTPase center is shown (DeltaG2046, C2047). Bottom: detail of the GTPase center with nucleotides implicated in termination encircled.
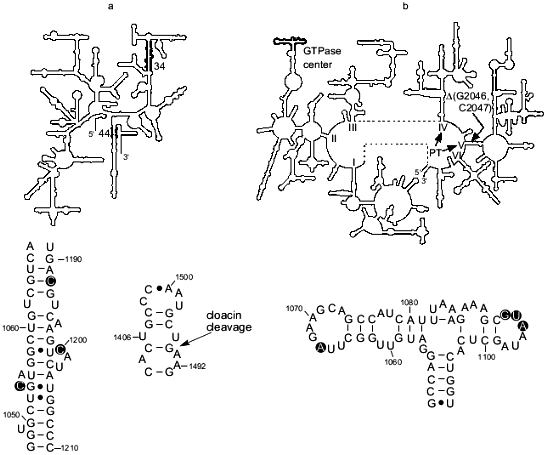
C1054 of helix 34, the A-site portion of helix 44, and the A-site codon are situated very close to each other in the 30S ribosomal subunit [23-25]. Therefore it is tempting to speculate that the C1054 region of helix 34, the A-site nucleotides of helix 44, and the three nucleotides of a stop codon form a structurally-unique surface that is specifically recognized by a small portion of RF. The strength of this recognition may be modulated by nucleotides adjacent to the stop codon [15, 26-29] and P-site bound tRNA [28, 30].
INVOLVEMENT OF LARGE-SUBUNIT rRNA IN TRANSLATION TERMINATION
The GTPase center. Genetic selections and screenings for E. coli 23S rRNA mutants that suppress UGA as well as detailed mutational analysis of specific rRNA regions and characterization of the rRNA mutants in a realistic in vitro termination system have implicated the GTPase center in termination (Fig. 1b). The GTPase center is a region involved in elongation factor G- and Tu-dependent hydrolysis of GTP [31, 32]. Specifically, the substitution of A for virtually universally conserved G1093 [9, 10] was isolated as a UGA-specific suppressor [33], namely, it did not suppress UAA and UAG nonsense mutations or several missense mutations, including UGG, under certain conditions. Therefore, as with C1054A of 16S rRNA, it was suggested that A1093 caused a defect in termination [33]. This suggestion was supported by analysis of the ribosomes with G1093A in realistic in vitro termination assays [14, 15]. In vitro this mutation severely decreased the effective association rate constant, kcat/Km, for RF2 and the ribosome. At UAA, the kcat/Km for mutant ribosomes was about 20 times lower than for wild-type ribosomes, and at UGA, it was 14 times lower [16]. As opposed to the large defect in RF2-dependent termination, G1093A caused just a 46% decrease in the kcat/Km for RF1 at UAA [16]. Furthermore the mutation decreased the catalytic rate of peptidyl-tRNA hydrolysis after RF2 was bound to the ribosome but RF1-mediated hydrolysis on the mutant ribosomes was very close to that on the wild-type ribosomes [17]. The major defect in RF2-dependent termination caused by G1093A in vitro was consistent with the specific suppression of UGA (RF2-specific stop codon) seen in vivo in the presence of the mutation [33]. As for C1054A in 16S rRNA discussed above, defects in RF1-dependent termination detected in vitro for the G1093A mutant might not be sufficiently large to cause UAG and UAA suppression under the same conditions where UGA suppression was seen [16]. The in vitro data indicated that G1093A affected binding of RF2 to the ribosome thereby suggesting that the G1093 region is a binding site for RF2 [16]. In addition to G1093, other nucleotides of the 1093-1098 loop in the GTPase center were implicated in RF2-dependent termination based on the UGA-specific suppression caused in vivo by different mutations in the loop (Fig. 1b) ([19, 34]; W. Xu and E. J. Murgola, unpublished data). Furthermore deletion of A1067 in another loop of the GTPase center as well as A1067C and A1067U (Fig. 1b) caused UGA-specific suppression also (W. Xu and E. J. Murgola, unpublished data). The recent determination of the crystal structure of the GTPase-center fragment bound to ribosomal protein L11 showed that both the 1093-1098 loop and the 1065-1073 loop are situated very close to each other [35, 36]. Considering that observation along with the results of functional studies in vivo ([33, 34]; W. Xu and E. J. Murgola, unpublished data) and in vitro [16, 17], it seems plausible that both loops interact with a compact region of RF2. Experiments in our laboratory are underway to test the hypothesis of direct interaction between the two loops of the GTPase center and RF2.
The UGA-specific suppression caused by the mutations in the GTPase center indicates that RF1 does not interact with the two loops of the GTPase center. Instead RF1 may interact with ribosomal protein L11 which binds to the GTPase center [35, 36]. An L11--RF1 interaction was suggested earlier [37, 38]. The small decrease in kcat/Km in the RF1-dependent reactions seen in vitro with G1093A-containing ribosomes [16] may be an indirect effect of this mutation on a hypothetical L11--RF1 complex.
Where is the hydrolytic site? An important question is where the site that directly catalyzes the hydrolysis of peptidyl-tRNA is located. Is this site on the RFs or/and on the ribosome? There is evidence that the ribosome contains such a site. An important discovery, which led to this point of view, was made by Caskey et al. [39], who showed that both bacterial and mammalian ribosomes can carry out the hydrolysis of peptidyl-tRNA without RFs in the presence of 30% acetone and tRNA. Mutations in rRNAs that substantially decrease catalysis of peptidyl-tRNA hydrolysis in the presence of just one RF, like C1054A [16S rRNA] and G1093A [23S rRNA], are not likely to affect the hydrolytic site. Mutations affecting such a hydrolytic site would be expected to cause decreases in the catalytic rates of peptidyl-tRNA hydrolysis in the presence of both RF1 and RF2 in vitro and readthrough of all stop codons in vivo. Therefore characterization of some non-specific rRNA suppressors of nonsense mutations in an in vitro termination system may lead to identification of the hydrolytic site. Meanwhile there is indirect evidence that the ribosomal peptidyl transferase is involved in hydrolysis of peptidyl-tRNA during termination [39-41]. Furthermore 23S rRNA has been shown to be the central element of peptidyl transferase [12, 42]. Therefore the 23S rRNA component of peptidyl-transferase (Fig. 1b) may be at least a part of the hydrolytic site. Involvement of the rRNA component of peptidyl transferase in termination has been proposed [43].
Under physiological conditions, association of either RF1 or RF2 with the ribosome activates the hydrolytic site. The mechanism of this activation is unknown. However there is genetic evidence for an interaction between the GTPase center in domain II and the entrance to domain V during termination that may be part of the activation process. More specifically, deletion of two nucleotides in the entrance to domain V, Delta(G2046, C2047), was found to compensate for the temperature-conditional lethality caused by G1093A in the GTPase center (Fig. 1b) [19, 44]. Furthermore, this deletion dramatically decreased the very effective UGA suppression caused by G1093A in vivo suggesting that Delta(G2046, C2047) was able to compensate for termination defects caused by G1093A. In addition, the deletion was found to cause weak UGA suppression on its own (A. L. Arkov and E. J. Murgola, unpublished data) implicating the entrance in the termination process. Collectively these data are consistent with the possibility of an interaction between the GTPase center and the entrance to domain V that may be important for proper termination. Since domain V has been implicated in peptidyl-transferase activity [12], one may speculate that an interaction between the GTPase center (domain II) and the entrance to domain V is important for the transformation of peptidyl transferase into peptidyl-tRNA hydrolase. Consistent with this hypothesis of a direct interaction between domain II and domain V is the proximity of these domains to each other [45, 46]. However, an indirect interaction between the GTPase center and the entrance to domain V, perhaps through RF2, is also possible.
“I fall to pieces...”A new approach to the study of rRNA involvement in termination [47, 48] has led to identification of fragments of E. coli 23S rRNA that caused UGA-specific suppression, namely, a domain I fragment, nucleotides 74-136, the antisense to that domain I fragment [47], and the antisense to a domain II segment, nucleotides 735-766 [48]. This approach included screening a library of rRNA fragments [49] to identify those whose expression caused suppression of UGA nonsense mutations in reporter genes. Since the identified fragments caused suppression of UGA but not UAA or UAG, it was suggested that they interfere with RF2-dependent termination [47, 48]. Precise mechanisms of UGA readthrough by the fragments await their discoveries, it seems likely however, that the fragment approach will be a valuable addition to the “tool box” for studying ribosome function [47-49].
CONCLUSION: rRNA INVOLVEMENT IN TRANSMISSION OF A STOP SIGNAL IN THE
PRESENCE OF RF2
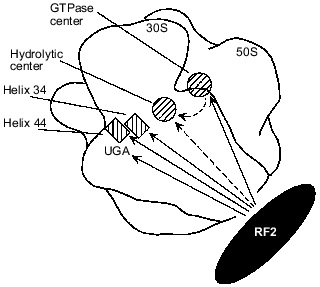
Available data make it possible to propose a model for the involvement of rRNAs in RF2-dependent termination. Figure 2 shows possible interactions between RF2 and different rRNA regions as well as an rRNA--rRNA interaction that may be involved in transmission of a stop signal (UGA) from the small subunit portion at helix 34 and helix 44 to the hydrolytic center of the large subunit. It is possible that when UGA enters the A site of the ribosome, RF2 directly binds to the UGA, as suggested by the results of cross-linking experiments [23, 53], and to helix 34 [6-8, 16, 17, 19, 20] and to helix 44 [20]. As discussed above, UGA, helix 34, and helix 44 together may form a unique structure specifically recognized by RF2. RF2 may also interact with the GTPase center as suggested by results obtained in vivo ([6, 19, 33, 34]; W. Xu and E. J. Murgola, unpublished data) and in vitro [16, 17]. After RF2 binds to the rRNA regions, the GTPase center may directly interact with the domain V of 23S rRNA ([19, 44]; A. L. Arkov and E. J. Murgola, unpublished data) which is a likely place for the hydrolytic center as discussed above. This RNA--RNA interaction may activate the hydrolytic center resulting in hydrolysis of peptidyl-tRNA. Alternatively RF2 may interact with and activate the hydrolytic center (Fig. 2).
Fig. 2. Possible functional interactions during transmission of a stop signal in the presence of RF2. Approximate ribosome locations of rRNA regions implicated in RF2-dependent termination are shown as either hatched rectangles (helix 34 and helix 44 of 16S rRNA) or hatched circles (23S rRNA regions, namely, the GTPase center and the hydrolytic center). A complete ribosome assembled from a 30S and a 50S subunit is viewed from the solvent side of the 30S subunit. Outlines of the 30S and 50S subunits are according to Frank et al. [50]. Locations of helix 34, helix 44, and the A-site UGA codon were deduced from their proximity to the anticodon of the A-site tRNA whose ribosome location has been presented ([51] and references therein). The GTPase center is shown according to [52]. The hydrolytic center is assumed to be situated near the acceptor end of the P-site tRNA whose location has been presented ([51] and references therein). Functional interactions between RF2 and RNA elements strongly suggested by the studies referred to in the text are shown as continuous arrows originating from RF2. Other interactions, for which there are few data available, are shown as dashed arrows.
PERSPECTIVES
While some progress in studying the involvement of rRNAs in translation termination has been made, we are yet very far from understanding the mechanism of the termination process. It is very unclear what exactly RF recognizes, what the hydrolytic center is, and how interaction of the RF with the ribosome triggers the hydrolytic center to release a polypeptide. However, satisfactory answers to these exciting problems should not be long in coming since genetic approaches to finding termination-defective mutants have proven to be successful, as indicated by the use of realistic in vitro termination assays.
We are very grateful to the former and present members of our laboratory, in particular Natalya Chernyaeva, Klas Hedenstierna, Frances Pagel, Wenbing Xu, and Song Zhao for sharing unpublished data and their support. We would also like to thank Möns Ehrenberg and members of his lab, in particular, David Freistroffer, Reza Karimi, and Michael Pavlov for very fruitful collaboration on rRNA mutants. We wish to thank Alexander Mankin for his great participation on identification of rRNA fragments. This work was supported by grant GM21499 from the National Institute of General Medical Sciences (USA) to E.J.M.
REFERENCES
1.Nakamura, Y., Ito, K., and Isaksson, L. A. (1996)
Cell, 87, 147-150.
2.Tate, W. P., Dalphin, M. E., Pel, H. J., and
Mannering, S. A. (1996) Genet. Eng., 18,157-182.
3.Tate, W. P., and Mannering, S. A. (1996) Mol.
Microbiol., 21,213-219.
4.Tate, W. P., Poole, E. S., and Mannering, S. A.
(1996) Prog. Nucleic Acids Res. Mol. Biol., 52,
293-335.
5.Buckingham, R. H., Grentzmann, G., and Kisselev, L.
(1997) Mol. Microbiol., 24, 449-456.
6.Murgola, E. J. (1996) in Ribosomal RNA:
Structure, Evolution, Processing, and Function in Protein
Biosynthesis, CRC Press, Boca Raton, pp. 357-369.
7.Murgola, E. J., Hijazi, K. A., Göringer, H.
U., and Dahlberg, A. E. (1988) Proc. Natl. Acad. Sci. USA,
85,4162-4165.
8.Pagel, F. T., Zhao, S. Q., Hijazi, K. A., and
Murgola, E. J. (1997) J. Mol. Biol., 267,1113-1123.
9.Maidak, B. L., Olsen, G. J., Larsen, N., Overbeek,
R., McCaughey, M. J., and Woese, C. R. (1996) Nucleic Acids
Res., 24, 82-85.
10.Van de Peer, Y., Chapelle, S., and De Wachter, R.
(1996) Nucleic Acids Res., 24, 3381-3391.
11.Zimmermann, R. A. (1996) in Ribosomal RNA:
Structure, Evolution, Processing, and Function in Protein
Biosynthesis, CRC Press, Boca Raton, pp. 277-309.
12.Garrett, R. A., and Rodriguez-Fonseca, C. (1996)
in Ribosomal RNA: Structure, Evolution, Processing, and Function in
Protein Biosynthesis, CRC Press, Boca Raton, pp. 327-355.
13.Chernoff, Y. O., Newnam, G. P., and Liebman, S.
W. (1996) Proc. Natl. Acad. Sci. USA, 93,2517-2522.
14.Freistroffer, D. V., Pavlov, M. Yu., MacDougall,
J., Buckingham, R. H., and Ehrenberg, M. (1997) EMBO J.,
16,4126-4133.
15.Pavlov, M. Yu., Freistroffer, D. V.,
Dinçbas, V., MacDougall, J., Buckingham, R. H., and Ehrenberg, M.
(1998) J. Mol. Biol., 284,579-590.
16.Arkov, A. L., Freistroffer, D. V., Pavlov, M.
Yu., Ehrenberg, M., and Murgola, E. J., submitted.
17.Arkov, A. L., Freistroffer, D. V., Ehrenberg, M.,
and Murgola, E. J. (1998) EMBO J., 17, 1507-1514.
18.Moine, H., and Dahlberg, A. E. (1994) J. Mol.
Biol., 243,402-412.
19.Murgola, E. J., Pagel, F. T., Hijazi, K. A.,
Arkov, A. L., Xu, W., and Zhao, S. Q. (1995) Biochem. Cell
Biol., 73, 925-931.
20.Brown, C. M., McCaughan, K. K., and Tate, W. P.
(1993) Nucleic Acids Res., 21, 2109-2115.
21.Caskey, C. T., Bosch, L., and Konecki, D. S.
(1977) J. Biol. Chem., 252,4435-4437.
22.Purohit, P., and Stern, S.(1994) Nature,
370,659-662.
23.Tate, W., Greuer, B., and Brimacombe, R. (1990)
Nucleic Acids Res., 18, 6537-6544.
24.Dontsova, O., Dokudovskaya, S., Kopylov, A.,
Bogdanov, A., Rinke-Appel, J., Jünke, N., and Brimacombe, R.
(1992) EMBO J., 11, 3105-3116.
25.Rinke-Appel, J., Jünke, N., Brimacombe, R.,
Dokudovskaya, S., Dontsova, O., and Bogdanov, A. (1993) Nucleic
Acids Res., 21, 2853-2859.
26.Buckingham, R. H., Sörensen, P., Pagel, F.
T., Hijazi, K. A., Mims, B. H., Brechemier-Baey, D., and Murgola, E. J.
(1990) Biochim. Biophys. Acta, 1050,259-262.
27.Poole, E. S., Brown, C. M., and Tate, W. P.
(1995) EMBO J., 14, 151-158.
28.Zhang, S., Rydén-Aulin, M., and Isaksson,
L. A. (1996) J. Mol. Biol., 261,98-107.
29.Poole, E. S., Major, L. L., Mannering, S. A., and
Tate, W. P. (1998) Nucleic Acids Res., 26, 954-960.
30.Arkov, A. L., Korolev, S. V., and Kisselev, L. L.
(1993) Nucleic Acids Res., 21, 2891-2897.
31.Thompson, J. (1996) Ribosomal RNA: Structure,
Evolution, Processing, and Function in Protein Biosynthesis, CRC
Press, Boca Raton, pp. 311-325.
32.Saarma, U., Remme, J., Ehrenberg, M., and Bilgin,
N. (1997) J. Mol. Biol., 272,327-335.
33.Jemiolo, D. K., Pagel, F. T., and Murgola, E. J.
(1995) Proc. Natl. Acad. Sci. USA, 92, 12309-12313.
34.Xu, W., and Murgola, E. J. (1996) J. Mol.
Biol., 264, 407-411.
35.Conn, G. L., Draper, D. E., Lattman, E. E., and
Gittis, A. G. (1999) Science, 284, 1171-1174.
36.Wimberly, B. T., Guymon, R., McCutcheon, J. P.,
White, S. W., and Ramakrishnan, V. (1999) Cell, 97,
491-502.
37.Tate, W. P., Dognin, M. J., Noah, M.,
Stöffler-Meilicke, M., and Stöffler, G. (1984) J. Biol.
Chem., 259, 7317-7324.
38.Tate, W. P., McCaughan, K. K., Ward, C. D.,
Sumpter, V. G., Trotman, C. N. A., Stöffler-Meilicke, M., Maly,
P., and Brimacombe, R. (1986) J. Biol. Chem., 261,
2289-2293.
39.Caskey, C. T., Beaudet, A. L., Scolnick, E. M.,
and Rosman, M. (1971) Proc. Natl. Acad. Sci. USA, 68,
3163-3167.
40.Vogel, Z., Zamir, A., and Elson, D. (1969)
Biochemistry, 8, 5161-5168.
41.Caskey, C. T., and Beaudet, A. L. (1972) in
Molecular Mechanisms of Antibiotic Action on Protein Biosynthesis
and Membranes, Elsevier Scientific Publishing Company,
Amsterdam-London-New York, pp. 326-336.
42.Noller, H. F., Hoffarth, V., and Zimniak, L.
(1992) Science, 256, 1416-1419.
43.Tate, W. P., and Brown, C. M. (1992)
Biochemistry, 31, 2443-2450.
44.Murgola, E. J., Xu, W., and Arkov, A. L. (1995)
Nucleic Acids Symp. Ser., 33, 70-72.
45.Stiege, W., Glotz, C., and Brimacombe, R. (1983)
Nucleic Acids Res., 11, 1687-1706.
46.Bogdanov, A. A., Dontsova, O. A., Dokudovskaya,
S. S., and Lavrik, I. N. (1995) Biochem. Cell Biol., 73,
869-876.
47.Arkov, A. L., Mankin, A., and Murgola, E. J.
(1998) J. Bact., 180, 2744-2748.
48.Chernyaeva, N. S., Murgola, E. J., and Mankin, A.
S. (1999) J. Bact., 181, 5257-5262.
49.Tenson, T., DeBlasio, A., and Mankin, A. (1996)
Proc. Natl. Acad. Sci. USA, 93, 5641-5646.
50.Frank, J., Penczek, P., Grassucci, R., and
Srivastava, S. (1991) J. Cell Biol., 115, 597-605.
51.Wilson, K. S., and Noller, H. F. (1998)
Cell, 92, 337-349.
52.Wilson, K. S., and Noller, H. F. (1998)
Cell, 92, 131-139.
53.Brown, C. M., and Tate, W. P. (1994) J. Biol.
Chem., 269, 33164-33170.