A Sensitive Assay of Translational Fidelity (Readthrough and Termination) in Eukaryotic Cells
T. M. M. Sogaard, C. G. Jakobsen, and J. Justesen*
Institute of Molecular and Structural Biology, University of Aarhus, C.F. Moellers Alle bldg. 130, DK-8000, Aarhus C, Denmark; fax: 45-89-42-26-37; E-mail: jj@mbio.aau.dk* To whom correspondence should be addressed.
Received June 2, 1999
The process of translation termination in eukaryotes has been monitored by different types of assays, each with its own merits. We have developed an in vivo system where the reporter protein is secreted from the cells in culture thus enabling continuous monitoring of translation termination activity by simple sampling of the cell culture media. Using this system, cell cultures can be challenged with various stimuli during growth and the cellular responses on the translational level can be investigated in vivo as well as in vitro. Sampling is rapid, easy, and non-destructive to the cells, which enables measurement of translational fidelity in real time during time-course experiments. In particular with this system it is possible to assess very low levels of stop codon suppression. The reporter enzyme, secreted alkaline phosphatase (SEAP), becomes tagged with the S-peptide when there is readthrough of a stop codon placed between the C-terminus of the SEAP and the S-peptide. The tagged SEAP is bound to a matrix and the bound SEAP activity is measured versus total SEAP activity in the medium as a reference. With this assay we have confirmed that eRF1 acts as an antisuppressor in cells transfected with a cognate suppressor tRNA as well as in control cells, where a small but significant level of readthrough (suppression) could be detected. We have also characterized suppression of the three stop codons individually, and especially UGA is prone for wobbling.
KEY WORDS: translation fidelity, readthrough, eukaryotic, stop codon suppression, peptide chain release factor, SEAP, S-peptide, eRF1, eRF3, assay
Abbreviations: AKV) murine leukemia virus from AKR mice; CMV) cytomegalovirus; DEAE) diethylaminoethyl; DMEM) Dulbecco's-modified Eagle's medium; ELISA) enzyme-linked immunosorbent assay; eRF1) eukaryotic class-1 release factor; eRF3) eukaryotic class-2 release factor; NCS) newborn calf serum; PVDF) polyvinyl difluoride; PBS) phosphate buffered saline; SEAP) secreted alkaline phosphatase; SSC) sodium salt citrate buffer.
The process of translation termination is of immense importance in
protein synthesis. The misinterpretation of a stop codon leads to
readthrough and translation beyond the natural end of the coding
sequence and into at best non-functional or even harmful additions to
the peptide chain. Obviously such illegitimate C-terminal extensions of
an amino acid chain can lead to misfolding and consequently to lack of
protein function. Potentially the protein could acquire a new and
perhaps deleterious activity resembling the way fusion proteins created
through chromosomal translocations can acquire novel carcinogenic
functions. In eukaryotes the peptide chain release factor protein eRF1
decodes all three stop codons and releases the nascent polypeptide
chain from the ribosome in a GTP independent process [1]. A second factor, eRF3, stimulates release factor
activity by binding to eRF1 [2, 3]. The association of the release factors with the
ribosome confers GTP dependence on the process of translation
termination [4].
The fidelity of translation and termination has been the focus of many investigations because of its potential to act as a regulator of protein expression [5-7]. In most cells [8] and in vitro [9] eRF1 competes with codon-specific nonsense suppressor tRNAs for the stop codon which acts as a cofactor in the termination reaction. Readthrough by suppression of stop codons is used by some viruses during the synthesis of the viral proteins [10]. Cellular modulation of the efficiency of stop codon recognition could then potentially serve as an antiviral defense.
In vivo as well as in vitro assay systems for eukaryotic cells have been developed to measure termination and readthrough. One in vitro system uses radioactively labeled E. coli fMet-tRNAfMet bound to rabbit reticulocyte ribosomes and the cofactors for the termination reaction are tetranucleotides encoding stop codons. The release of fMet from the ribosome is then a measure of the termination activity (see [1]). In vivo systems to measure translation efficiency have been developed by several groups [8, 11-13]. Some of the systems are based on readthrough mediated rescue from a nonsense mutation to achieve a signal from the reporter protein. These systems generally lack an easily measurable internal control of protein synthesis. Other systems include an internal control but require actual harvesting of cells to conduct measurements on readthrough or frameshifting in the system. In the case of time-course experiments a measuring system that is non-destructive to the cells would be useful.
We set out to design a new in vivo and in vitro applicable translation termination assay system that would include the merits of the earlier systems without including the drawbacks. The system is based upon the heat-stable secreted alkaline phosphatase (SEAP) in a context where suppression of at stop codon will tag it with an S-peptide (15 N-terminal amino acid residues of ribonuclease A). We have been able to measure precisely the basal suppression of individual stop codons in human cells and study the effect of suppressor tRNAs. Furthermore the system has allowed a determination of the effects of the peptide chain release factors eRF1 and eRF3 in vivo. Finally, we have examined the effect of ethanol and paromomycin on translation termination in cultured cells.
MATERIALS AND METHODS
Cloning of the reporter constructs. The constructs are based on the ptk-SAP plasmid [14] as modified by A. L. Nielsen expressing SEAP under the control of an AKV promoter. The S-peptide was inserted by a PCR approach utilizing two unique restriction sites (BssHII and MunI, positions 2623 and 2871 in the vector, respectively) on either side of the SEAP stop codon. Two primers specific for the vector sequence surrounding the natural SEAP stop codon were designed. The forward primer carried a 5´ extension coding for the S-peptide sequence followed by three different in-frame stop codons and the reverse primer carried a 5´ extension coding for a redundant stop codon sequence (TRR, where each R may be A or G). Additionally each primer also carried a 5´ XhoI site. PCR was run against primers specific for sequences up- and downstream of the unique restriction sites in the vector. The PCR product was purified using the Qiagen PCR purification kit and digested with XhoI (New England Biolabs, USA). The PCR products were divided into two pools and digested with BssHII and MunI, respectively. The resulting BssHII--XhoI and XhoI--MunI fragments were inserted into the BssHII/MunI digested pAKV-SEAP vector in a three fragment ligation reaction resulting in a mixture of the plasmids pRTA-UGA, pRTA-UAG, pRTA-UAA, and pRTA-UGG. The constructs are shown in Fig. 1. Individual clones were isolated and proper sequence was confirmed by sequencing. Thus the positive control for protein synthesis is the plasmid pRTA-UGG which codes for tryptophan inserted in between the SEAP and the S-peptide sequence. To be able to assess the degree of noise generated as a result of unspecific binding of SEAP to the Maxisorp plates, a negative control construct was made by cutting out the S-peptide sequence from the reporter construct harboring the TAG codon with subsequent re-ligation of the vector. The resulting construct pRTA-neg (Fig. 1) codes for SEAP followed by two in-frame stop codons.
Plasmids pCMVhRF1 and pCMVhRF3 expressing eRF1 and eRF3 (under the control of a CMV promoter), respectively, as well as the suppressor tRNA-expressing plasmids ptRNAochre, ptRNAamber, and ptRNAopal have been previously described [8]. The release factor expressing plasmids and the suppressor tRNA expressing plasmids were generous gifts from Dr. X. le Goff and Dr. L. Yu. Frolova.
DNA transfection of human HeLa and AMA cells. The procedure was carried out in a 24-well tissue culture dish format. Cells were seeded at mid confluence and grown overnight. All transfections were done in triplicate. For each transfection, reporter constructs and release factor expressing plasmids were added at 0.3 µg DNA per well and plasmids expressing the compatible suppressor tRNA were added at 0.5 µg DNA per well.
For transfection of HeLa cells Lipofectamine was used according to the manufacturer's instructions. Serum-free medium was mixed with plasmid DNA to a total of 50 µl. Lipofectamine (2 µl) was added to 48 µl of serum-free medium and gently mixed with the DNA solution and the DNA/Lipofectamine mixture was incubated at room temperature for 45 min. The cells were washed in serum-free medium and the supernatant was aspirated. The DNA/Lipofectamine solution was supplemented with 400 µl serum-free medium and the resulting 500 µl transfection mixture was added to the wells and the cells were incubated for 2 h at 37°C. After incubation, 500 µl of medium (DMEM with 20% NCS) was added to each well. After overnight incubation, the transfection medium was aspirated and 700 µl new medium (DMEM supplemented with 10% NCS and 1% penicillin--streptomycin mixture) was added to each well. Required stimulating agents were also added at this point.
For transfection of AMA cells, plasmid DNA was mixed with medium (DMEM with 10% NCS and 1% penicillin--streptomycin mixture) to a total volume of 320 µl and 3 µl chloroquine (10 mM) and 12 ml DEAE-dextran (10 mg/ml) was added. Cells were washed in 1× PBS and the supernatant was aspirated. The resulting 335 µl transfection mixture was added to the wells and the cells were incubated for 3 h at 37°C. After incubation the transfection mixture was aspirated and 500 µl of dimethylsulfoxide (10% in 1× PBS) was added and the cells were incubated for 2 min at room temperature. The solution of dimethylsulfoxide was aspirated and 700 µl of new medium was added to each well. Required stimulating agents were added at this point.
After 72 h of incubation the medium was sampled and assayed for SEAP activity.
Western blotting. AMA cells transfected with pCMVhRF1 and pCMVhRF3 were harvested and spun down. The pellet was dissolved in loading buffer and 10 µl was loaded onto 10% sodium dodecyl sulfate (SDS)-acrylamide gels. The gels were electroblotted to PVDF membranes (Immobilon-P, Millipore, USA) and incubated overnight in blocking buffer (BLOTTO: 5% low fat dry milk in 1× PBS) at 4°C. The membranes were incubated for 1 h with primary rabbit polyclonal antibodies against eRF1 and eRF3, respectively. After washing three times in washing buffer (1% low fat dry milk, 0.5% Tween 20 in 1× PBS), the membranes were incubated for 1 h with horseradish-peroxidase-conjugated goat-anti-rabbit antibodies diluted 1:5000 in 1× PBS (DAKO, Denmark). Final detection was done using the Enhanced Chemiluminescence kit (ECL, Amersham, U.K.) according to the manufacturer's instruction.
Northern blotting. Northern blotting was done on total RNA extracted from transfected cells using the RNA isolation kit (Genosys, U.K.) according to manufacturer's instruction. RNA was loaded on a 10% polyacrylamide--formamide gel (15 µg per lane). The gel was run at 15 W for 90 min. The RNA was electroblotted onto Zeta-probe membrane (Zeta-probe GT, Biorad) according to the manufacturer's instruction using a Trans-Blot cell (Biorad). The electroblotting was done at 20 V for 1 h and then at 50 V for 2 h. RNA was fixed to the membrane by baking under vacuum at 80°C for 30 min in a Biorad gel dryer. The Zeta-probe membrane was prehybridized in hybridization buffer (0.5 M Na2HPO4, pH 7.5, 7% SDS, 1% BSA, 1 mM EDTA) at 43°C for 15 min. Oligonucleotide probe (50 pmoles) was end-labeled using T4 polynucleotide kinase in labeling buffer supplied with [gamma-32P]ATP.
After the 1-h incubation at 37°C the probe was purified on a spin column (Qiagen Nucleotide Removal kit) and eluted in Tris-HCl, pH 8.0. Zeta-probe membrane was hybridized with the probe in hybridization buffer overnight at 43°C. After a quick rinse in 2× SSC the membrane was washed for 15 min in 2× SSC containing 0.1% SDS, then for 15 min in 0.5× SSC containing 0.1% SDS, and finally for 15 min in 0.1× SSC containing 0.1% SDS. All washing was done at room temperature. The membrane was sealed in a plastic bag and autoradiography was performed.
ELISA measurements of tagged and un-tagged SEAP activity. Preparation of S-protein-coated 96-well microtiter plates (Nunc-Immuno Maxisorp Surface, Nunc, Denmark) was done by incubating the plates at 4°C overnight in 50 µg/ml S-protein (Sigma, USA) in 1× PBS (100 µl per well). The S-protein solution was aspirated and wells were blocked in 1% low fat milk in 1× PBS for 2 h at room temperature. Finally wells were washed 2 times in washing buffer (1× PBS, 0.1% Tween 20).
Measurement of the total expression level of SEAP. This was done by sampling the cell medium. Samples were heat inactivated at 65°C for 20 min. All samples were centrifuged for 5 min. Aliquots of 50 µl were mixed with 150 µl SEAP substrate (5 mM p-nitrophenyl phosphate, 50 mM Tris-HCl, pH 8.2) in a 96-well microtiter plate and incubated at 37°C. To detect colorimetric change, we employed an ELISA microtiter plate reader (Bio-Tek, USA) reading at 405 nm. Measurements were started at the time of addition of the substrate and continued through 10 time points. Linear regression was performed using only time points where the rate of change was constant and maximal. The calculated slope is proportional to the expression level of the reporter protein.
Measurement of the level of readthrough of the stop codon. Aliquots (100 µl) of the culture medium were placed into microtiter wells pre-coated with S-protein. The plates were incubated overnight at 4°C and washed twice in washing buffer (0.1% Tween 20 in 1× PBS) and once in 1× PBS. SEAP substrate (150 µl) was added to each well and measurements and calculations were carried out as described. The ratio between the calculated expression level of SEAP and the calculated activity of the bound fusion protein indicates the level of readthrough of the stop codon on the reporter plasmid.
RESULTS
Our assay is based on the readthrough of a stop codon placed in-frame between two genes. The construct is shown in Fig. 1. The first gene codes for secreted alkaline phosphatase (SEAP) derived by a C-terminal truncation of alkaline phosphatase from human placental cells so that the enzyme is actively secreted from the cells [15]. SEAP is stable at 65°C, which enables heat inactivation of endogenous phosphatases improving the signal/noise ratio. Because of the existence of very sensitive substrates for SEAP, many systems have utilized this enzyme as an easily detectable reporter [15-18]. The second gene codes for a 15-amino acid-peptide also known as an S-peptide which is identical to an N-terminal proteolytic fragment of ribonuclease A. The remainder of the degraded ribonuclease A is termed the S-protein. The S-peptide binds tightly and with high specificity to the S-protein. This interaction has been thoroughly investigated [19, 20]. If the stop codon is correctly read, the SEAP protein will be expressed and secreted to the medium without any tag. In the case of readthrough of the stop codon the secreted SEAP protein will be fused C-terminally to the S-peptide. The total activity of SEAP serves as an internal control of protein expression levels. To measure the proportion of S-peptide-tagged SEAP, we bind the molecules by their S-peptide tag to S-protein-coated maxisorp microtiter plates before SEAP detection. The handling steps of the procedure are outlined in Fig. 2.
Fig. 1. Reporter constructs and control constructs. The stop codon is TRR, where each R designates either A or G.
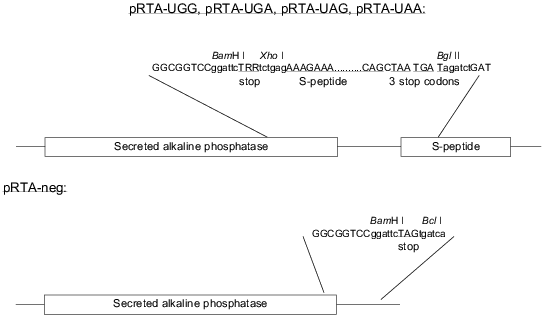
Confirmation of expression of transfected genes. The transfection level of the reporter plasmid was confirmed by total SEAP activity in all experiments. Expression of eRF1 and eRF3 was confirmed by western blots on crude cell extracts (results not shown). Expression of the three different suppressor tRNAs was confirmed by northern blotting (results not shown).Fig. 2. Schematic description of the whole process involved in the assay. Activity of SEAP with or without an S-tag is measured by estimating the slope (alpha) of the linear part of the curve; alphatotal, after inactivation; alphaRT, at room temperature.
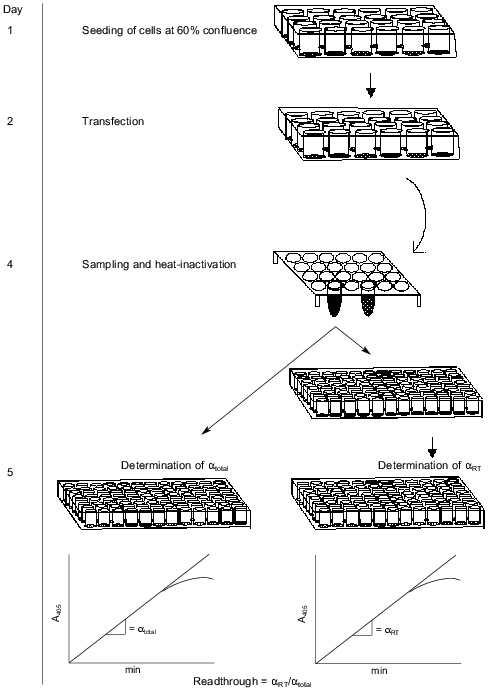
Confirmation of a linear relationship between binding and concentration of fusion protein. The SEAP/S-peptide fusion protein binds tightly to the S-protein in the coated microtiter wells. To ensure linearity of fusion protein binding we performed a dilution experiment. AMA cells were transfected with the pRTA-UGG construct expressing the wild-type SEAP fused to the S-peptide. A dilution series of medium samples was applied to the S-protein-coated wells and activity was measured (Fig. 3). The result shows the activity to be proportional to the dilution indicating that at the range of concentrations applied the bound S-protein never saturates with tagged SEAP/S-peptide fusion protein.
Endogenous readthrough. To measure the extent of endogenous readthrough, we transfected different pools of cells with each of the four reporter constructs as well as the negative control construct without the S-peptide. Table 1 shows the extent of readthrough normalized to the wild-type UGG level. Readthrough is clearly higher than the level of noise (pRTA-neg) and UGA seems to be suppressed more efficiently than UAA, which has a readthrough slightly higher than UAG.Fig. 3. A dilution experiment with medium sampled from transfected AMA cells. Increasing amounts of fusion protein was applied to an S-protein-coated microtiter plate. Bound activity as a function of the concentration of fusion protein applied to the wells.
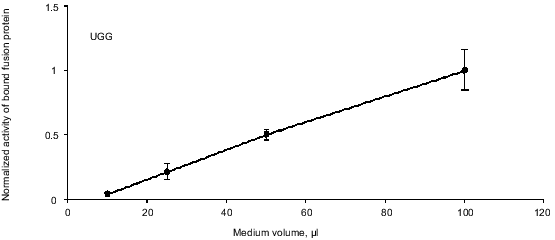
Table 1. Endogenous readthrough of the
reporter constructs transfected into AMA cells
Enhanced readthrough by expression of suppressor tRNAs. The peptide chain release factors compete with suppressor tRNAs for decoding of stop codons and our assay measures the outcome of such a competition. Therefore we wanted to see if we could enlarge the dynamic range of the assay by co-transfecting the reporter constructs with plasmids that express the compatible specific suppressor tRNAs. Readthrough was expected to be proportional to the amount of transfected suppressor tRNA expressing plasmid. Figure 4 shows the result of this experiment. The UGA and UAA suppressors achieve more suppression than the UAG suppressor and generally we see a tendency of the UGA and UAA suppressors to show a suppression level of up to 45% while the UAG suppressor reaches about 20% suppression compared to the pRTA-UGG construct (data not shown).
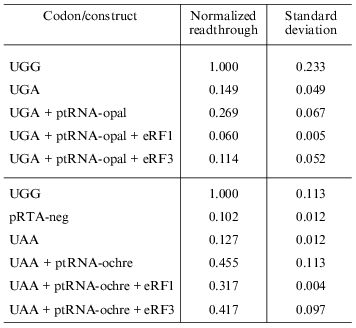
Overexpression of eRF1 and eRF3. To investigate the effect of overexpression of release factors on our system, the reporter construct was co-transfected into the cells along with the construct expressing the appropriate suppressor tRNA as well as constructs that express either eRF1 or eRF3 under the control of a CMV promoter. Results are summarized in Table 2. All values are normalized to the activity of the pRTA-UGG construct. Overexpression of eRF1 lowers the level of stop codon suppression by the expressed suppressor tRNAs indicating its ability to function as a release factor on its own and in good agreement with previous results [1, 8, 9]. The expression of eRF3 hardly results in significant antisuppression, which agrees with previous results [8, 9]. However the UGA codon seems to be somewhat suppressed.Fig. 4. Co-transfection of AMA cells with reporter and compatible tRNA suppressor constructs. Chart of readthrough activity as a function of amount of tRNA suppressor transfected.
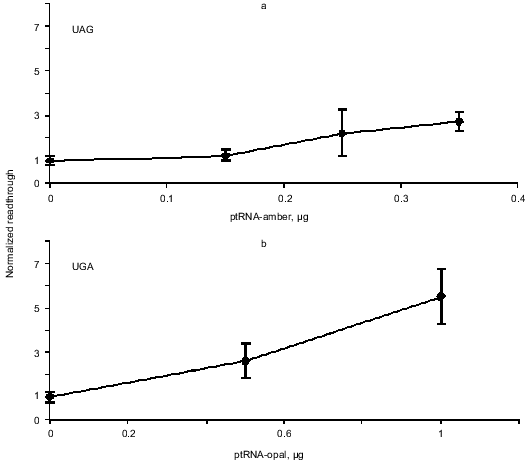
Table 2. Effect of overexpression of release
factors on readthrough activity in HeLa cells
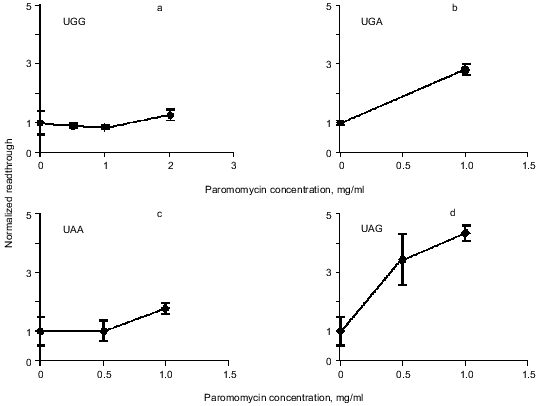
Readthrough induced by “wobbling” agents. Ethanol has previously been observed to reduce translation fidelity [21]. We have grown cells at different concentrations of ethanol to investigate its effect in our system. Figure 5 shows the extent of readthrough induced by ethanol. This effect is more pronounced when the stop codon is UGA than with UAA and UAG. Previous workers have seen a mistranslation effect induced by aminoglycosides like paromomycin and streptomycin [13, 22, 23]. We have applied paromomycin in our system as shown in Fig. 6. Panel (a) shows the negligible effect of paromomycin on the general level of translation. In contrast paromomycin is a potent inducer of UGA readthrough (Fig. 6b) while the effect is less pronounced on UAA and UGA (Fig. 6, c and d).
Fig. 5. AMA cells were grown at different concentrations of ethanol. The chart illustrates readthrough activity as a function of amount of ethanol.
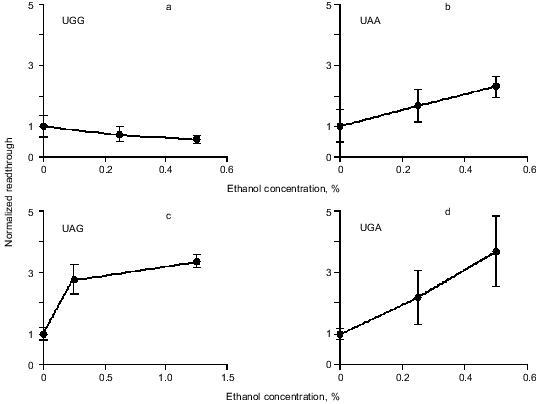
Fig. 6. AMA cells were grown at different concentrations of paromomycin. The chart illustrates readthrough activity as a function of amount of paromomycin.
DISCUSSION
The proper decoding of stop codons is of immense importance in the process of protein synthesis. The misinterpretation of a stop codon by readthrough results in C-terminally extended proteins carrying novel peptide ends that can affect both structural and functional aspects of the proteins. By controlled readthrough of in-frame stop codons or by frameshifting the same RNA sequence can code for proteins with different C-termini. This mode of translation is known to occur naturally for a number of cases investigated [5]. The stop codon context is important for translation termination efficiency [24-26]. Strong termination codons preferably have A or G as the neighboring 3´ base whereas weaker stop codons are often followed by U or C. As we wanted to measure any changes in general stop codon recognition we opted for the context that would make the stop codon as weak as possible using TRRC (where each R is either A or G) as the model stop codon.
Endogenous readthrough. The extent of natural readthrough is normally below 12% [5] and in our system with AMA cells we have found 6-11% readthrough depending on stop codon (Table 1). Aminoglycosides like streptomycin and paromomycin are known to augment translational misreading [22, 23, 27, 28]. They act by interfering with the ribosome whereby near-cognate tRNAs are allowed to deliver their amino acids to the growing peptide chain. All cells used in this study were grown in the presence of 100 µg/ml streptomycin to inhibit bacterial growth. We therefore cannot claim to precisely model the natural state of the cells in this experiment. Still, streptomycin is known to be a weaker inducer of readthrough than paromomycin [23] and since we still see a significant effect of paromomycin the assay system is clearly not saturated with suppression agents at this concentration of streptomycin.
Readthrough by expression of suppressor tRNAs. The UGA-specific suppressor tRNA seems to be able to compete with the release factor with greater success than the UAG-specific suppressor tRNA. This could in fact be explained by wobbling of the third base in the anticodon allowing near-cognate UGG-specific tRNA to enter the A-site on the ribosome. This natural readthrough enhances the total readthrough compared to the readthrough of UAG, which could never wobble into a sense codon. The lower level of suppression of the UAG codon could possibly be caused by a higher efficiency of eRF1 in the recognition of UAG codons.
Readthrough is reduced by overexpression of eRF1. Competition between suppressor tRNAs and release factors has been shown to occur in eukaryotic cells [9]. Overexpression of eRF1 in the cells leads to a clear drop in readthrough, which is in good agreement with previous in vivo results [8]. Overexpression of eRF3 has a much less pronounced effect on the system indicating eRF1 to be the limiting factor in this system. It has been shown in vitro that at high intracellular concentration of stop codon eRF1 is capable of functioning as release factor without the help of eRF3 [1, 2, 4]. Only in the case of low abundance of stop codons do eRF3 and GTP significantly stimulate the activity of eRF1.
Readthrough induced by “wobbling” agents. The tendency for UGA to be read through more efficiently than UAG under the influence of ethanol or paromomycin probably reflects a greater tendency for the translational machinery to misread the third base in the stop codon, in agreement with the general theory of wobbling [5].
As mentioned above, all cells were grown in the presence of 100 µg/ml streptomycin. This antibiotic does have a small effect on translation termination like paromomycin [23], which could account for part of the basal readthrough.
To precisely determine the extent of endogenous readthrough of each individual stop codon we need to repeat the experiments with cultured cells grown without streptomycin. Most cultured cell lines are grown routinely with streptomycin as an antibacterial agent and this aspect should be taken into consideration when attempting to assess the extent of basal readthrough. Probably the best measure of basal endogenous readthrough would be obtained by applying the assay on primary cells that have not yet evolved far from a natural state.
In the future we would like to apply our assay system to the investigation of frameshift events. On the other hand, this system can be applied to characterize a number of cell lines with respect to translational readthrough as part of an effort to shed light on a possible role of eukaryotic peptide chain release factors in neoplastic transformation. Finally, we plan to examine the effects of agents like cytokines, including interferons, on translational readthrough.
REFERENCES
1.Frolova, L., Le Goff, X., Rasmussen, H. H.,
Cheperegin, S., Drugeon, G., Kress, M., Arman, I., Haenni, A. L.,
Celis, J. E., Philippe, M., Justesen, J., and Kisselev, L. L. (1994)
Nature, 372, 701-703.
2.Zhouravleva, G., Frolova, L., Le Goff, X., Le
Guellec, R., Inge-Vechtomov, S., Kisselev, L., and Philippe, M. (1995)
EMBO J., 14, 4065-4072.
3.Kisselev, L. L., and Frolova, L. Y. (1995)
Biochem. Cell Biol., 73, 1079-1086.
4.Frolova, L., Le Goff, X., Zhouravleva, G.,
Davydova, E., Philippe, M., and Kisselev, L. (1996) RNA,
2, 334-341.
5.Atkins, J. F., and Gesteland, R. F. (1996) in
Translational Control (Hershey, J. W. B., Mathews, M. B., and
Sonenberg, N., eds.) Cold Spring Harbor Laboratory Press, New-York, pp.
653-684.
6.Cao, J., and Geballe, A. P. (1998) RNA,
4, 181-188.
7.Valle, R. P., and Morch, M. D. (1988) FEBS
Lett., 235, 1-15.
8.Le Goff, X., Philippe, M., and Jean Jean, O. (1997)
Mol. Cell Biol., 17, 3164-3172.
9.Drugeon, G., Jean Jean, O., Frolova, L., Le Goff,
X., Philippe, M., Kisselev, L., and Haenni, A. L. (1997) Nucleic
Acids Res., 25, 2254-2258.
10.Li, G., and Rice, C. M. (1993) J. Virol.,
67, 5062-5067.
11.Firoozan, M., Grant, C. M., Duarte, J. A., and
Tuite, M. F. (1991) Yeast, 7, 173-183.
12.Grentzmann, G., Ingram, J. A., Kelly, P. J.,
Gesteland, R. F., and Atkins, J. F. (1998) RNA, 4,
479-486.
13.Stansfield, I., Jones, K. M., Herbert, P.,
Lewendon, A., Shaw, W. V., and Tuite, M. F. (1998) J. Mol.
Biol., 282, 13-24.
14.Tolstrup, A. B., Bejder, A., Fleckner, J., and
Justesen, J. (1995) J. Biol. Chem., 270, 397-403.
15.Berger, J., Hauber, J., Hauber, R., Geiger, R.,
and Cullen, B. R. (1988) Gene, 66, 1-10.
16.Yang, T. T., Sinai, P., Kitts, P. A., and Kain,
S. R. (1997) Biotechniques, 23, 1110-1114.
17.Bronstein, I., Fortin, J. J., Voyta, J. C., Juo,
R. R., Edwards, B., Olesen, C. E., Lijam, N., and Kricka, L. J. (1994)
Biotechniques, 17, 172-174, 176-177.
18.Cullen, B. R., and Malim, M. H. (1992) Meth.
Enzymol., 216, 362-368.
19.Kim, J. S., and Raines, R. T. (1993) Protein
Sci., 2, 348-356.
20.Karpeisky, M. Y., Senchenko, V. N., Dianova, M.
V., and Kanevsky, V. Y. (1994) FEBS Lett., 339,
209-212.
21.Laughrea, M., Latulippe, J., and Filion, A. M.
(1984) Biochemistry, 23, 753-758.
22.Wilhelm, J. M., Jessop, J. J., and Pettitt, S. E.
(1978) Biochemistry, 17, 1149-1153.
23.Eustice, D. C., and Wilhelm, J. M. (1984)
Biochemistry, 23, 1462-1467.
24.McCaughan, K. K., Brown, C. M., Dalphin, M. E.,
Berry, M. J., and Tate, W. P. (1995) Proc. Natl. Acad. Sci. USA,
92, 5431-5435.
25.Farabaugh, P. J. (1993) Cell, 74,
591-596.
26.Tate, W. P., Poole, E. S., Horsfield, J. A.,
Mannering, S. A., Brown, C. M., Moffat, J. G., Dalphin, M. E.,
McCaughan, K. K., Major, L. L., and Wilson, D. N. (1995) Biochem.
Cell Biol., 73, 1095-1103.
27.Tai, P. C., Wallace, B. J., and Davis, B. D.
(1978) Proc. Natl. Acad. Sci. USA, 75, 275-279.
28.Palmer, E., and Wilhelm, J. M. (1978)
Cell, 13, 329-334.