Mini-REVIEW: The [URE3] Yeast Prion: from Genetics to Biochemistry
A. A. Komar1*, R. Melki2*, and C. Cullin1*
1Centre de Genetique Moleculaire, Centre National de la Recherche Scientifique, 91198 Gif-sur-Yvette Cedex, France; E-mail: anton.komar@cgm.cnrs-gif.fr, christophe.cullin@cgm.cnrs-gif.fr2Laboratoire d'Enzymologie et Biochimie Structurales, Centre National de la Recherche Scientifique, 91198 Gif-sur-Yvette Cedex, France; E-mail: ronald.melki@lebs.cnrs-gif.fr
* To whom correspondence should be addressed.
Received June 15, 1999
[URE3] is a non-Mendelian genetic element of the yeast Saccharomyces cerevisiae, an altered prion form of Ure2 protein. We show that recombinant Ure2p is a soluble protein that can assemble in vitro into dimers, tetramers, and octamers or form insoluble fibrils observed for PrP in its filamentous form or for Sup35p upon self-assembling, suggesting a similar mechanism for all prions. Computational, genetic, biochemical, and structural data allow us to specify a new boundary between the so-called prion-forming and nitrogen regulator (catalytic) domains of the protein and to map this boundary to Met-94. We bring strong evidence that the COOH-terminal (94-354) part of the protein forms a tightly folded domain, while the NH2-terminal (1-94) part is unstructured. These domains (or various parts of these domains) were shown (by means of the two-hybrid system approach and affinity binding experiments) to interact with each other (both in vivo and in vitro). We bring also evidence that the COOH-terminal (94-354) catalytically active part of the protein can be synthesized (both in vitro and in vivo) via an internal ribosome-binding mechanism, independently of the production of the full-length protein. We finally show that Ure2p aggregation in vivo (monitored by fluorescence of Ure2p--GFP fusion) does not necessarily give rise to [URE3] phenotype. The significance of these findings for the appearance and propagation of the yeast prion [URE3] is discussed.
KEY WORDS: [URE3], Ure2p, prion, yeast, translation, expression, structure, function, aggregation
Several aberrant genetic elements have been described in the yeast Saccharomyces cerevisiae. The pioneering work of Brian Cox and Francois Lacroute at first lead to the discovery of the so-called [PSI] and [URE3] yeast non-Mendelian genetic elements (or phenotypes) [1-4]. These two phenotypes reveal similar genetic characteristics. First, both are dominant (the phenotype is maintained in a diploid cell that results from a cross between a haploid mutant and haploid wild-type yeast cell). Second, the diploid cells (when allowed to sporulate) may form tetrads among which many show a 4/0 segregation (all four spores display the mutant phenotype). Third, both [PSI] and [URE3] elements can be transmitted from a mutant cell to a wild-type cell by a cytoduction procedure (transfer of cell cytoplasm), which excludes the possibility of the transfer of nuclear genetic information.
Surprisingly, both [PSI] and [URE3] phenotypes can be lost (or reverted) in cells growing in the presence of 5 mM guanidine hydrochloride [1-4]. The later observation suggests a common mechanism underlying the appearance and maintenance of these two mutant phenotypes. However, it should be emphasized that these two phenotypes are not related. [PSI] arises spontaneously and independently of [URE3] and vice versa [3, 4].
In this review we will mainly focus on the yeast non-Mendelian genetic element [URE3] and Ure2p, the putative yeast prion protein involved in the appearance and propagation of this phenotype. We will describe genetic properties of [URE3], describe selected features of Ure2p translation, and discuss specific structural properties of recombinant Ure2p (which was purified after expression in E. coli host cells).
THE URE2 GENE AND THE [URE3] PHENOTYPE
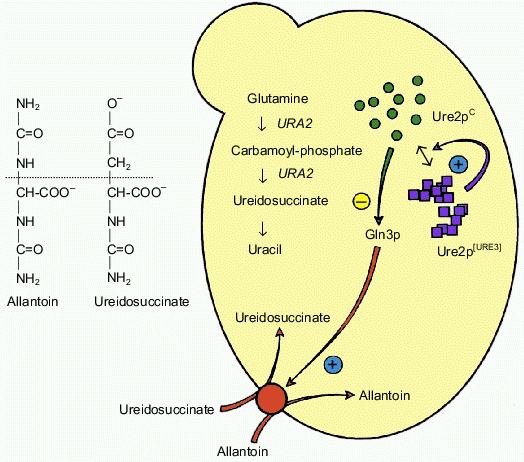
The URE2 gene is non-essential in yeast [5]. It was initially characterized during an indirect genetic screen by F. Lacroute and co-workers searching for yeast mutants unable to produce the ureidosuccinate metabolite [4]. Such mutants were isolated as yeast colonies able to grow on a selective medium containing ureidosuccinate. Many of these mutants represented the same complementary group which defines the URE2 locus. More recently, it has been shown that the deletion of the yeast URE2 gene leads to the same phenotype. It follows that the [Ureidosuccinate+] phenotype arises due to a complete loss of function of the Ure2 protein (the product of the URE2 gene). The uptake of ureidosuccinate in yeast cells is in fine related to nitrogen catabolic repression (NCR). The permease responsible for the uptake of ureidosuccinate is under transcriptional control of the Gln3 protein. Gln3p itself is a target of Ure2p action and is widely assumed to be inactivated upon interaction with Ure2p. In the presence of a rich nitrogen source (such as NH4+), the Ure2p protein prevents the activation of many target genes by Gln3p. This type of regulation is abolished if cells grow in the presence of a poor nitrogen source such as allantoin. Since ureidosuccinate enters the cell due to allantoin permease, its uptake is under the control of the NCR pathway [6-8]. The inactivation of URE2 prevents the inhibition of Gln3p, which activates constitutively numerous target genes (which are under positive control of this transcriptional factor) without any requirement for the nitrogen source. Surprisingly, it turned out that various URE2 mutants are genetically different. The [URE3] mutant was selected among those which form a group displaying very unstable [Ureidosuccinate+] phenotype. More than twenty years later the prion hypothesis appeared to explain the unusual behavior of these mutants [9]. It has been suggested that Ure2p can exist in two distinct structural states. Ure2pC in its native conformation represents the functional form, while the Ure2p[URE3] is suggested to be a non-functional prion-like isoform. The later isoform has been proposed to acquire a new feature that is a characteristic of a prion; it can convert Ure2pC into the altered non-functional Ure2p[URE3] conformation. These features of URE2, [URE3], and Ure2p are summarized in Fig. 1 (see colored insert).
Fig. 1. The prion hypothesis for [URE3]. The [URE3] phenotype corresponds to a loss of function of Ure2pC but the genetic experiments argue for a model in which no mutations are involved. The protein would exist in a functional form (Ure2pC) and also in a non-functional form (Ure2p[URE3]). This altered form would have acquired the remarkable property of being able to convert the wild-type protein to the abnormal form, similar to the mechanism described for mammalian prions.
Ure2p SYNTHESIS
Prokaryotic and eukaryotic in vitro translation systems have now become the focus of increasing interest for solving fundamental problems of molecular biology and biochemistry. Cell-free systems are widely used to study the assembly and folding of proteins and viral particles and the import of proteins into various cellular compartments and organelles; they allow production and analysis of proteins and their mutants (for review see [10]). We attempted to express Ure2p in various eukaryotic cell-free systems with the aim of performing preliminary structural studies of this protein [11]. Our results showed that depending on the factors present in the cell-free translation systems, the newly synthesized Ure2p differs regarding its structural properties [11]. Interestingly, translation of Ure2p in a cell-free system results in two polypeptide products: full-length Ure2p and a truncated 30-kD product. At first we suggested that the 30-kD fraction could be ascribed to an .abortive. polypeptide (apparently, the product of premature termination and release of the incomplete nascent Ure2p chains) [11]; however, it appeared that mutation of AUG94 into CUU (Leu) in the URE2 open reading frame (ORF) abolished production of the 30-kD polypeptide [12]. This suggested the existence of Ure2p mRNA of an alternative ribosome-binding pathway downstream from the first initiator AUG codon, and it prompted us to look for a possible internal ribosome entry site (IRES).
Initiation of translation of the majority of eukaryotic mRNAs is accomplished by a cap- and 5´-end-dependent mechanism involving scanning of 5´-untranslated regions (UTRs) by small ribosomal subunits (for review see [13]). Eukaryotes have acquired a group of initiation factors which seems to have no analogs in prokaryotes and which includes special mRNA (cap)-binding proteins (in particular eIF-4E) and mRNA-unwinding proteins which are known to facilitate initiation of translation. A set of eukaryotic mRNAs, however, appeared to exploit a different initiation mode based on a poorly understood cap- (and eIF-4E) and 5´-end-independent internal ribosome-binding mechanism, a process which was initially thought to be restricted to picornaviral RNAs (for review see [14]). Such mRNAs contain the IRES which can mediate translation initiation in the absence of a free 5´-end in the mRNA, probably by direct recruitment of ribosomal subunits.
Studies of Ure2p translation in vitro in various cell-free systems as well as in vivo allowed us to suggest that the large COOH-terminal part of the Ure2 protein (starting from AUG94) can be synthesized via a cap-independent internal ribosome entry mechanism [12]. Concurrent inhibition of initiation of Ure2p translation in vitro with cap-analog m7GDP showed significant decrease in the level of synthesis of the full-length protein, while the level of expression of the COOH-terminal (94-354) part was found to be unchanged. It was also demonstrated that depletion from the yeast cell-free translation system of the major cap-recognition factor eIF-4E significantly enhanced the translation of the C-terminal Ure2p part, but not the full-length protein. These observations further supported the suggestion that alternative initiation of translation of Ure2p at AUG94 could be 5´-end and cap-independent. The Ure2p IRES is presumably located within the 200 bp surrounding AUG94. Dicistronic vectors were used to test this suggestion. The dicistronic reporter CAT/IRES/LUC construct containing 164 bp fragment comprising nucleotides 203-367 of the URE2 ORF was shown to direct in vitro cap-independent translation of firefly luciferase. A similar result was obtained for beta-galactosidase translation in vivo in yeast cells in case when the same URE2 gene fragment was placed in the dicistronic CAT/IRES/lacZ vector. However, the increase in lacZ activity in vivo was observed mainly in 4--2 ts yeast strain which bears a temperature sensitive mutation in the eIF-4E gene after eIF-4E was shut off at 37°C [15]. This could mean that Ure2p IRES is mild and can be inefficient under normal conditions of cell-growth. Yeast cell-free translation data revealed that the COOH-terminal Ure2p (94-354) part mediated by IRES translation could represent (under normal conditions) not more than 5-7% of total Ure2p translation products (Fig. 2, colored insert).
In yeast strains with disrupted alleles of the URE2 gene, it was reported that a fragment smaller that the entire gene (apparently, as they suggested, encoding the polypeptide starting at AUG94) could fully complement URE2 [5]. We confirmed this observation by complementation analysis using the COOH-terminal (94-354) part of Ure2p [12]. It follows that internal translation initiation allows the production of the truncated protein which is still catalytically active, but lacks the so-called prion inducing (or forming) domain. Thus, present findings allow us to redefine the boundaries of the prion-forming and catalytic (nitrogen repression) Ure2p domains and assign this boundary to Met-94 instead of Arg-65, initially specified on the ground of URE2 deletion analysis [16].Fig. 2. Prion-forming domain (PFD) and nitrogen repression (catalytic) domains of Ure2p shown in red and blue as defined by R. Wickner [16]. At the bottom: Ure2p cell-free translation products (yeast extract)--full-length protein and 30-kD catalytic fragment. Arrows show the start points of translation initiation.

Some cellular mRNAs including those for immunoglobulin heavy chain binding protein (BiP), Drosophila antennapedia protein, fibroblast growth factor 2 (FGF-2), insulin-like growth factor II (IGF-II), initiation factor 4G (eIF-4G), platelet-derived growth factor B (PDGF-B), c-myc, vascular endothelial growth factor (VEGF) [17], human estrogen receptor ERalpha [18] were shown to contain an IRES. The exact physiological role of most of these IRESs is not yet clear [17], but it is evident that most of these genes encode important regulatory proteins. It is possible that some capped mRNAs can be translated both by a 5´-end-dependent scanning mechanism and by the internal ribosome-binding mechanism. Such mRNAs may contain IRES elements in their 5´-UTR thus mediating internal initiation at a time when eIF-4E is not available and can not bind the cap structure. Alternatively, an IRES located within the coding region of an mRNA could render the mRNA functionally polycistronic, and translation could result in the production of several protein products, like happens in case of fibroblast growth factor 2 [19] and human estrogen receptor ERalpha [18]. Alternative initiation of translation of the human FGF-2 results in the synthesis of four FGF-2 isoforms differing in their intracellular localization [19]. Three of them are nuclear and their constitutive expression is able to induce cell immortalization, whereas the other product is mostly cytoplasmic and can generate cell transformation. Very recently it was found that alternative translation initiation (due to a ribosome leaky scanning or, possibly, also due to an IRES) of histone H4 at AUG85 leads to the synthesis of the truncated product, which is osteogenic growth peptide [20]. Thus, alternative initiation of translation also increases diversity of gene products.
The presence of an IRES elements in particular mRNAs may reflect the need to maintain the translation of key vitality important proteins (or various parts of these proteins) under stress, condition when normal cap-dependent translation is compromised. It also seems very likely that translation initiation by internal binding can be used at particular times during cell development [17]. Cap-dependent initiation is known to be severely impaired during mitosis in mammalian cells because of underphosphorylation of the eIF-4E [21].
There is increasing awareness of the importance of the internal initiation of translation in the control of eukaryotic gene expression [14, 17, 22]. However, the biological relevance of the Ure2p IRES is not yet clear. Overexpression of Ure2p66-354 COOH-terminal part can cure [URE3] prion in [URE3] cells, possibly due to disruption of the Ure2p aggregates [16, 23]. Thus, the ratio between the full-length Ure2p and its truncated COOH-terminal part may affect the propagation of [URE3] in yeast cells and the balance between the full-length and truncated Ure2p products can be important for normal cell function. Our findings also suggest that cells may have developed a natural mechanism for curing the [URE3] prion. It follows that under conditions when cap-dependent initiation in yeast is compromised [24] (and thus more 94-354 Ure2p product can be produced due to internal initiation) [URE3] phenotype can be naturally cured. It ought also to be mentioned that a 60-nucleotide-long RNA termed IRNA, isolated from the yeast Saccharomyces cerevisiae, was shown to selectively block IRES-mediated translation [25, 26]. Up to now no natural target for this IRNA has been found in yeast cells. IRNA's ability to bind cellular transacting proteins believed to be required for IRES-mediated translation and its ability to block IRES-mediated translation [25, 26]. It is possible that different yeast strains contain different amounts of this IRNA and thus could reveal different activity in supporting IRES-mediated translation. Thus cells in which transacting proteins are blocked by IRNA could be apparently more prone to become [URE3].
Further studies will help to answer questions concerning the role of Ure2p IRES and the truncated Ure2p94-354 product in nitrogen regulation in yeast cells, the mechanism of Ure2p aggregation, as well as the propagation of the [URE3] prion.
STRUCTURAL PROPERTIES OF RECOMBINANT Ure2p
Ure2p autoassembles in vitro. It is now widely accepted that the basis for partitioning between different conformers of prion protein is provided by its particular, not yet fully understood structural properties. Thus, it is of keen interest to characterize and compare the structure of various proteins displaying prion properties and to compare them with that of mammalian prions. In an attempt to understand the features that can provide the basis for possible structural plasticity of Ure2 protein and to elucidate the mechanism by which [URE3] can arise de novo, we have carried out purification and characterization of Ure2p after its expression in Escherichia coli. The codon bias of the Ure2p gene has been optimized in order to overcome the problems due to differences in codon usage between prokaryotes and eukaryotes, which affected efficient translation of Ure2p in E. coli host cells [27]. The recombinant protein was then efficiently expressed and purified to near homogeneity and was found to be soluble [28]. Extreme care must be brought in order to get the full-length Ure2p, since the protein is prone to degradation during the purification procedure yielding a product that has a molecular mass of 38 kD (that corresponds to Ure2p devoid of its 20 first amino acid residues). The properties of this degradation product may be different from that of the authentic protein.
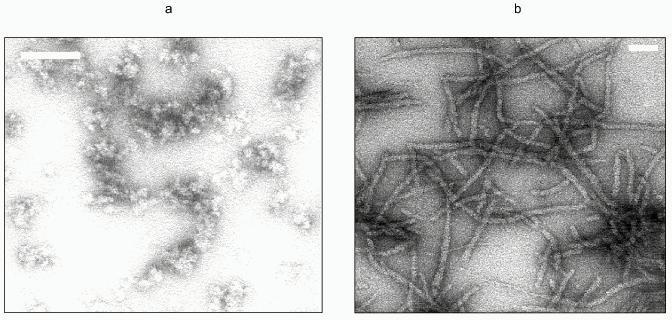
Full-length Ure2p is a monomeric protein that has a tendency to oligomerize into either soluble or insoluble oligomers [28]. The soluble oligomers may be composed of dimers, tetramers, or octamers of Ure2p. The proportion of each oligomer depends on the total concentration of Ure2p, which indicates that the oligomers are in equilibrium. Ure2p insoluble oligomers are very high-molecular-weight particles. Assembly of the soluble form of Ure2p into high-molecular-weight, insoluble, oligomers occurs spontaneously in vitro at pH 7.5. These oligomers form fibrils from 15- to 20-nm in width and over 10 µm in length; fibrils often associate laterally (Fig. 3, colored insert). These fibrils are obtained irrespectively of whether the solution of Ure2p is subjected to rotatory shaking or not. They are also obtained at various temperatures. Ure2p fibrils are similar to the fibrils observed upon assembly of the soluble form of the PrP91-231 [29] fragment as well as to Sup35p fibrils made of recombinant Sup35p that has been refolded in vitro after its solubilization from inclusion bodies [30]. Ure2p assembly into high-molecular-weight oligomers can also be induced by adjusting the pH of the solution to 6.4, the theoretical isoelectric point of the protein. In the latter case, assembly is instantaneous and generates aggregates that are very heterogeneous in size and shape (Fig. 3) that disappear upon increasing or lowering the pH of the solution. It is not clear whether these oligomeric forms of Ure2p are related to [URE3] phenotype. Indeed none of these oligomers has ever been observed in vivo.
Ure2p assembly into fibrils can be monitored by light scattering or Congo red binding. Binding of Congo red to amyloid fibrils is accompanied by a spectral shift of its absorbance maximum toward the UV. Assembly of Ure2p is a cooperative process. One can distinguish in the assembly kinetics a lag phase, where nuclei are formed, an acceleration phase where elongation of the nuclei occurs preceding the establishment of a plateau when the system reaches equilibrium. The amount of soluble form of Ure2p at equilibrium corresponds to the critical concentration for assembly. It is lower than 1 µM. The kinetics of Ure2p assembly are slowed down when assembly temperature is reduced from 28 to 4°C, which indicates that Ure2p oligomerization is a hydrophobic process. Finally, the lag phase preceding the onset of Ure2p assembly into insoluble oligomers disappears upon addition to the soluble form of Ure2p of preformed Ure2p fibrils. This strongly suggests a seeded polymerization model for Ure2p aggregation.Fig. 3. Electron micrographs of negatively stained Ure2p oligomers. a) Ure2p assembly into high-molecular-weight heterogeneous oligomers induced by adjusting the pH of the solution from 7.5 to 6.4, the theoretical isoelectric point of the protein. b) Ure2p fibrils obtainedupon autoassembly at pH 7.5. The white scale bar is 100 nm (a) and (b).
A number of proteins (actin, tubulin, RecA) self-assemble into filamentous structures. In all these cases assembly is followed by hydrolysis, an irreversible reaction that leads to destabilization of the polymers and their subsequent depolymerization. The first stage of assembly is a step where a conformational change occurs leading to an activated state of the protein that becomes capable of interacting with another activated monomer. Very little is known about the assembly of Ure2p. Nevertheless, the fact that assembly proceeds with a lag time is indicative of a rate-limiting step consistent with an activation of Ure2p monomer or dimer, i.e., a change in the conformation of the protein.
Limited proteolysis is a widely used technique to probe conformational changes in proteins. However, because protease protection is a relatively insensitive indicator for conformational changes, it is essential to carry out complementary measurements such as monitoring of the fluorescence of aromatic amino acid residues and circular dichroism. When such measurements are carried out with both soluble and insoluble forms of Ure2p, only discreet conformational changes are detected. The major conclusion from protease treatment data of the soluble and insoluble forms of Ure2p is that assembly protects the protein from proteolysis. Given that the first aromatic amino acid residue in Ure2p is located at position 99, intrinsic fluorescence measurements can only indicate conformational changes occurring within the Ure2p sequence stretching from 99 to 354. Slight conformational changes occur in this part of the molecule resulting in a shift of the fluorescence maximum from 339 to 336 nm. Finally, because circular dichroism measurements are difficult to interpret when a molecule undergoes assembly into highly structured polymers, we only measured conformational changes that can be induced by a number of solvents. The most interesting finding is that solvents such as trifluoroethanol that favor alpha-helical structures only increases the alpha-helical content of Ure2p by 8%. This suggests that Ure2p, which is rich in alpha-helices, has a low degree of flexibility.
Ure2p is at least a two-domain protein. Once evidences for conformational changes have been brought, the obvious question that arises is whether Ure2p is a multidomain protein and if this is the case whether the changes are located within a given domain. There are numerous definitions of protein domains (reviewed in [31]). The most widely accepted definition is that domains are substructures of a polypeptide chain which can be disconnected by limited proteolysis. Other definitions are based either on specific functional properties or on genetic units. Based on genetic and functional evidence, Ure2p is predicted to be a two-domain protein. When soluble, authentic Ure2p is subjected to treatment using various proteases, a product corresponding to the Ure2p94-354 moiety that has a molecular mass of 30 kD is generated. Interestingly, the same product was shown to arise due to alternative initiation of Ure2p translation. Proteolysis data indicate that the COOH-terminal part of Ure2p matches the definition of a protein domain [31]. Moreover, Ure2p1-94 as well and Ure2p94-354 fragments behave differently when subjected to proteolysis. While the NH2-terminal part of the molecule is quite sensitive to protease treatment, the COOH-terminal part resists proteolysis. After prolonged periods of incubation it is degraded into polypeptides that have molecular masses of around 20 kD. This strongly suggest that the Ure2p94-354 domain is not only compactly folded but also organized into subdomains, while the NH2-terminal part of the molecule (Ure2p1-94) has a looser conformation. At present, the slight conformational changes we documented cannot be attributed to a specific domain although they seem to involve the COOH-terminal part of Ure2p. Nevertheless the conformational changes we observed are not comparable to that induced upon reduction of disulfide bonds in the human PrP91-231 fragment [29].
Further characterization of Ure2p assembly into insoluble oligomers and the reversibility of this process, combined with the identification of Ure2p--Ure2p interface in the insoluble oligomers must be achieved in order to gain a full grasp of the mechanism of Ure2p invasive propagation.
Ure2p AND [URE3]: INTERACTION OF Ure2p DOMAINS
The prion paradigm postulates the existence of an unusual (so-called snow-ball) mechanism of interaction between prion molecules, which initiates the propagation of 'inactivated' protein [32]. The same holds true for Ure2p. It was therefore challenging to try to locate the interacting parts of Ure2p molecules [33]. The two-hybrid system approach which was chosen for this purpose revealed the existence in Ure2p of at least two polypeptide regions which might be involved in such intermolecular interaction. The first one (domain A) was located in the first third of the protein (spanning from residues 1 to 97). The second interacting part (domain B) was found to be located in the second two-thirds catalytic part of Ure2p (between residues 152 and 354). This observation was supported by affinity binding experiments in which corresponding N- and C-terminal parts of the protein were synthesized in an in vitro cell-free system [33].
It has been previously shown that the frequency of the spontaneous occurrence of [URE3] can be significantly enhanced upon overexpression of the URE2 gene, or its NH2-terminal part encoding first 65 amino acid residues of Ure2p [16] (which was defined as the prion-forming domain). However, a chimeric protein containing the first 65 residues of Ure2p fused in frame to E. coli ß-galactosidase was not found to induce [URE3] [16]. Interestingly, at the same time the overproduction of the catalytic (66-354) COOH-terminal domain induced a dramatic decrease in the frequency of [URE3] appearance [16, 23]. The above mentioned experiments allowed the authors to conclude that the catalytic domain can stabilize the NH2-terminal (prion forming) part of the protein thus preventing its conversion into the prion conformation. It was also postulated that destabilization of the COOH-terminal domain requires a covalent link between the NH2- and COOH-terminal parts of the protein [33]. These conclusions, however, do not provide any explanation for the effects due to over-expression of the full-length Ure2p. In the latest case, the relative moiety of both domains is unaltered; however, the COOH-domain seems not to prevent any more the formation of [URE3]. Unfortunately, in the experiments mentioned above the effective level of Ure2p synthesis and the amount of protein produced upon overexpression in yeast cells was not measured (e.g., by western blotting) and compared to that of nontransformed cells. As the concentration of the Ure2 protein is suggested to play a critical role in the formation and propagation of [URE3], one should be cautious in accepting the conclusions which were drawn, especially regarding the statement accepting the idea that the prion properties of Ure2p are solely provided by its first 65 amino acid residues.
Recently several point mutations located in the catalytic domain of Ure2p were mapped. These mutations were found to increase drastically the frequency of occurrence of [URE3] [35]. No difference in the intracellular concentration of the mutant proteins in comparison with the wild-type protein was noticed in yeast cells by western blotting analysis with anti-Ure2p antibodies. This finding clearly indicated that the C-terminal Ure2p part can also play a crucial role in the propagation of the [URE3] prion.
IS [URE3] PRION A RESULT OF Ure2p AGGREGATION in vivo?
Several recent studies have ascribed [PSI] and [URE3] phenotypes to an in vivo aggregation of the proteins implicated in the propagation of these prions [23, 36-41]. It was shown that Sup35p--green fluorescent (GFP) fusion protein is aggregated in cells carrying [PSI+] but the fluorescence was found to be evenly distributed in cells lacking the prion [36]. The same observation was recently made for Ure2p--GFP fusion [23]. These data supported a seeded polymerization model for the inheritance of yeast prions. In case of [PSI+] this model was also supported by size fractionation of the cell lysates carrying [PSI+], which showed that the prion activity was associated solely with Sup35p[PSI+] aggregates (characterized as fast-sedimenting material upon centrifugation) [37, 38]. However, no similar data is yet available for [URE3]. Moreover, the data on Ure2p--GFP aggregation [23] look not as conclusive as that of Sup35p--GFP [36].
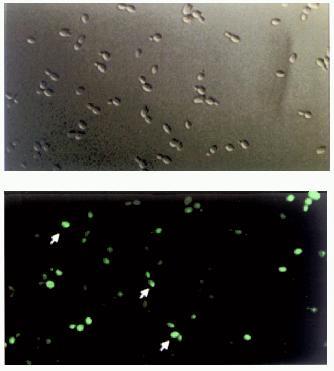
To verify more accurately the idea that the appearance of [URE3] prion involves in vivo self-propagating aggregation of Ure2p, a set of high and low copy number vectors bearing chimeric URE2--GFP fusion under control of various promoters was constructed and the aggregation state of the fusion protein was tested after cell transformation (C. Cullin, to be published elsewhere). Surprisingly, it was found that characteristic fluorescent clumps ascribed previously to Ure2p[URE3]--GFP aggregates [23] can be found throughout the cytoplasm of many wild-type yeast cells transformed with the chimeric URE2--GFP construct (Fig. 4, colored insert). We were unable to detect any difference in the amount and distribution of these fluorescent clumps between wt and [URE3] cells. Moreover, the relative number of wt and [URE3] cells displaying these clumps was the same. In addition when Ure2p--GFP fusion was expressed in cells cured of [URE3] the general picture of Ure2p--GFP fluorescence was found to be almost the same. However, it was indeed found that the number of Ure2p--GFP aggregates (fluorescent clumps) is increasing upon cell transformation with high-copy-number vectors bearing URE2--GFP under control of a strong PGK promoter. The increase in intracellular Ure2p--GFP concentration was confirmed by western blotting analysis, but even under these conditions cells did not necessarily display [URE3] phenotype. These findings allowed us to conclude that although the increase in Ure2p concentration is in fact leading to its aggregation in vivo, this does not necessarily give rise to [URE3]. Unfortunately, this unexpected result could not be directly compared with the previously reported observations on Ure2p[URE3]--GFP aggregation, as neither statistical data, nor quantitative Ure2p measurements are available [23]. However, taking into account all the facts and considerations mentioned above, one cannot exclude the aggregation mechanism leading to [URE3] formation. In vitro data showing that Ure2 protein is able to polymerize forming amyloid fibrils is in favor of a seeded mechanism similar among all prions. However, we believe that more experiments both in vivo and in vitro must be carried out to confirm this model.
Fig. 4. Cellular distribution of Ure2--GFP fusion proteins. Yeast wild-type cells (strain CC30) were transformed with multicopy vectors allowing strong overproduction of chimeric proteins. Top, cells examined by light microscopy. Bottom, the same cells examined by fluorescent microscopy. Arrows point to cells bearing Ure2p--GFP aggregates.
CONCLUSIONS
1. Computational, genetic, biochemical, and structural data allow us to redefine the boundary between the so-called prion-forming and nitrogen regulator (catalytic) domains of the protein and assign this boundary to Met-94 instead of Arg-65 initially specified by R. B. Wickner on the ground of URE2 deletion analysis.
2. We bring evidence that the COOH-terminal (94-354) catalytically active part of the protein can be synthesized (in vitro and in vivo) via an internal ribosome-binding mechanism, independently of the production of the full-length protein.
3. We show that the NH2- and COOH-terminal parts of the protein can interact with each other during protein expression in vivo.
4. We bring evidence that the COOH-terminal (94-354) part of the protein forms a compact domain, while the NH2-terminal (1-94) part is unstructured.
5. We show that Ure2p is a soluble protein that can assemble into dimers, tetramers, and octamers, or form insoluble fibrils observed for PrP in its filamentous form or for Sup35p upon self-assembly.
6. We bring evidence that only slight conformational changes (mainly located into Ure2p COOH-terminal domain) accompany its self-assembly into insoluble high-molecular-weight oligomers.
7. Finally, we show that Ure2p aggregation in vivo (monitored by fluorescence of Ure2p--GFP fusion) does not necessarily give rise to [URE3] prion phenotype.
We would like to thank Drs. A. Baudin-Baillieu, L. Bousset, E. Fernandez-Bellot, E. Guillemet, T. Lesnik, C. Reiss, and C. Thual for their indispensable help and fruitful discussions. Dr. M. Altmann is gratefully acknowledged for his generous help and valuable advice in connection with Ure2p yeast extract cell-free translation experiments. Special thanks to Prof. Dr. F. Lacroute for his critical comments and stimulating discussion.
REFERENCES
1.Cox, B. S. (1965) Heredity (Edinburgh),
20, 505-521.
2.Cox, B. S. (1971) Heredity (Edinburgh),
26, 211-232.
3.Aigle, M., and Lacroute, F. (1975) Mol. Gen.
Genet., 136, 327-335.
4.Lacroute, F. (1971) J. Bacteriol.,
106, 519-522.
5.Coschigano, P. W., and Magasanik, B. (1991) Mol.
Cell. Biol., 11, 822-832.
6.Turoscy, V., Chisholm, G., and Cooper, T. G. (1984)
Genetics, 108, 827-831.
7.Turoscy, V., and Cooper, T. G. (1979) J.
Bacteriol., 140, 971-979.
8.Turoscy, V., and Cooper, T. G. (1987) J.
Bacteriol., 169, 2598-2600.
9.Wickner, R. B. (1994) Science, 264,
566-569.
10.Jermutus, L., Ryabova, L. A., and Pluckthun, A.
(1998) Curr. Opin. Biotechnol., 5, 534-548.
11.Komar, A. A., Lesnik, T., Cullin, C., Guillemet,
E., Ehrlich, R., and Reiss, C. (1997) FEBS Lett., 415,
6-10.
12.Komar, A. A., Lesnik, T., Thual, C., Cullin, C.,
Reiss, C., and Altmann, M. (2000) J. Biol. Chem., submitted.
13.Merrick, W. C., and Hershey, J. W. B. (1996) in
Translational Control (Hershey, J. W. B., Mathews, M. B., and
Sonenberg, N., eds.) Cold Spring Harbor Laboratory Press, New York, pp.
31-70.
14.Sachs, A. B., Sarnow, P., and Hentze, M. W.
(1997) Cell, 89, 831-838.
15.Altmann, M., Sonenberg, N., and Trachsel, H.
(1989) Mol. Cell. Biol., 10, 4467-4472.
16.Masison, D. C., and Wickner, R. B. (1995)
Science, 270, 93-95.
17.Van der Velden, A. W., and Thomas, A. A. (1999)
Int. J. Biochem. Cell. Biol., 1, 87-106.
18.Barraille, P., Chinestra, P., Bayard, F., and
Faye, J. C. (1999) Biochem. Biophys. Res. Commun., 257,
84-88.
19.Vagner, S., Gensac, M. C., Maret, A., Bayard, F.,
Amalric, F., Prats, H., and Prats, A. C. (1995) Mol. Cell.
Biol., 1, 35-44.
20.Bab, I., Smith, E., Gavish, H., Attar-Namdar, M.,
Chorev, M., Chen. Y.-C., Muhlrad, A., Birnbaum, M. J., Stein, G., and
Frenkel, B. (1999) J. Biol. Chem., 274, 14474-14481.
21.Duncan, R., Milburn, S. C., and Hershey, J. W.
(1987) J. Biol. Chem., 262, 380-388.
22.Altmann, M., and Trachsel, H. (1993) Trends.
Biochem. Sci., 18, 429-432.
23.Edskes, H. K., Gray, V. T., and Wickner, R. B.
(1999) Proc. Natl. Acad. Sci. USA, 96, 1498-1503.
24.Altmann, M., Schmitz, N., Berset, C., and
Trachsel, H. (1997) EMBO J., 16, 1114-1121.
25.Das, S., Coward, P., and Dasgupta, A. (1994)
J. Virol., 68, 7200-7211.
26.Venkatesan, A., Das, S., and Dasgupta, A. (1999)
Nucleic Acids Res., 27, 562-572.
27.Komar, A. A., Guillemet, E., Reiss, C., and
Cullin, C. (1998) Biol. Chem., 379, 1295-1300.
28.Thual, C., Komar, A. A., Bousset, L.,
Fernandez-Bellot, E., Cullin, C., and Melki, R. (1999) J. Biol.
Chem., 274, 13666-13674.
29.Jackson, G. S., Hosszu, L. L. P., Power, A.,
Hill, A. F., Kenney, J., Saibil, H., Craven, C. J., Waltho, J. P.,
Clarke, A. R., and Collinge, J. (1999) Science, 283,
1935-1937.
30.Glover, J. R., Kowal, A. S., Schirmer, E. C.,
Patino, M. M., Liu, J. J., and Lindquist, S. (1997) Cell,
89, 811-819.
31.Jaenicke, R. (1999) Prog. Biophys. Mol.
Biol., 71, 155-241.
32.Prusiner, S. B. (1998) Proc. Natl. Acad. Sci.
USA, 95, 13363-13383.
33.Fernandez-Bellot, E., Guillemet, E.,
Baudin-Baillieu, A., Gaumer, S., Komar, A. A., and Cullin, C. (1999)
Biochem. J., 338, 403-407.
34.Masison, D. C., Maddelein, M. L., and Wickner, R.
B. (1997) Proc. Natl. Acad. Sci. USA, 94,
12503-12508.
35.Fernandez-Bellot, E., et al. (2000), in
preparation.
36.Patino, M. M., Liu, J. J., Glover, J. R., and
Lindquist, S. (1996) Science, 273, 622-626.
37.Paushkin, S. V., Kushnirov, V. V., Smirnov, V.
N., and Ter-Avanesyan, A. M. (1996) EMBO J., 15,
3127-3134.
38.Paushkin, S. V., Kushnirov, V. V., Smirnov, V.
N., and Ter-Avanesyan, A. M. (1997) Science, 277,
381-383.
39.Kochneva-Pervukhova, N. V., Paushkin, S. V.,
Kushnirov, V. V., Cox, B. S., Tuite, M. F., and Ter-Avanesyan, M. D.
(1998) EMBO J., 17, 5805-5810.
40.DePace, A. H., Santoso, A., Hillner, P., and
Weissman, J. S. (1998) Cell, 93, 1241-1252.
41.Taylor, K. L., Cheng, N., Williams, R. W., Steven, A. C., and
Wickner, R. B. (1999) Science, 283, 1339-1343.