REVIEW: Molecular Mechanisms of Alpha-Fetoprotein Gene Expression
N. L. Lazarevich
Institute of Carcinogenesis, Blokhin Cancer Research Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, Moscow, 115478 Russia; fax: (7-095) 324-1205; E-mail: abelev@mx.iki.rssi.ru
Received October 13, 1999
Alpha-fetoprotein (AFP) is the main component of mammalian fetal serum. It is synthesized by visceral endoderm of the yolk sac and by fetal liver. Immediately after birth AFP level in blood decreases dramatically. AFP synthesis is reactivated in liver tumors and germinogeneous teratoblastomas, in a lesser degree after chemical and mechanical liver injuries followed by regeneration (i.e., acute viral hepatitis). AFP blood level change is an important marker for liver tumors that is widely used in clinical practice. Therefore, the study of the molecular and cellular mechanisms participating in regulation of the oncoembryonal protein AFP is an important task. On various experimental models it has been shown that the expression is regulated mainly on the transcriptional level, the AFP gene having a 7 kb regulatory region upstream. Within this region a tissue-specific promoter, three independent enhancers, and a silencer that is at least partially responsible for AFP gene expression decrease in adult liver have been defined. Some ubiquitous and some tissue-specific transcription factors, including hepatocyte nuclear factors (HNFs), which mediate the transcription of most of the liver-specific genes, have been shown to bind to the promoter. However, the mechanisms determining drastic changes of AFP synthesis level in the course of ontogenesis and carcinogenesis remain incompletely clarified. Also, little is known about negative regulators of AFP gene expression in cells of non-hepatic origin and in adult liver.
KEY WORDS: alpha-fetoprotein, hepatocyte nuclear factors, transcriptional regulation, transcription factors, hepatocytes, differentiation, hepatocarcinogenesis
Abbreviations: AFP) alpha-fetoprotein; GRC) glucocorticoid hormone--receptor complex; HNF) hepatocyte nuclear factor; SA) serum albumin.
Alpha-fetoprotein (AFP) was first detected [1] by
electrophoresis of human fetal serum in the first
(alpha1) position next to serum albumin (SA). In
normal adult serum this protein was not detected.
In 1960 G. Abelev and collaborators discovered a murine hepatoma alpha-globulin that was absent in the liver, blood, and other tissues of normal adult mice. It was proved that this protein is one of the main components of the murine fetal serum and, furthermore, appears in the adult liver during regeneration [2]. Later an embryo-specific alpha-globulin was detected in the serum of hepatocellular carcinoma [3] and teratoblastoma patients [4]. Within the next few years it became clear that the protein, which was named AFP, is an important marker for differential diagnostics of these tumors.
AFP is a polypeptide of about 600 amino acids and consisting of 4% carbohydrate residues. It is a secretory protein with structure and physicochemical properties similar to SA.
AFP functions and its role in development and carcinogenesis are not still incompletely investigated (for review see [5, 6]). The main properties of AFP are high affinity for polyunsaturated fatty acids (105 times higher than SA) and ability to bind estrogens. Some data indicate that AFP participates in immune response regulation.
AFP GENE STRUCTURE AND PRODUCTS OF ITS TRANSCRIPTION
The AFP gene belongs to albumin gene family along with SA, a group-specific component, also known as vitamin D-binding protein [7, 8], and alpha-albumin [9] genes. As well as their genes, these proteins are highly homologous in primary structure [9]. All of them are synthesized in liver and secreted into blood serum, providing delivery of their bound ligands to different tissues.
All the albumin family genes are located on the same chromosome. The AFP, SA, and alpha-albumin genes are positioned near each other and have a common direction of transcription (Fig. 1). Albumin genes are located on chromosome 5 of mouse [10], 14 of rat [9], and on the long arm of chromosome 4 of human (4q11-q13) [11].
The rat AFP gene size is 19 kilobase pairs (kb). Like the SA and alpha-albumin genes, it consists of 15 exons and 14 introns; exons 1 and 15 are not coding. This structure is conserved among mouse [12], rat [13], and human [14].Fig. 1. The rat albumin gene cluster and AFP gene 5´-regulatory region structure (P, promoter; S, silencer; EI-EIII, minimal enhancers of AFP gene; alphaALB, alpha-albumin gene).
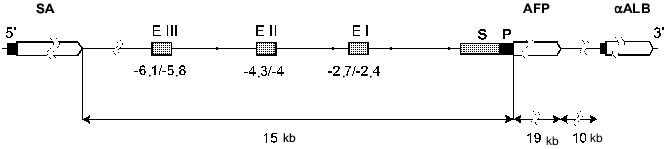
All the albumin proteins consist of three homologous domains. This suggests that the coding genes originate from common predecessor, which, in turn, arose as a result of triplication of a primary gene corresponding to one domain of the protein [15, 16].
The data about the clustered localization and about the evolution of the albumin genes, the dynamics of their synthesis during ontogenesis and carcinogenesis, as well as the structure of their regulatory regions, which will be discussed below, suggest that the expression of these genes is interconnected and has common principles of regulation.
The main product of AFP gene transcription in fetal liver is a 2.1 kb mRNA. Besides, 1.7, 1.4, and 1.0 kb mRNAs were detected in fetal and regenerating liver and in carcinogenesis [17, 18]. The shorter forms of mRNA--1.4 and 1.0 kb--dominate in adult liver [19]. Apparently, expression levels of the multiple mRNA forms are controlled by different mechanisms and can be changed independently [19, 20].
All the products of AFP gene transcription can be translated. The 2.1 kb mRNA corresponds to polypeptides weighing 68 and 70 kD [18, 19]. Functions of the different AFP forms are insufficiently studied; however, it is known that the shortened translation products maintain the transport properties [19].
AFP SYNTHESIS IN NORM AND PATHOLOGY
During embryogenesis AFP can be detected in visceral endoderm of the yolk sac (in 6-7 days of murine gestation) [21]; at this stage AFP is a dominant serum protein. Later the maximum level of its expression is observed in fetal liver (for review see [6]) and, at significantly lower levels, in embryonic gut [22] and in some other organs [23, 24].
At the end of the embryonic period of development, at the same time as the morphological restructuring of the liver, a drastic decrease in AFP blood level and reduction of AFP-producing cell number take place. Simultaneously SA blood level increases, and the main adult serum protein substitutes the embryonic one. This switch is carried out on the transcriptional level [25]. At the same time, it has been shown that both genes can be expressed in the same cell simultaneously [26]. Shortly after birth AFP concentration in blood decreases 104-fold [26, 27].
In parallel with the increase in SA level, induction of alpha-albumin [9] and group-specific component [24] synthesis takes place. Coordinated regulation of expression levels has been described for some other systems, e.g., for the globin gene cluster [28] and liver-specific apolipoprotein genes AI-CIII-AIV [29]. Perhaps the coordinated change in expression levels of clustered genes in the course of development is regulated by common elements localized in the intergenic space.
AFP gene expression is repressed reversibly in adult liver. It can be restored during the course of liver regeneration induced by partial hepatectomy, when up to 2/3 of the organ is removed surgically, or by acute CCl4 intoxication that causes necrosis of the hepatocytes bordering central veins. Simultaneously with AFP induction, SA synthesis decreases. The most significant effect is observed in mice. Hepatocyte localization within the liver plate or outside it is the defining factor that regulates the activity of AFP synthesis on a cellular level [30]. An elevation of AFP serum level is observed in the case of acute viral hepatitis and, to a lesser extent, in liver cirrhosis.
AFP blood level elevation is observed in primary liver tumors, teratocarcinomas, and gut tumors [4, 31]. In the case of embryonic carcinomas, teratocarcinomas, yolk sac tumors, and hepatoblastomas an increase in AFP level is observed in 80-90% of cases and appears to be an important diagnostic marker. The cellular aspects of AFP regulation are discussed in detail in reviews [6, 32].
Many teratocarcinomas and hepatomas are characterized by a decrease in SA synthesis along with an increase in AFP. At the same time a number of cell cultures, e.g., human hepatoma Hep G2, produce AFP and SA simultaneously [33, 34]. As in normal tissues, in most of the teratocarcinomas and hepatomas a dependence of SA and AFP synthesis on the differentiation state is observed [34-36].
MECHANISMS OF AFP GENE REGULATION
Transcription is the determining level for AFP gene regulation. This is confirmed by correlation of AFP mRNA amount and the protein synthesis level in various systems [26, 27].
However, it is impossible to exclude completely the participation of posttranscriptional mechanisms in the regulation of AFP level in some cases. It was supposed by a number of authors that some posttranscriptional mechanisms occur in vitro [37] or play some role in the mRNA increase in murine yolk sac [38] and in rat hepatoma cells [39].
Two genetic loci have been found in mice, Afr1 (raf) and Afr2 (Rif), that participate in regulation of AFP gene expression after birth [40, 41]. A possible mechanism of their action will be discussed below. It was shown that Afr1 can influence AFP transcript stability and/or its processing. Perhaps an element of the AFP mRNA 3'-end secondary structure, a stem-loop which is highly conservative and is also found in human AFP mRNA, is involved in this process [42].
Studies on the correlation of the methylation level of the AFP gene or its regulatory region and the gene expression level have given contradictory results. No clear correlation between the expression level and the level of the structural gene demethylation in embryonic and adult hepatocytes has been observed [43].
A few critical sites the methylation level of which correlates with possible or real activity of the AFP gene were mapped at the 5'-end region and the first intron [44, 45]. On the other hand, during AFP gene expression decrease in the course of development or under dexamethasone treatment the methylation level of the 5'-end region is changed not immediately but after a few weeks. 5-Azacytidine treatment of embryonic carcinoma cells or newborn rat liver cells does not lead to an activation of AFP gene expression [44]. It seems, that methylation is not a key mechanism for AFP gene regulation but reflects the gene activity status and can stabilize it in an inactive state.
REGULATORY cis-ELEMENTS OF THE AFP GENE
Transfectional analysis of hepatoma cell lines [46-49] and transgenic mouse studies [22, 50-52] have shown that the elements defining the AFP gene transcription level are located within a region from -7.6 kb to the transcription start site. There is a high homology between the AFP gene 5'-regulatory regions of mouse, rat, gorilla, and human [53, 54].
Within the mouse and rat AFP regulatory region three distal enhancers, a promoter element [46-49], and a silencer have been defined [49-51] (Fig. 1).
Promoter. A sequence of -200/0 base pairs (bp) of the AFP gene is characterized by tissue-specific promoter activity and contains multiple overlapping binding sites for ubiquitous and tissue-specific transcription factors. In vivo in the absence of the enhancer the AFP promoter is inactive [52]. A considerable similarity of AFP and other albumin gene promoter organization can be noted, in particular, the presence and localization of CCAAT-box and HNF1 binding sites within [9, 55]. The structure of the promoter region will be discussed in detail below.
Enhancers. Within the mouse and rat AFP gene regulatory region three independent enhancers (EI-EIII) of size 200-300 bp have been revealed (Fig. 1) [48, 49, 56, 57].
All of these elements are typical enhancers and are able to stimulate AFP promoter as well as heterological promoters [56]. Like AFP promoter, the enhancers are tissue-specific and are not active in non-hepatic cells [56]. Each of the enhancers is able to stimulate SA as well as AFP promoter. Probably, at some stages of development and in some hepatoma lines, in which the upstream SA enhancer is non-active, the intergenic enhancers control the expression of the two genes independently, and the corresponding promoters do not compete with each other due to their interaction with the different enhancers sites [58].
All the three enhancers of the rat AFP gene exert an additive action on the promoter, the highest level of the expression being achieved in presence of all the enhancers [49]. However, neither the promoter nor the enhancers are sufficient for postnatal repression of this gene [49, 59].
The enhancers of the mouse AFP gene in vitro do not differ in their activity and do not cause an additive effect [48, 60]. However, in transgenic mouse studies it has been shown that in vivo each of the enhancers is characterized by various activities with its own promoter as well as with a heterological one in any of the tissues (yolk sac, fetal liver and gut), where AFP gene is expressed [52, 61]. Murine EIII is also active in the brain cells [61]. All three enhancers are potentially active in adult mouse liver cells. EI and EII are most active in hepatocytes surrounding a central vein, and EIII is active exclusively in a layer of hepatocytes surrounding a central vein [61]. These data confirm a hypothesis that one of the factors that affects AFP gene expression in adult liver is a hepatocyte position in a liver lobule [30]. The lack of EIII activity in most of the adult hepatocytes is possibly connected with the existence of some negative factors binding this region [61].
The results obtained in AFP gene regulatory element studies on transgenic mice have been reviewed in detail [62].
Within enhancer EIII multiple binding sites for regulatory proteins have been revealed, including sites for hepatocyte nuclear factors HNF3 and C/EBP (see below) and nuclear receptor COUP-TF [63], which are necessary for complete stimulation of AFP promoter [57, 60, 64].
Within the human AFP gene regulatory region two enhancer elements have been revealed: -4.0/-3.7 and -3.7/-3.3 kb. In the proximal enhancer the HNF1 binding site is localized [65].
Silencer elements. In all the studied species the silencer sequences have been identified. They are localized between the promoter and enhancers of the AFP gene. They probably play the critical role in suppression of the AFP gene transcription after birth [49-51, 65]. Removal of silencer sequence -800/-250 bp in transgenic mice leads to maintenance of AFP gene expression controlled by the enhancers and the promoter in liver and gut of the adult mice [51]. The transgene expression in this system is of zonal distribution: it is observed only in hepatocytes surrounding a central vein [66] and, probably, is determined by enhancer EIII activity.
In the regulatory region of the rat the silencer is also linked with the promoter region [49, 56]. Deletion of the silencer leads to restoration of expression of the reporter gene controlled by the AFP promoter in human hepatoma cell line H4C3, usually non-expressing AFP [49]. This silencer is able to suppress AFP promoter stimulation by heterological SV40 enhancer.
At least two silencers, -1822/-951 and -402/-169 bp, have been revealed in the human AFP gene regulatory region. The distal silencer is more powerful; it inhibits the activity of homologous and heterologous enhancers according to their localization and independently from their orientation but does not actually influence the AFP promoter functioning [65].
Within the murine AFP gene promoter, a sequence at -57/-43 bp has been identified that contributes to the suppression of AFP gene activity in cells of non-hepatic origin. In a reaction with nuclear extracts from rat embryonic fibroblast cell culture, this sequence forms three complexes specific for the cell type; one of these complexes appears in extracts from AFP non-producing lines of both non-hepatic and hepatic origin [67]. Yet another presumable silencer sequence is localized in the region of -608/-371 bp [67]. The negative factors that bind these sequences are not yet identified.
Thus, the 5'-end AFP region is highly conservative. It contains the promoter, the enhancer, and the silencer that contain specific binding sequences for the transcription factors and provide precisely regulated AFP gene transcription. Before proceeding to the description of the transcription factors, let's mention some common rules of liver gene regulation.
HEPATOCYTE NUCLEAR FACTORS AND THEIR ROLE IN THE REGULATION OF
GENES IN LIVER
Regulation of liver-specific gene expression is a complex and multi-stage process. This is due to the need for continuous changes and coordination of expression of a great number of structural, secretory, and transport proteins and enzymes.
Hepatocyte nuclear factors (HNF) play a critical role in the regulation, which is carried out mainly on the transcriptional level [27, 68]; this section is dedicated to a short review of them.
To date several families of regulatory proteins (HNF1, C/EBP, HNF3, HNF4, and HNF6) are attributed to HNFs, their binding sites are identified in the regulatory elements of the numerous liver-specific genes. Although, the expression of these factors is not restricted to liver cells, they occur in maximum amounts in liver. Apparently, the tissue-specificity of the expression of each liver gene is achieved by the simultaneous participation of several hepatocyte transcription factors in the regulation of this process.
HNF1 family. The members of this family--HNF1 (HNF1alpha, LFB1) [69] and vHNF1 (HNF1beta, LFB3) [70, 71]--according to their DNA-binding domain structure are related to the superfamily of homeoproteins, the members of which play an important role in differentiation of various cell types [72]. According to their structure the HNF1 proteins are distinguished in a separated group of homeobox-containing factors (for review see [150]).
Apparently, HNF1 family proteins are the most widely distributed regulators of liver-specific gene expression; their potential binding sites have been found in regulatory regions of more than one hundred genes. Most often these sites are localized in promoter regions and form clusters with binding sites of other transcription factors [73].
HNF1 and vHNF1 recognize the same sequence and in contrast to other homeoproteins interact with DNA as homo- or heterodimers [71, 74]. A small protein DCoH plays an important role in stabilization of the dimer. Binding to HNF1 dimer, two DCoH molecules form a more stable tetramer, thus providing an optimal transactivational effect [75].
Several forms of HNF1 and vHNF1 exist. They possess various transactivational properties and are found at different levels in different organs [76, 77]. The transactivational properties may also vary when the dimer is stabilized at various DCoH protein concentrations [78].
HNF1 gene expression is controlled by a complex of both tissue-specific and ubiquitous factors. Within the HNF1 gene promoter binding sites for HNF3, HNF4, and AP-1 have been revealed [79, 80]. vHNF1 promoter can be activated by nuclear receptors COUP-TFI/Ear3 and COUP-TFII/Arp1, retinoic acid receptors, and, in contrast to HNF1, it does not depend on HNF4alpha [81].
In the course of embryonic development vHNF1 appears earlier than HNF1. In mice vHNF1 mRNA begins to be expressed in visceral endoderm of the yolk sac not later than between the fifth and sixth day of gestation [82]; on the tenth day vHNF1 is revealed in primary gut and liver diverticulum, in later stages of development its synthesis is localized in liver, kidney, and lung. HNF1 appears for the first time in liver diverticulum on the eleventh day of gestation; by this time AFP and SA mRNAs are clearly detected in this organ [82, 83]. HNF1 localizes similarly to vHNF1, but its expression level is significantly lower. After birth the situation changes dramatically: HNF1 mRNA appears in liver, kidney, gut, and pancreas, and in any tissue besides kidney in amount much greater than vHNF1 [70, 84, 85]. vHNF1 (but not HNF1) is expressed also in esophagus, lung, and thyroid [71].
A significant amount of HNF1 (but not vHNF1) usually appears in differentiated hepatoma lines. Hepatoma dedifferentiation and suppression of hepato-specific gene expression usually is accompanied by a decrease in HNF1 expression and an increase in vHNF1 [69, 86, 87]. Differentiation of embryonic carcinoma F9 in visceral endoderm induced by retinoic acid is accompanied by AFP gene activation; this is connected with significant increase in vHNF1 but not HNF1 expression [81, 82].
HNF1 gene inactivation is not lethal for the mouse embryo, but after birth it causes growth retardation, phenylketonuria, and hepatic, renal, and pancreas dysfunction and leads to 75% death of the homozygous animals within 20-40 days. It should be emphasized that in these animals a significant decrease of SA gene transcription rate is detected [88].
Mutation of HNF1, its main regulator HNF4alpha (see below), and vHNF1 in humans leads to insulin secretion system failure and causes the development of three forms of the non-insulin-dependent diabetes, which develops in young people (MODY3, MODY1, and MODY5, respectively) [89-91]. An insulin secretion failure is observed in mice with inactivated HNF1 also [92]. vHNF1 gene inactivation leads to embryonic death between the seventh and eighth day of mouse gestation, probably because of visceral endoderm development failure. It is important to emphasize that in these mice suppression of visceral endoderm markers such as AFP and HNF4alpha expression occurs (S. Cereghini, personal communication).
C/EBP family. The discovery of the first member of this family, C/EBPalpha [93], gave the opportunity to characterize a novel class of transcription factors that bind to a CCAAT-box. They contain a basic DNA-recognizing domain (so-called "bZIP"), an amino terminal trans-activation domain, and a helical structure of leucine zipper type providing dimerization. Members of the C/EBP family described so far possess similar structure, form homo- and heterodimers, and bind to a common DNA sequence [70, 94]. A decrease in expression level of one of the C/EBP genes can be compensated by variation in the concentration of other factors of the family [95]. C/EBP protein binding sites have been identified in regulatory elements of AFP [96], SA [97], C/EBPalpha [98], and other liver genes.
Two C/EBP proteins--the positive regulator C/EBPbeta and the transcription inhibitor LIP--are products of the same gene, their ratio in the cell being regulated by two mechanisms: either on the translational level [99] or by C/EBPalpha-dependent proteolytic cleavage of C/EBPbeta [100]. Heterodimers of other C/EBP proteins with shortened protein LIP lacking the trans-activational domain are able to suppress transcription.
C/EBPalpha mRNA appears for the first time on the thirteenth day of murine gestation. In the adult organism it is identified in different tissues of endodermal and mesodermal origin, whereas the corresponding protein has been revealed in liver and adipose tissue only. A similar situation is observed on analysis of the expression of other family members [101].
The so-called "PAR proteins", which lack a leucine zipper and are not capable of dimerization, are distinguished in an separated group within the C/EBP family. One of these proteins, DBP, is synthesized in adult liver only [102]. During liver regeneration the expression of this gene is completely downregulated. DBP expression follows a circadian rhythm that is controlled on the transcriptional level [103].
As a significant amount of C/EBP family proteins is revealed in non-proliferating cells only, while in regenerating liver cells, primary hepatocyte cultures, and hepatomas their level is low, these factors were supposed to possess anti-proliferative action [104]. It was found that sequential expression of a few factors of the C/EBP family is a necessary condition for terminal differentiation of a preadipocyte culture under hormonal treatment [105]. Hyperexpression of C/EBPalpha causes preliminary cessation of cell division [104].
Mice with an inactivated C/EBPalpha gene die of a glycogen synthesis and storage system failure within a few hours after birth [106]. The primary hepatocyte cultures obtained from these mice are characterized by a significant increase in proliferation rate and transformation frequency in comparison to the hepatocyte cultures of normal newborn mice [107]. These cultures preserve epithelial phenotype and SA expression.
Thus, C/EBPalpha is apparently an important switch for processes of maintenance of differentiation, proliferation control, and maintenance of non-transformed phenotype. Besides, there are some indications that C/EBP family proteins participate in hematopoietic stem cell differentiation, which takes place in liver [108].
HNF3 family. HNF3 family members contain a DNA-binding domain of the "winged helix" type, which is homologous to a corresponding region of fork head protein of Drosophila and contains a segment of a helix-loop-helix and two "wings" interacting with DNA [109, 110]. According to its tertiary structure, the HNF3 DNA-binding domain possesses a remarkable structural similarity to non-related DNA-packing histone H5. This implies that HNF3 factors participate in alteration of nucleosomal organization of chromatin during gene activation [94].
The family members--proteins HNF3alpha, beta, and gamma [111, 112]--recognize the same DNA sequences and interact with them as monomers [110]. These factors activate transcription of a number of liver-specific genes, e.g., SA [113], HNF1 [80], and also HNF3alpha and HNF3beta [114], in this way forming an autoregulatory loop.
HNF3beta is first detected on the seventh day of murine gestation in the primitive streak and node [115]. HNF3alpha follows the HNF3beta dynamics but at a lower concentration level. HNF3gamma starts to be expressed on the twelfth day of gestation [116].
In the adult organism HNF3alpha, beta, and gamma are localized in liver, gut, lung, and stomach, besides HNF3beta and gamma in ovary and HNF3gamma in testicles [101]. Increase of HNF3alpha level is observed in primary hepatocyte cultures grown on an extracellular matrix. Apparently, HNF3alpha participates in extracellular signal transduction that determines hepatocyte differentiation [83, 117].
HNF3beta gene inactivation leads to embryonic death between the tenth and eleventh day of murine gestation due to foregut formation defects [118, 119]. HNF3beta gene inactivation using Cre recombinase under the control of SA regulatory elements does not affect expression levels of the other HNFs--HNF1, HNF4alpha, HNF3alpha and gamma [120]. HNF3alpha deletion considerably decreases mouse growth rate after birth and causes death within the first week of life due to glucose homeostasis failure because of proglucagon downregulation in the pancreas [121]. Mice with inactivated HNF3gamma have no visible developmental abnormalities and differ from normal animals by increased expression level of HNF3alpha and beta and decreased transcription of some hepatospecific genes [122].
HNF4 family. So far few members of the HNF4 family have been identified [123, 124]. HNF4alpha is the most completely characterized. A homologous gene expressed in tissues corresponding to the HNF4-expressing tissues of mammals has been described in Drosophila [125]. A Xenopus laevis HNF4 homolog apparently is a preexisting maternal component spread gradually in the egg [126].
According to their structure HNF4 factors are related to the nuclear receptors superfamily. Like other nuclear receptors, they contain two DNA-binding "zinc-finger" domains and a spacious carboxy-terminal region providing dimerization and ligand binding. No ligand clearly shown to activate HNF4alpha in vivo has yet been identified.
As with the majority of nuclear receptors, HNF4alpha must form a homodimer in order to bind to DNA [123]. Other nuclear receptors do not dimerize with HNF4alpha, but they can participate in the regulation of HNF4alpha-controlled genes through competition for the common binding sites. Nuclear receptors COUP-TF1 and COUP-TF2 are HNF4 co-factors that intensify its action by orienting optimally its activational domain [127].
Among non-tissue-specific factors numerous possible HNF4alpha co-activators have been described [128, 129]. Multiple HNF4alpha forms that appear due to alternative splicing are characterized by various capacities to interact with co-activators which modulate their transactivational abilities [130].
HNF4alpha is capable to activate the expression of numerous hepatospecific genes, and it also appears to be the main HNF1 gene expression regulator [79, 80].
HNF4alpha is one of the earliest primary endoderm markers. In mouse embryo HNF4alpha mRNA first appears on the fifth day of gestation in a single layer of the cells facing the blastocoele. Till ninth day of gestation HNF4alpha expression is restricted to the extraembryonic visceral endoderm, then HNF4alpha mRNA appears in the liver and the gut primordium [131].
In the adult organism HNF4alpha is expressed in liver, kidney, gut, and pancreas [101, 123, 132].
In dedifferentiated hepatoma cell lines and somatic cell hybrids a loss of the hepatic phenotype usually correlates with HNF4alpha downregulation [87, 133]. A similar correlation was observed in vivo on comparative characterization of transplantable mouse hepatomas of various differentiation state (Lazarevich et al., in preparation). Exogenous HNF4alpha expression in dedifferentiated hepatoma H5 leads not only to re-expression of a number of the liver-specific genes, but also to a partial restoration of epithelial morphology and acquisition of the capability to respond to differentiation stimuli [134, 135].
Targeted disruption of the HNF4alpha gene leads in homozygous mice to embryonic lethality between the tenth and eleventh day of gestation [136] due to impaired gastrulation caused by visceral endoderm development failure [137].
It is important to note that a similar phenotype is observed in mice with inactivated GATA6 gene [138]. Transcription factors of the GATA family play an important role in the regulation of cell differentiation and embryonic morphogenesis. In the embryoid bodies obtained from the embryonic stem cells with inactivated GATA6 gene the expression of endodermal markers HNF4alpha, HNF1, HNF3beta, GATA4, and AFP is suppressed. Exogenous GATA6 expression in the non-endodermal cells activates HNF4alpha promoter.
HNF4alpha and other visceral endoderm marker expression can also be induced by factors of the TGFbeta superfamily [139, 140].
HNF6 family. HNF3 binding sites in promoters of some of the hepatospecific genes can be bound by one more factor, HNF6, which relates to a novel family of HNFs [141, 142]. This factor appeared to be the first characterized member of a novel homeoprotein class named ONECUT.
HNF6 first appears in the mouse embryo on the ninth day of gestation [143]. In the adult organism it is expressed in liver, pancreas, spleen, brain, and testicle. Probably at some stages of development HNF6 participates in the regulation of HNF4alpha and HNF3beta genes. HNF6 gene expression in adult liver is sex-dependent and regulated by growth hormone [144].
Regulational hierarchy of HNFs. There is a regulational hierarchy among HNFs. An interconnection between HNF1 and HNF4alpha expression is studied the most detailed. It is confirmed by studies on mice with a deletion in the hsdr-1 locus on chromosome 7 (so-called C14CoS albino deletion mice). In animals with this deletion the expression of numerous liver-specific genes is significantly reduced, and these animals die soon after birth. The transcription rates of HNF1 and HNF4alpha genes in these mice are 20 and 10% of the norm, respectively, and the C/EBP transcription rate is twofold decreased. The transcription of other HNFs is changed insignificantly, indicating that their expression is regulated independently [145].
Within the HNF1 gene promoter binding sites for HNF4, HNF3, and AP-1 have been revealed [79]. Mutations in the HNF4 binding site lead to 95% reduction of HNF1 expression level, while damage to HNF3 sites causes 33% reduction. In dedifferentiated hepatoma cell lines and in somatic cell hybrids a loss of the hepatic phenotype usually correlates with HNF4alpha and HNF1 extinction, whereas transfection of HNF4alpha expression vector restores HNF1 and other liver-specific gene expression [80, 87, 134, 135]. HNF1 gene expression in these cells is not connected with expression of the HNF3 factors. In some of the cell lines induction of AP-1 transcriptional activity elevates HNF4alpha activation severalfold [80].
An autoregulatory mechanism has been described according to which HNF1 can decrease the transcription rate of its own gene [146]. Since HNF1 has been shown not to bind its own promoter, the negative regulation should be governed by an indirect mechanism. Most likely HNF1 blocks an HNF4alpha activation effect, but it is not yet clear on what level.
A positive autoregulatory mechanism has been described for the C/EBP [98] and HNF3 [114] genes.
There are some data indicating that transfection of the HNF1 expression vector in some hepatoma cell lines can restore HNF4alpha transcription that was suppressed earlier [87]. Indeed, in the promoter region of the HNF4alpha gene an HNF1 binding site has been found, and HNF1 expression increases a reporter gene expression controlled by HNF4alpha promoter [147]. However, this observation is not true for all cell lines [80] and is not confirmed on transgenic mice [147]. On HNF1 gene inactivation in vivo HNF4alpha expression level decreases insignificantly [88]. All of these data as well as the dynamics of the expression of HNFs during ontogenesis indicate that even if HNF1 participates in HNF4alpha regulation in vivo, then it happens only at the late stages of development at HNF4alpha expression level establishment in the adult organism.
Some rules of HNF regulation have been investigated in an expression spectrum analysis in embryoid bodies obtained from embryonic stem cells with inactivated HNF3alpha or HNF3beta genes [148]. With this system the participation of HNF3beta in the maintenance of HNF4alpha and HNF1 expression levels and its necessity for HNF3alpha gene expression has been demonstrated.
HNF3alpha gene inactivation in embryoid bodies does not affect expression levels of vHNF1 and HNF3beta and gamma but leads to upregulation of HNF4alpha and HNF1 genes.
Thus, it has been shown that in course of development HNF3beta appears to be a potent activator of HNF4alpha and HNF1 expression, while HNF3alpha, possessing much weaker transactivational properties, acts as a negative regulator of transcription through its competition for binding sites with HNF3beta. The HNF3beta/HNF3alpha ratio in a cell may be increased on addition of insulin [148].
Available data on the interconnections of different HNF groups are summarized in Fig. 2.
Unfortunately, little is known about the interaction of the HNF cascade with non-tissue-specific regulatory pathways that provide control of cell proliferation and differentiation. It seems that HNF4, which not only controls liver-specific gene expression but also determines epithelial morphogenesis, possibly, via E-cadherin activation [135], could play a key role in the integration of the ubiquitous and the tissue-specific pathways.
Thus, several HNF families have now been described that in some degree control the expression of the majority of the known liver-specific genes. These families are related to the earlier described broader superfamilies of transcription factors and are highly conserved among different species. The factors within one family recognize the same DNA sequence and often are able to form heterodimers, modulating their individual properties. Besides, several forms of the same protein differing in transactivational abilities might be expressed within one cell. As far as these forms compete for the binding sites on a regulated gene, the efficacy of a transcription depends on which form binds more successfully.Fig. 2. Transcriptional hierarchy of hepatocyte nuclear factors. The interconnections possessing the most universal character are denoted by continuous lines, and dotted lines show regulation revealed at certain stages of development.
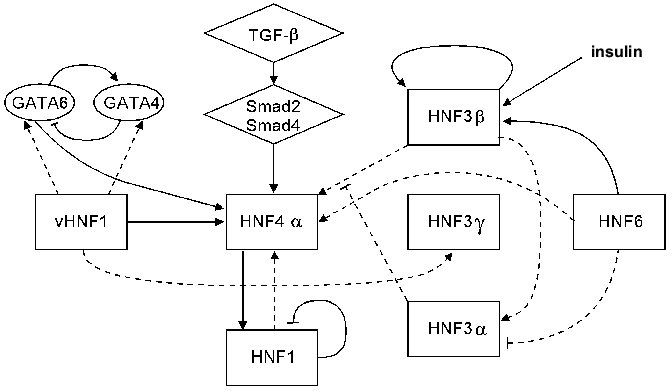
The factors related to different families modify each others effect by acting either synergically or in competition for overlapping binding sites.
Is seems that a significant portion of the transcription factors determining liver-specific gene expression have now been revealed. Almost all are characterized as transcription activators. However, this does not exclude the possible existence of negative regulatory mechanisms. Negative regulation could be fulfilled by the action of repression factors and forms with lowered transcriptional activity and by a competition between the proteins for common or overlapping binding sites or by variations of chromatin conformation which prohibit an activator protein from binding to a recognized sequence.
A number of the genes regulated by HNFs are significantly greater than the number of factors. The deciding mechanism for the fine regulation of expression of a great number of liver genes at different stages of development is apparently due to both the presence of a certain set of hepatocyte and ubiquitous factors and their concentration ratio.
TRANSCRIPTION FACTORS PARTICIPATING IN AFP GENE REGULATION
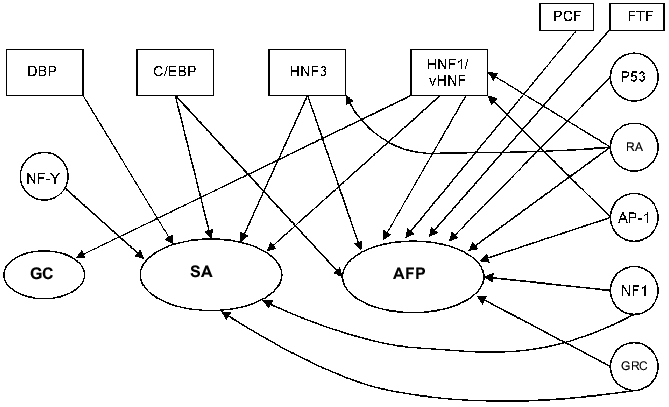
HNF1. Within the AFP gene promoter two HNF1 binding sites have been revealed, at -64/-49 and -131/-117 bp [96] (Fig. 3). The distal HNF1 site binds HNF1 in hepatic cells only and does it more efficiently than the proximal site [67, 149]. Deletion of this site inhibits the transcription of the AFP gene [47].
HNF1 binding sites have been found in the promoters of SA (-65/-53 bp) [69], alpha-albumin (-171/-159 and -101/-89 bp) [9], and group-specific component [55] genes, which confirms a hypothesis that this group of factors takes part in regulation of the expression of clustered liver genes. The HNF1 binding site localization is highly conservative in AFP and SA promoter regions among mouse, rat, and human [9, 150].Fig. 3. A possible model of AFP gene promoter regulation. TATA is the TATA-box. I) Promoter activation. The factors above the line indicating AFP promoter activate the transcription. AP-1 can activate the promoter in the absence of GRC (glucocorticoid hormone--receptor complex) and vice versa. II) Promoter suppression. The factors below the line indicating AFP promoter inhibit the expression. In the case of a simultaneous presence of AP-1 complex and GRC, their competition takes place on a level of either protein--protein interaction or DNA binding. Some of the C/EBP heterodimers and shortened C/EBP form LIP can suppress the promoter at different stages of development.
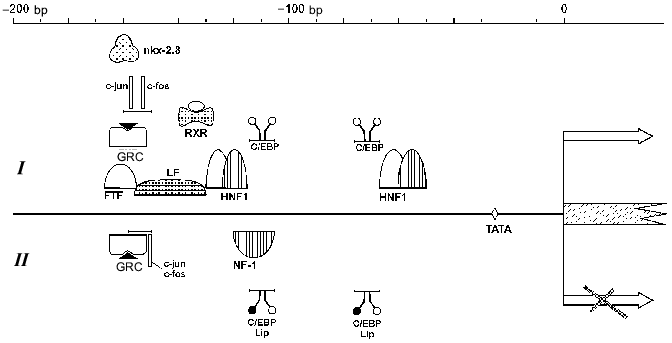
HNF1 and vHNF1 are capable of AFP gene activation both in vitro and in vivo [149]. In hepatic cells vHNF1, which appears in the course of embryogenesis earlier than HNF1, stimulates AFP but not SA promoter more efficiently than HNF1. In turn, HNF1 renders a significant stimulating effect on other albumin gene promoters, e.g., on group-specific component in human hepatoma HepG2 cells. In this case vHNF1 acts as a transdominant inhibitor [55]. These data indicate that HNF1/vHNF1 balance may considerably influence AFP and the other albumin gene product ratio (at least in some systems). In non-hepatic cells a transfection of HNF1 and vHNF1 expressing vectors induces a drastic increase of AFP and SA gene expression, and in this case the HNF1 activation effect is higher [151].
ATBF1. Within the AFP gene enhancer a sequence has been described that is similar to the HNF1 binding site; however, HNF1 binding efficacy in this region is significantly lower than in the promoter elements [149]. One more transcription factor binds this region--ATBF1 [152]. HNF1 binding sites in AFP and SA promoters are able to bind ATBF1, although much less efficiently. ATBF1 is expressed both in hepatomas and in cell cultures of non-hepatic origin [152]. In human hepatoma cells, ATBF1 suppresses the activity of AFP enhancer and promoter, apparently by competition with HNF1 for the common binding site [153].
C/EBP. Two C/EBP binding sites have been revealed within the AFP gene promoter at -77/-68 and -115/-106 bp (Fig. 3). Some more binding sites are found in enhancer element EI [96]. The distal promoter site binds C/EBP family proteins more efficiently than the proximal, while binding affinity of these factors to the enhancer sites is higher than to the promoter's sites [96, 151].
At least four transcription activators related to the C/EBP family (C/EBPalpha, C/EBPbeta, C/EBPgamma, and DBP) and negative regulator LIP can take part in AFP gene regulation [96, 151]. Although C/EBPalpha and C/EBPbeta can activate AFP promoter in vitro, no correlation between AFP promoter activity and an activity of this family of proteins in the course of development has been disclosed. After birth, while AFP synthesis decreases dramatically, C/EBPalpha and C/EBPbeta concentrations increase remarkably and LIP/C/EBPbeta ratio decreases. In contrast to the promoter sites, C/EBP sites localized in the enhancer in the course of late embryogenesis and after birth are being activated by C/EBPalpha. This enhancer remains active in adult liver and, possibly, influences SA promoter [151].
NF-1. Within the AFP gene promoter a binding site for non-tissue-specific factor NF-1 has been localized at -123/-108 bp that partially overlaps with HNF1 and C/EBP sites [47, 96] (Fig. 3). At low concentration NF-1 weakly stimulates AFP promoter; in a high concentration it suppresses the promoter activity, while SA promoter is activated by NF-1 proportionally to the concentration of the latter [151].
FTF. A promoter sequence of -166/-155 bp, which is a canonical binding motif for nuclear receptors related to the Drosophila fushi tarazu family (Ftz-F1), is necessary for binding of a recently identified member of this family--FTF protein [154] (Fig. 3).
FTF isoforms possessing similar transcriptional activity bind DNA as monomers and do not require a ligand binding. FTF is specific for liver of newborn and adult rats, and lesser amounts are found in embryonic liver and pancreas. It also appears in some AFP-producing hepatomas [154, 155].
FTF apparently binds the promoter cooperatively with one more factor, LF, the binding site of which partially overlaps with the FTF site: -155/-134 bp [154]. FTF binding sites have been revealed in other liver-specific gene promoters, including the HNF3beta promoter [155].
nkx-2.8. A sequence at -166/-153 bp, which is conservative among mouse, rat, and human [156], is necessary for the stimulation of AFP promoter by the remote enhancers (Fig. 3). Deletion of this region leads to a 70% reduction of expression level of a reporter gene [157], whereas shortening of the distance between the promoter and the enhancers or in the absence of the enhancers this deletion does not effect the expression level [158].
At screening of expression library from human hepatoma a new factor nkx-2.8 has been cloned that binds the mentioned site [159]. Apparently, nkx-2.8 provides a contact of AFP gene promoter with one or few enhancers interacting with associated with them regulatory protein complexes.
nkx-2.8 is expressed only in embryonic liver and AFP-expressing hepatoma lines [157]. As nkx-2.8 expression is clearly associated with AFP expression, it is supposed that it may play an important role in the maintenance of AFP expression in embryonic liver, while its absence might be a cause of the silence of the gene [157]. It is also possible that reduction of AFP gene expression might be due to a competition of nkx-2.8 with other transcription factors for overlapping binding sites.
SA promoter does not contain an nkx-2.8 binding site, and it probably interacts with AFP enhancers via other trans-factors and cis-elements, thus not competing for interaction with the same sequences within AFP enhancers [58, 157].
Nuclear receptor response elements. At certain stages of development some nuclear receptors, especially glucocorticoid and retinoid acid receptors, can take part in the modulation of AFP gene expression. At different stages of ontogenesis and carcinogenesis the same hormone--receptor complex may render either an activating or repressing influence on the same gene expression.
Glucocorticoid hormones. The glucocorticoids and their synthetic analog (dexamethasone) considerably accelerate reduction of AFP gene expression in rodent liver immediately after birth. The hormonal regulation, which does not influence synthesis of SA in liver and of AFP in kidney [25, 160], appears to be tissue- and stage-specific in this case. Dexamethasone suppresses AFP gene expression in rat hepatoma McA-RH 7777 and induces it in hepatoma McA-RH 8994, which is characterized by relatively low AFP synthesis level [161].
Within the AFP gene regulatory region several sequences have been described that are recognized by glucocorticoid hormone--receptor complex (GRC) [47]. Two of them are localized in the promoter region: -166/-154 bp and -224/-219 bp [47, 96] (Fig. 3), they determine the sensitivity for dexamethasone in McA-RH 7777 cells [162]. The promoter GRC binding sites partially overlap with binding sites of the other transcription factors, e.g., nkx-2.8, FTF, LF, AP-1 [154, 158], which may compete with GRC for a site binding.
Retinoic acid. Retinoic acid regulates AFP gene expression in embryonic carcinoma and hepatoma cells. It induces a differentiation of mouse teratocarcinoma F9 cells into visceral endoderm, followed by activation of AFP and vHNF1 gene expression [82, 163]. In rat hepatomas McA-RH 7777 and McA-RH 8994 retinoic acid activates AFP gene expression on the transcriptional level [164-166], and in McA-RH 8994 cells a simultaneous induction of the SA gene has been shown. At the same time retinoic acid causes downregulation of AFP and SA genes in some human hepatomas [167] and of the SA gene in a primary rat hepatocyte culture [168].
The influence of retinoic acid on AFP gene expression can be carried out both by means of HNF induction (see above) and through the hormone--receptor complex binding to the corresponding sites in AFP regulatory elements. Within the AFP gene regulatory region three elements determining sensitivity for retinoic acid have been revealed. One of them is localized in promoter (-139/-127 bp) and overlaps with other transcription factor binding sites [165]. Expression of nuclear receptor COUP-TF1, which can bind to the same promoter sequence, blocks the retinoic acid activation of AFP promoter.
The diversity of AFP gene regulation mechanisms under glucocorticoid and retinoic acid treatment may be determined by the presence of several forms of the corresponding receptors with different transactivation properties, by competition for overlapping binding sites, and by protein--protein interactions with other transcription factors.
The mechanisms of AFP gene regulation by glucocorticoid and retinoic receptors are discussed in more detail in review [169].
Influence of oncoproteins on AFP gene expression. Within the AFP gene promoter a binding site for transcription complex AP-1 has been localized (-160/-152 bp) (Fig. 3). This site is bound by products of the oncogene families jun and fos [162] both as heterodimers Jun-Fos and as homodimers Jun-Jun, in the latter case the binding efficacy and activation properties being considerably reduced [170].
Protooncogenes participating in cell differentiation and proliferation processes are expressed in liver at certain stages of embryonic development. After birth their synthesis decreases significantly. In in vivo hepatic proliferation in adult regenerating liver the expression level of c-jun, c-fos, and c-myc oncogenes increases considerably, high levels of expression being detected in some hepatoma cell lines [171]. The kinetics of nuclear oncogene activation in normal hepatocyte cultures under EGF induction correlates with the AFP gene activation, and AFP mRNA appears several hours later than oncogene mRNAs. On the basis of these observations, the hypothesis of the possible participation of the oncoproteins in AFP gene regulation has been suggested [171, 172]. As in some hepatoma cell lines with a high level of AFP synthesis and in non-producing AFP cultures the nuclear oncogene expression levels do not differ, it is supposed that at least in transformed hepatocytes the nuclear oncogenes do not render a direct activation effect on AFP gene expression [171].
In AFP gene promoter, the AP-1 binding site partially overlaps with the GRC binding site (Fig. 3). Also, in various cell lines a functional antagonism between GRC and AP-1 has been shown.
In monkey kidney CV-1 and mouse teratocarcinoma F9 cells the AFP promoter can be activated either by transfection with c-jun and c-fos oncogenes or with glucocorticoid receptor in the absence of c-jun/c-fos products. A simultaneous expression of the nuclear oncogenes and glucocorticoid receptor leads to AFP gene expression downregulation [162]. In human hepatoma HuH-7 cells transfection of c-jun and c-fos oncogenes suppresses AFP promoter activity and decreases the activation effect that was observed under dexamethasone treatment of non-transfected cells [170].
The functional antagonism between AP-1 and GRC may come either from partial overlapping of the corresponding binding sites in the AFP promoter region, or from protein--protein interactions [162, 170]. Functional antagonism takes place in the regulation of some other genes [169]. A reciprocal suppression of transcriptional activity is also described for nuclear oncogenes and retinoic acid receptors [173].
The AP-1 complex does not influence the activity of the SA gene promoter and enhancers [170], this suggesting a possible contribution of these factors in the differential regulation of the AFP and SA genes in embryonic and postnatal stages of ontogenesis.
p53. Recently a p53-dependent mechanism of AFP gene repression that is due to competition of this protein with activation factor HNF3 for overlapping binding sites in the silencer region of AFP gene has been suggested [174]. The supposed mechanism probably takes place in regulation of AFP gene expression in some stages of development; however, it apparently is not universal, since in primary mouse hepatocyte culture an induction of AFP gene expression is fulfilled independently from p53 activity and in rat hepatoma clones differing in AFP expression level (see below) a correlation of this property with p53 activity has not been observed (Kudryavtseva and Lazarevich, unpublished data).
Participation of factors Afr1 and Afr2 in AFP gene expression. When comparing murine lines differing in AFP expression levels in adult liver both in norm and during regeneration, two unlinked trans-acting loci, Afr1 and Afr2, were revealed [40, 41]. Afr1 controls (at least in part) the low level of AFP gene expression in adult liver, while Afr2 determines the increase of the expression during regeneration [41]. The effect of both genes is pleiotropic and controls at least one more liver gene--H19 [175].
The Afr1 gene is mapped to chromosome 15, 2-3 centimorgans proximal from the c-myc gene [176]. Two alleles have been revealed: dominant Afr1a and recessive Afr1b, providing 15-20-fold increase of AFP gene expression level in adult liver [40, 41]. The influence of the Afr1-locus on the AFP and H19 genes is specific for liver and is not connected with their repression in gut [175, 177]. SA gene expression does not depend on Afr1 phenotype [42].
For the Afr2 gene, mapped to human chromosome 11 [178], the dominant allele is Afr2b, which determines a tenfold weaker induction of the expression at regeneration in adult liver than Afr2a allele, present in the majority of mouse lines [41].
The Afr1 and Afr2 targets appear to be cis-elements localized within the silencer region, which determines AFP mRNA level decrease during the first weeks of mouse life [42, 179, 180].
Some data indicate that the Afr1 gene product may take part in a posttranscriptional regulation interacting with the 5´- and 3´-end untranslatable regions of the AFP structural gene that control the mRNA stability [42]. However, the expression of mouse histocompatibility class I H-2D gene under control of the AFP EI enhancer, the promoter, and the silencer in the corresponding tissues is suppressed after birth and activated during regeneration in parallel with the endogenous AFP gene, indicating that the regulation is not carried out through the structural gene sequences [179].
Apparently, the Afr1 product acts as a factor of negative regulation immediately after birth. The Afr1 gene product binds DNA in the silencer region and probably interacts either directly or indirectly with one of the basal transcription factors, a component of the preinitiation RNA-polymerase II complex. This interaction may alter some of the RNA-polymerase II activities. As the transcription rates of the AFP and H19 genes in transgenic mice do not depend on Afr1 phenotype, the repression of the transcription is probably performed not at an initiation stage but influences the elongation or mRNA 3´-end formation [42]. This might affect AFP transcript stability. A basal factor of transcription that could serve as a link between transcriptional and post-transcriptional mechanisms is of a special interest in this scheme [179].
The proposed mechanism does not contradict a suggestion about post-transcriptional regulation of AFP expression by Afr1 factor but, on the contrary, supplements it. Probably in this case there is no clear border between the transcriptional and the post-transcriptional regulation levels and, apparently, some non-characterized factors exist that couple it into a united mechanism.
The AFP gene expression level rise during liver regeneration, which is probably connected to an action of the positive regulator Afr2. Cis-elements necessary for this regulation are localized within the region -1010/-838 bp [180].
Thus, the proteins coded by Afr2 and Afr1 bind different sequences and act independently [180]. The cloning of Afr1 and Afr2 will help considerably to clarify the mechanisms of AFP gene repression in adult liver.
A set of results obtained in the author's laboratory within recent years gives strong evidence for the existence of some additional mechanisms of AFP gene negative regulation. A collection of rat hepatoma McA-RH 7777 differing in AFP synthesis level which was obtained by T. L. Eraiser [181] (Laboratory of Immunochemistry, Institute of Carcinogenesis, Cancer Research Center, Russian Academy of Medical Sciences) served as a model for studies of AFP gene regulation mechanisms. AFP expression levels in these clones differ by ~1000-fold and are controlled on the transcriptional level.
With this model we have demonstrated a correlation of AFP, SA, HNF4, and HNF1 gene expression.
On the transfection of AFP-non-producing clones with HNF4 expression vector HNF1 synthesis activates in all of the cells and SA mRNA level increases significantly. No considerable increase in AFP synthesis in the HNF4 transfected cells occurs (Lazarevich et al., in preparation). This implies that either HNF4 is necessary but insufficient for AFP gene expression activation in the tumor cells or HNF4 does not participate in AFP gene regulation but there is some common regulatory factor that participates in the regulation of these genes.
The increase in HNF1 level in a cell is sufficient for SA but not for AFP gene activation. It appears to be HNF1 that determines the correlation of the levels of SA and AFP the in the described system.
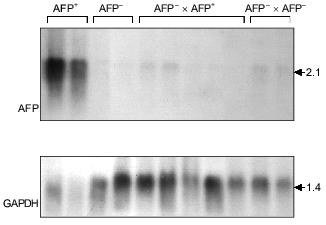
In the somatic cell hybrids of AFP-producing and AFP-non-producing clones AFP gene expression is suppressed completely (Fig. 4) (Kustova and Lazarevich, unpublished data). At the same time, in the hybrid cells HNF1 protein appears, which has not been identified in AFP-non-producing clones; thus, AFP gene expression decrease is not connected with the absence of HNF1, which is known to be a potential AFP promoter regulator. These data support the notion that some factor(s) exists in the AFP-non-producing clones that inhibits AFP gene expression. As this factor(s) has not been identified yet, the described system could appear to be a valuable model for its identification. It seems that only in the case if the negative regulators are absent in a cell, the functioning promoter activity is determined by a set of other transcription factors and a ratio of their forms with different activation properties.
Fig. 4. A somatic hybridization of AFP-producing (AFP+) and AFP-non-producing (AFP-) clones of rat hepatoma. Northern-blot hybridization of total cellular RNA from the obtained hybrids (AFP+ × AFP- and AFP- × AFP-) and the original clones with AFP and GAPDH probes. The sizes of the revealed transcripts in kb are indicated on the right.
A POSSIBLE MODEL OF AFP GENE REGULATION
On the basis of the described data, the hypothesis has been suggested that the AFP promoter is activated by a complex of HNF1/FTF transcription factors [154]. The authors presume that FTF interacts with HNF1 via LF factor, so far as the interaction does not occur without formation of the FTF/LF complex. The HNF1/FTF complex may compete with GRC and NF-1 for the binding sites.
According to this conception AFP promoter activity considerably depends on HNF1/NF-1 molar ratio in the nucleus, which varies in the different stages of hepatocyte development [151, 154]. HNF1/NF-1 balance depends on an expression spectrum of other specific and common transcription factors and, possibly, serves as a promoter activity modulator on the different stages of development [154]. The AFP gene regulation might be realized through formation of HNF1 dimers with various transcriptional activity and, especially due to ratio modulation of HNF1 and vHNF1, which possess different transcriptional properties. This ratio may play a crucial role in coordinated regulation of the albumin cluster genes in the course of development.
The importance of HNF1/NF-1 balance and their competition is confirmed in vivo for the human AFP gene promoter. A mutation at -119 bp affecting NF-1 binding site determines a hereditary high AFP synthesis level in adults [181].
In the promoter region a competition exists between AP-1, HNF1/FTF, GRC transcription complexes and retinoic hormone--receptor complex. Depending on the presence of other transcription factors, GRC, retinoic hormone--receptor complex and AP-1 may function both as positive and negative transcription regulators. Furthermore, GRC binding to the promoter leads to a chromatin structure modification which influences the binding of other transcription factors, e.g., NF-1.
Apparently, C/EBP family proteins do not compete for binding sites with other factors; however, they may modulate promoter activity due to formation of homo- and heterodimers with different transcriptional properties.
C/EBP dimers bind EI enhancer more efficiently than AFP promoter [96]. This intergenic enhancer is capable of both AFP and SA promoter activation, which is localized at a distance of 27 kb and contains a C/EBPalpha binding site [151, 154]. C/EBP family proteins may be an important chain in tandem SA and AFP regulation in the course of development, especially taking into account that at early stages of embryogenesis the SA enhancer is not functional, while the intergenic enhancer is extremely active. EI activity is preserved in adult liver also, whereas AFP expression is suppressed. In some cell types this activity correlates more strictly with SA expression than with that of AFP [154]. SA upregulation after birth is accompanied by C/EBP level increase [154].
Thus, to date a considerable number of transcription factors that can modulate the expression of AFP and other albumin genes in different systems has been revealed (Fig. 5). It seems that the most suitable candidate for the role of the key AFP gene transcription activator could be a factor: 1) whose binding sites could be revealed in the most important elements of the AFP gene regulatory region; 2) whose expression tissue-specificity would maximally cover the spectrum of AFP gene expression; 3) which would be already expressed in the tissues when AFP mRNA was detected there; 4) whose expression dynamics during development would be similar to that of the AFP gene; 5) which would be able to reduce other albumin gene expression specific for the differentiated states of hepatocytes; 6) whose expression levels would be considerably increased during hepatocarcinogenesis.
Even taking into account that the criteria provided above describe an ideal situation not considering some aspects (e.g., the difference of the regulatory mechanisms at specific stages of development; the complicated and multi-component nature of those mechanisms; the variety of forms of some regulatory factors), it is easy to note that vHNF more than other characterized AFP gene activators corresponds to the above criteria and is probably the most important AFP gene regulator in the course of development.Fig. 5. Transcription factors participating in the regulation of albumin gene expression (RA, retinoic acid; GC, group specific component).
The highly specific AFP factor nkx-2.8 is able to significantly activate the expression of this gene providing an interaction of the promoter with distant enhancers. Due to clear correlation of nkx-2.8 and AFP expression levels both in the course of development and in tumor cells, this factor also seems to be one of the key AFP transcription activators.
Thus, AFP gene regulation is governed mainly on the transcriptional level. In this process a wide spectrum of both tissue-specific and ubiquitous transcription factors takes part, the expression of which is precisely regulated and is strictly dependent on the developmental stage. The factors may compete with each other for binding to overlapping DNA sites, enter into protein--protein interactions, or form complexes to bind DNA. The activation properties of the nuclear factors may be modulated by homo- and heterodimer formation.
At the same time, it seems that the mechanisms studied so far are not able to explain the highly tissue- and stage-specific AFP gene suppression that, at our opinion, should be accomplished by means of not yet identified negative transcription regulators, the identification of which seems to be one of the main promises for opening in this field.
One more of the most important directions of studies of AFP gene transcription mechanisms seems to be the search for the factors by means of which its regulation by extracellular matrix components is realized [183] and its zonal expression is controlled.
The author is deeply grateful to G. I. Abelev and E. V. Varga for their useful remarks and to A. V. Bosyi for the help in preparing the illustrations.
The experimental data discussed in this review were obtained in the Institute of Carcinogenesis, Cancer Research Center, Russian Academy of Medical Sciences with support from the Russian Foundation for Basic Research (grants 99-04-49068, 99-04-49227), the program "Leading Scientific Schools" (grant 96-15-98082), subprogram "Frontiers in Genetics" of the priority direction "Technology of Living Systems" (grant 99-2-024), and Federal aim program "Integration" (direction 1.5/99, project No. 12c/99).
REFERENCES
1.Bergstrand, C. G., and Czar, B. (1956) Scand. J.
Clin. Lab. Invest., 8, 174-179.
2.Abelev, G. I., Perova, S., Khramkova, N. I.,
Postnikova, Z. A., and Irlin, I. (1963) Transplant. Bull.,
1, 174-180.
3.Tatarinov, S. (1964) Vopr. Med. Khim.,
2, 584-589.
4.Abelev, G. I., Assecritova, I. V., Kraevsky, N. A.,
Perova, S. D., and Perevodchikova, N. I. (1967) Int. J. Cancer,
2, 551-558.
5.Deutsch, H. F. (1991) Adv. Cancer Res.,
56, 253-312.
6.Abelev, G. I. (1993) Sov. Sci. Rev. D.
Physicochem. Biol., 11, 85-109.
7.Cooke, N. E., and David, E. V. (1985) J. Clin.
Invest., 76, 2420-2424.
8.Yang, F., Brune, J. L., Naylor, S. L., Cupples, R.
L., Naberhaus, K. H., and Bowman, B. H. (1985) Proc. Natl. Acad.
Sci. USA, 82, 7994-7998.
9.Belanger, L., Roy, S., and Allard, D. (1994) J.
Biol. Chem., 269, 5481-5484.
10.Guan, X. J., Arhin, G., Leung, J., and Tilghman,
S. M. (1996) Mamm. Genome, 7, 103-106.
11.Song, Y. H., Naumova, A. K., Liebhaber, S. A.,
and Cooke, N. E. (1999) Genome Res., 9, 581-587.
12.Kioussis, D., Eiferman, F., van de Rijn, P.,
Gorin, M. B., Ingram, R. S., and Tilghman, S. M. (1981) J. Biol.
Chem., 256, 1960-1967.
13.Sargent, T. D., Jagodzinski, L., Yang, M., and
Bonner, J. (1981) Mol. Cell. Biol., 1, 871-883.
14.Dugaiczyk, A., Harper, M. E., and Minghetti, P.
P. (1985) Kroc. Found. Ser., 19, 181-188.
15.Gorin, M. B., Cooper, D. L., Eiferman, F., van de
Rijn, P., and Tilghman, S. M. (1981) J. Biol. Chem., 256,
1954-1959.
16.Gibbs, P. E., Witke, W. F., and Dugaiczyk, A.
(1998) J. Mol. Evol., 46, 552-561.
17.Petropoulos, C. J., Yaswen, P., Panzica, M., and
Fausto, N. (1985) Cancer. Res., 45, 5762-5768.
18.Wan, Y. J., and Chou, J. Y. (1989) Arch.
Biochem. Biophys., 270, 267-276.
19.Lemire, J. M., and Fausto, N. (1991) Cancer
Res., 51, 4656-4664.
20.Watanabe, T., Jimenez-Molina, J. L., and Chou, J.
Y. (1992) Biochem. Biophys. Res. Commun., 185,
648-656.
21.Dziadek, M., and Adamson, E. (1978) J.
Embryol. Exp. Morphol., 43, 289-313.
22.Tyner, A. L., Godbout, R., Compton, R. S., and
Tilghman, S. M. (1990) J. Cell. Biol., 110, 915-927.
23.Nahon, J. L., Tratner, I., Poliard, A., Presse,
F., Poiret, M., Gal, A., Sala-Trepat, J. M., Legres, L., Feldmann, G.,
and Bernuau, D. (1988) J. Biol. Chem., 263,
11436-11442.
24.Cooke, N. E., McLeod, J. F., Wang, X. K., and
Ray, K. (1991) J. Steroid Biochem. Mol. Biol., 40,
787-793.
25.Belanger, L., Frain, M., Baril, P., Gingras, M.
C., Bartkowiak, J., and Sala-Trepat, J. M. (1981) Biochemistry,
20, 6665-6672.
26.Sala-Trepat, J. M., Dever, J., Sargent, T. D.,
Thomas, K., Sell, S., and Bonner, J. (1979) Biochemistry,
18, 2167-2178.
27.Tilghman, S. M., and Belayew, A. (1982) Proc.
Natl. Acad. Sci. USA, 79, 5254-5257.
28.Jane, S. M., Ney, P. A., Vanin, E. F., and
Gumucio, D. L. (1992) EMBO J., 11, 2961-2969.
29.Vergnes, L., Taniguchi, T., Omori, K., Zakin, M.
M., and Ochoa, A. (1997) Biochim. Biophys. Acta, 1348,
299-310.
30.Abelev, G. I. (1978) in Cell Differentiation
and Neoplasia (Saunders, G. G., ed.) Raven Press, N. Y., pp.
257-269.
31.Abelev, G. I. (1971) Adv. Cancer Res.,
14, 295-358.
32.Abelev, G. I., and Eraiser, T. L. (1999)
Semin. Cancer Biol., 9, 95-107.
33.Knowles, B. B., Howe, C. C., and Aden, D. P.
(1980) Science, 209, 497-499.
34.Schulz, W. A., Crawford, N., and Locker, J.
(1988) Exp. Cell. Res., 174, 433-447.
35.Latchman, D. S., Brzeski, H., Lovell-Badge, R.,
and Evans, M. J. (1984) Biochim. Biophys. Acta, 783,
130-136.
36.Deschartrette, J., Fouger-Deschartrette, C.,
Corcos, L., and Schimke, R. T. (1985) Proc. Natl. Acad. Sci.
USA, 82, 765-769.
37.Nahon, J. L. (1987) Biochimie, 69,
445-459.
38.Andrews, G. K., Janzen, R. G., and Tamaoki, T.
(1982) Dev. Biol., 89, 111-116.
39.Innis, M. A., and Miller, D. L. (1979) J.
Biol. Chem., 254, 9148-9154.
40.Olsson, M., Lindahl, G., and Ruoslahti, E. (1977)
J. Exp. Med., 145, 819-827.
41.Belayew, A., and Tilghman, S. M. (1982) Mol.
Cell. Biol., 2, 1427-1435.
42.Vacher, J., Camper, S. A., Krumlauf, R., Compton,
R. S., and Tilghman, S. M. (1992) Mol. Cell. Biol., 12,
856-864.
43.Vedel, M., Gomez-Garcia, M., Sala, M., and
Sala-Trepat, J. M. (1983) Nucleic Acids Res., 11,
4335-4354.
44.Turcotte, B., Guertin, M., Chevrette, M., LaRue,
H., and Belanger, L. (1986) Nucleic Acids Res., 14,
9827-9841.
45.Opdecamp, K., Riviere, M., Molne, M., Szpirer,
J., and Szpirer, C. (1992) Nucleic Acids Res., 20,
171-178.
46.Widen, S. G., and Papaconstantinou, J. (1986)
Proc. Natl. Acad. Sci. USA, 83, 8196-8200.
47.Guertin, M., LaRue, H., Bernier, D., Wrange, O.,
Chevrette, M., Gingras, M. C., and Belanger, L. (1988) Mol. Cell.
Biol., 8, 1398-1407.
48.Godbout, R., Ingram, R. S., and Tilghman, S. M.
(1988) Mol. Cell. Biol., 8, 1169-1178.
49.Wen, P., Groupp, E. R., Buzard, G., Crawford, N.,
and Locker, J. (1991) DNA Cell. Biol., 10, 525-536.
50.Camper, S. A., and Tilghman, S. M. (1989)
Genes Dev., 3, 537-546.
51.Vacher, J., and Tilghman, S. M. (1990)
Science, 250, 1732-1735.
52.Hammer, R. E., Krumlauf, R., Camper, S. A.,
Brinster, R. L., and Tilghman, S. M. (1987) Science, 235,
53-58.
53.Chevrette, M., Guertin, M., Turcotte, B., and
Belanger, L. (1987) Nucleic Acids Res., 15,
1338-1339.
54.Ryan, S. C., Zielinski, R., and Dugaiczyk, A.
(1991) Genomics, 9, 60-72.
55.Song, Y. H., Ray, K., Liebhaber, S. A., and
Cooke, N. E. (1998) J. Biol. Chem., 273, 28408-28418.
56.Mathew, J. K., Knoll, B. J., Pilla, A., Durham,
D., and Sell, S. (1994) J. Tumor Marker Oncol., 9,
21-28.
57.Groupp, E. R., Crawford, N., and Locker, J.
(1994) J. Biol. Chem., 269, 22178-22187.
58.Jin, J. R., Wen, P., and Locker, J. (1995) DNA
Cell. Biol., 14, 267-272.
59.Camper, S. A., Godbout, R., and Tilghman, S. M.
(1989) Progr. Nucleic Acid Res. Mol. Biol., 36,
131-143.
60.Millonig, J. H., Emerson, J. A., Levorse, J. M.,
and Tilghman, S. M. (1995) Mol. Cell. Biol., 15,
3848-3856.
61.Ramesh, T. M., Ellis, A. W., and Spear, B. T.
(1995) Mol. Cell. Biol., 15, 4947-4955.
62.Spear, B. T. (1999) Semin. Cancer Biol.,
9, 109-116.
63.Qiu, Y., Krishnan, V., Pereira, F. A., Tsai, S.
Y., and Tsai, M. J. (1996) J. Steroid Biochem. Mol. Biol.,
56, 81-85.
64.Thomassin, H., Bois-Joyeux, B., Delille, R.,
Ikonomova, R., and Danan, J. L. (1996) DNA Cell. Biol.,
15, 1063-1074.
65.Nakabayashi, H., Hashimoto, T., Miyao, Y., Tjong,
K. K., Chan, J., and Tamaoki, T. (1991) Mol. Cell. Biol.,
11, 5885-5893.
66.Emerson, J. A., Vacher, J., Cirillo, L. A.,
Tilghman, S. M., and Tyner, A. L. (1992) Dev. Dyn., 195,
55-66.
67.Henriette, M. F., Gabant, P., Dreze, P. L.,
Szpirer, C., and Szpirer, J. (1997) Folia Biol., 43,
5-13.
68.Powell, D. J., Friedman, J. M., Oulette, A. J.,
Krauter, K. S., and Darnell, J. E., Jr. (1984) J. Mol. Biol.,
179, 21-35.
69.Cereghini, S., Blumenfeld, M., and Yaniv, M.
(1988) Genes Dev., 2, 957-974.
70.De Simone, V., De Magistris, L., Lazzaro, D.,
Gestner, J., Monaci, P., Nicosia, A., and Cortese, R. (1991) EMBO
J., 5, 1435-1443.
71.Rey-Campos, J., Chouard, T., Yaniv, M., and
Cereghini, S. (1991) EMBO J., 10, 1445-1457.
72.Wright, C. V. E. (1991) Curr. Opin. Cell
Biol., 3, 976-982.
73.Tronche, F., Ringeisen, F., Blumenfeld, M.,
Yaniv, M., and Pontoglio, M. (1997) J. Mol. Biol., 266,
231-245.
74.Mendel, D. B., Hansen, L. P., Graves, M. K.,
Conley, P. B., and Crabtree, G. R. (1991) Genes Dev., 5,
1042-1056.
75.Mendel, D. B., Khavari, P. A., Conley, P. B.,
Graves, M. K., Hansen, L. P., Admon, A., and Crabtree, G. R. (1991)
Science, 254, 1762-1767.
76.Bach, I., and Yaniv, M. (1993) EMBO J.,
12, 4229-4242.
77.Ringeisen, F., Rey-Campos, J., and Yaniv, M.
(1993) J. Biol. Chem., 268, 25706-25711.
78.Hansen, L. P., and Crabtree, G. R. (1993)
Curr. Opin. Genet. Dev., 3, 246-253.
79.Tian, J. M., and Schibler, U. (1991) Genes
Dev., 5, 2225-2234.
80.Kuo, C. J., Conley, P. B., Chen, L., Sladek, F.
M., Darnell, J. E., Jr., and Crabtree, G. R. (1992) Nature,
355, 457-461.
81.Power, S. C., and Cereghini, S. (1996) Mol.
Cell. Biol., 16, 778-791.
82.Cereghini, S., Ott, M. O., Power, S., and Maury,
M. (1992) Development, 116, 783-797.
83.Cascio, S., and Zaret, K. S. (1991)
Development, 113, 217-225.
84.Blumenfeld, M., Maury, M., Chouard, T., Yaniv,
M., and Condamine, H. (1991) Development, 113,
589-599.
85.Ott, M. O., Rey-Campos, J., Cereghini, S., and
Yaniv, M. (1991) Mech. Dev., 36, 47-58.
86.Baumhueter, S., Courtois, G., and Crabtree, G. R.
(1988) EMBO J., 7, 2485-2493.
87.Bulla, G. A., and Fournier, R. E. (1994) Mol.
Cell. Biol., 14, 7086-7094.
88.Pontoglio, M., Barra, J., Hadchouel, M., Doyen,
A., Kress, C., Bach, J. P., Babinet, C., and Yaniv, M. (1996)
Cell, 84, 575-585.
89.Yamagata, K., Oda, N., Kaisaki, P. J., Menzel,
S., Furuta, H., Vaxillaire, M., Southam, L., Cox, R. D., Lathrop, G.
M., Boriraj, V. V., Chen, X., Cox, N. J., Oda, Y., Yano, H., Le Beau,
M. M., Yamada, S., Nishigori, H., Takeda, J., Fajans, S. S.,
Hattersley, A. T., Iwasaki, N., Hansen, T., Pedersen, O., Polonsky, K.
S., and Bell, G. I. (1996) Nature, 384, 455-458.
90.Yamagata, K., Furuta, H., Oda, N., Kaisaki, P.
J., Menzel, S., Cox, N. J., Fajans, S. S., Signorini, S., Stoffel, M.,
and Bell, G. I. (1996) Nature, 384, 458-460.
91.Guazzarotti, L., Bartolotta, E., and Chiarelli,
F. (1999) J. Pediatr. Endocrinol. Metab., 12,
487-497.
92.Pontoglio, M., Sreenan, S., Roe, M., Pugh, W.,
Ostrega, D., Doyen, A., Pick, A. J., Baldwin, A., Velho, G., Froguel,
P., Levisetti, M., Bonner-Weir, S., Bell, G. I., Yaniv, M., and
Polonsky, K. S. (1998) J. Clin. Invest., 101,
2215-2222.
93.Landschulz, W. H., Johnson, P. F., and McKnight,
S. L. (1988) Science, 240, 1759-1764.
94.Zaret, K. S. (1994) in The Liver: Biology and
Pathobiology (Arias, I. M., Boyer, J. L., Fausto, N., Jakoby, W.
B., Schachter, D. A., and Shafritz, D. A., eds.) Raven Press, N. Y.
95.Croniger, C., Trus, M., Lysek-Stupp, K., Cohen,
H., Liu, Y., Darlington, G. J., Poli, V., Hanson, R. W., and Reshef, L.
(1997) J. Biol. Chem., 272, 26306-26312.
96.Thomassin, H., Hamel, D., Bernier, D., Guertin,
M., and Belanger, L. (1992) Nucleic Acids Res., 20,
3091-3098.
97.Costa, R. H., Grayson, D. R., Xanthopoulos, K.
G., and Darnell, J. E., Jr. (1988) Proc. Natl. Acad. Sci. USA,
85, 3840-3844.
98.Legraverend, C., Antonson, P., Flodby, P., and
Xanthopoulos, K. G. (1993) Nucleic Acids Res., 21,
1735-1742.
99.Descombes, P., Chojkier, M., Lichtsteiner, S.,
Falvey, E., and Schibler, U. (1990) Genes Dev., 4,
1541-1551.
100.Welm, A. L., Timchenko, N. A., and Darlington,
G. J. (1999) Mol. Cell. Biol., 19, 1695-1704.
101.Xanthopoulos, K. G., and Mirkovitch, J. (1993)
Eur. J. Biochem., 216, 353-360.
102.Mueller, C. R., Maire, P., and Schibler, U.
(1990) Cell, 61, 279-291.
103.Wuarin, J., Falvey, E., Lavery, D., Talbot, D.,
Schmidt, E., Ossipow, V., Fonjallaz, P., and Schibler, U. (1992) J.
Cell. Sci. Suppl., 16, 123-127.
104.Umek, R. M., Friedman, A. D., and McKnight, S.
L. (1991) Science, 251, 288-292.
105.Samuelsson, L., Stromberg, K., Vikman, K.,
Bjursell, G., and Enerback, S. (1991) EMBO J., 10,
3787-3793.
106.Wang, N. D., Finegold, M. J., Bradley, A., Ou,
C. N., Abdelsayed, S. V., Wilde, M. D., Taylor, L. R., Wilson, D. R.,
and Darlington, G. J. (1995) Science, 269, 1108-1112.
107.Soriano, H. E., Kang, D. C., Finegold, M. J.,
Hicks, M. J., Wang, N. D., Harrison, W., and Darlington, G. J. (1998)
Hepatology, 27, 392-401.
108.Zhang, D. E., Zhang, P., Wang, N. D.,
Hetherington, C. J., Darlington, G. J., and Tenen, D. G. (1997)
Proc. Natl. Acad. Sci. USA, 94, 569-574.
109.Weigel, D., and Jackle, H. (1990) Cell,
63, 455-456.
110.Clark, K. L., Halay, E. D., Lai, E., and
Burley, S. K. (1993) Nature, 364, 412-420.
111.Costa, R. H., Grayson, D. R., and Darnell, J.
E., Jr. (1989) Mol. Cell. Biol., 9, 1415-1425.
112.Lai, E., Prezioso, V. R., Smith, E., Litvin,
O., Costa, R. H., and Darnell, J. E., Jr. (1990) Genes Dev.,
4, 1427-1436.
113.Jackson, D. A., Rowader, K. E., Stevens, K.,
Jiang, C., Milos, P., and Zaret, K. S. (1993) Mol. Cell. Biol.,
13, 2401-2410.
114.Pani, L., Quian, X. B., Clevidence, D., and
Costa, R. H. (1992) Mol. Cell. Biol., 12, 552-562.
115.Ang, S. L., Wierda, A., Wong, D., Stevens, K.
A., Cascio, S., Rossant, J., and Zaret, K. S. (1993)
Development, 119, 1301-1315.
116.Kaestner, K. H., Hiemisch, H., Luckow, B., and
Schutz, G. (1994) Genomics, 20, 377-385.
117.DiPersio, C. M., Jackson, D. A., and Zaret, K.
S. (1991) Mol. Cell. Biol., 11, 4405-4414.
118.Weinstein, D. C., Ruiz i Altaba, A., Chen, W.
S., Hoodless, P., Prezioso, V. R., Jessell, T. M., and Darnell, J. E.,
Jr. (1994) Cell, 78, 575-588.
119.Ang, S. L., and Rossant, J. (1994) Cell,
78, 561-574.
120.Sund, N., Katz, J., Liu, Y., Drucker, D.,
Schutz, G., Ang, S.-L., Magnuson, M., and Kaestner, K. (1999) in
Liver Development, Gene Regulation and Disease, Orvieto, Italy,
p. 123.
121.Kaestner, K. H., Katz, J., Liu, Y., Drucker, D.
J., and Schutz, G. (1999) Genes Dev., 13, 495-504.
122.Kaestner, K. H., Hiemisch, H., and Schutz, G.
(1998) Mol. Cell. Biol., 18, 4245-4251.
123.Sladek, F. M., Zhong, W. M., Lai, E., and
Darnell, J. E., Jr. (1990) Genes Dev., 4, 2353-2365.
124.Holewa, B., Zapp, D., Drewes, T., Senkel, S.,
and Ryffel, G. U. (1997) Mol. Cell. Biol., 17,
687-694.
125.Zhong, W., Sladek, F. M., and Darnell, J. E.,
Jr. (1993) EMBO J., 12, 537-544.
126.Holewa, B., Strandmann, E. P., Zapp, D.,
Lorenz, P., and Ryffel, G. U. (1996) Mech Dev., 54,
45-57.
127.Ktistaki, E., and Talianidis, I. (1997) Mol.
Cell. Biol., 17, 2790-2797.
128.Green, V. J., Kokkotou, E., and Ladias, J. A.
(1998) J. Biol. Chem., 273, 29950-29957.
129.Wang, J. C., Stafford, J. M., and Granner, D.
K. (1998) J. Biol. Chem., 273, 30847-30850.
130.Sladek, F. M., Ruse, M. D., Jr., Nepomuceno,
L., Huang, S. M., and Stallcup, M. R. (1999) Mol. Cell. Biol.,
19, 6509-6522.
131.Duncan, S. A., Manova, K., Chen, W. S.,
Hoodless, P., Weinstein, D. C., Bachvarova, R. F., and Darnell, J. E.,
Jr. (1994) Proc. Natl. Acad. Sci. USA, 91, 7598-7602.
132.Taraviras, S., Monaghan, A. P., Schutz, G., and
Kelsey, G. (1994) Mech Dev., 48, 67-79.
133.Faust, D. M., Boshart, M., Imaizumi-Scherrer,
T., Schutz, G., and Weiss, M. C. (1994) Cell Growth. Differ.,
5, 47-53.
134.Spath, G. F., and Weiss, M. C. (1997) Mol.
Cell. Biol., 17, 1913-1922.
135.Spath, G. F., and Weiss, M. C. (1998) J.
Cell. Biol., 140, 935-946.
136.Chen, W. S., Manova, K., Weinstein, D. C.,
Duncan, S. A., Plump, A. S., Prezioso, V. R., Bachvarova, R. F., and
Darnell, J. E., Jr. (1994) Genes Dev., 8, 2466-2477.
137.Duncan, S. A., Nagy, A., and Chan, W. (1997)
Development, 124, 279-287.
138.Morrisey, E. E., Tang, Z., Sigrist, K., Lu, M.
M., Jiang, F., Ip, H. S., and Parmacek, M. S. (1998) Genes Dev.,
12, 3579-3590.
139.Coucouvanis, E., and Martin, G. R. (1999)
Development, 126, 535-546.
140.Nastos, A., Pogge von Strandmann, E., Weber,
H., and Ryffel, G. U. (1998) Nucleic Acids Res., 26,
5602-5608.
141.Samadani, U., and Costa, R. H. (1996) Mol.
Cell. Biol., 16, 6273-6284.
142.Lemaigre, F. P., Durviaux, S. M., Truong, O.,
Lannoy, V. J., Hsuan, J. J., and Rousseau, G. G. (1996) Proc. Natl.
Acad. Sci. USA, 93, 9460-9464.
143.Landry, C., Clotman, F., Hioki, T., Oda, H.,
Picard, J. J., Lemaigre, F. P., and Rousseau, G. G. (1997) Dev.
Biol., 192, 247-257.
144.Lahuna, O., Fernandez, L., Karlsson, H.,
Maiter, D., Lemaigre, F. P., Rousseau, G. G., Gustafsson, J., and Mode,
A. (1997) Proc. Natl. Acad. Sci. USA, 94,
12309-12313.
145.Tonjes, R. R., Xanthopoulos, K. G., Darnell, J.
E., Jr., and Paul, D. (1992) EMBO J., 11, 127-133.
146.Kritis, A. A., Ktistaki, E., Barda, D., Zannis,
V. I., and Talianidis, I. (1993) Nucleic Acids Res., 21,
5882-5889.
147.Zhong, W., Mirkovitch, J., and Darnell, J. E.,
Jr. (1994) Mol. Cell. Biol., 14, 7276-7284.
148.Duncan, S. A., Navas, M. A., Dufort, D.,
Rossant, J., and Stoffel, M. (1998) Science, 281,
692-695.
149.Feuerman, M. H., Godbout, R., Ingram, R. S.,
and Tilghman, S. M. (1989) Mol. Cell. Biol., 9,
4204-4212.
150.Tronche, F., and Yaniv, M. (1992)
Bioessays, 14, 579-587.
151.Bois-Joyeux, B., and Danan, J. L. (1994)
Biochem. J., 301 (Pt. 1), 49-55.
152.Morinaga, T., Yasuda, H., Hashimoto, T.,
Higashio, K., and Tamaoki, T. (1991) Mol. Cell. Biol.,
11, 6041-6049.
153.Yasuda, H., Mizuno, A., Tamaoki, T., and
Morinaga, T. (1994) Mol. Cell. Biol., 14, 1395-1401.
154.Bernier, D., Thomassin, H., Allard, D.,
Guertin, M., Hamel, D., Blaquiere, M., Beauchemin, M., LaRue, H.,
Estable-Puig, M., and Belanger, L. (1993) Mol. Cell. Biol.,
13, 1619-1633.
155.Galarneau, L., Pare, J. F., Allard, D., Hamel,
D., Levesque, L., Tugwood, J. D., Green, S., and Belanger, L. (1996)
Mol. Cell. Biol., 16, 3853-3865.
156.Buzard, G., and Locker, J. (1990) DNA
Seq., 1, 33-48.
157.Wen, P., and Locker, J. (1994) Mol. Cell.
Biol., 14, 6616-6626.
158.Wen, P., Crawford, N., and Locker, J. (1993)
Nucleic Acids Res., 21, 1911-1918.
159.Apergis, G. A., Crawford, N., Ghosh, D.,
Steppan, C. M., Vorachek, W. R., Wen, P., and Locker, J. (1998) J.
Biol. Chem., 273, 2917-2925.
160.Turcotte, B., Guertin, M., Chevrette, M., and
Belanger, L. (1985) Nucleic Acids Res., 13,
2387-2398.
161.Dong, J. M., Nordloh, P. W., and Chiu, J. F.
(1989) Mol. Cell. Endocrinol., 66, 109-114.
162.Zhang, X. K., Dong, J. M., and Chiu, J. F.
(1991) J. Biol. Chem., 266, 8248-8254.
163.Hogan, B. L., Taylor, A., and Adamson, E.
(1981) Nature, 291, 235-237.
164.Wan, Y. J., and Wu, T. C. (1992)
Differentiation, 50, 107-111.
165.Liu, Y., and Chiu, J. F. (1994) Nucleic
Acids Res., 22, 1079-1086.
166.Varga, E. V., Kustova, I. F., and Lazarevich,
N. L. (1999) Mol. Biol., 33, 273-281.
167.Yamada, Y., Shidoji, Y., Fukutomi, Y.,
Ishikawa, T., Kaneko, T., Nakagama, H., Imawari, M., Moriwaki, H., and
Muto, Y. (1994) Mol. Carcinog., 10, 151-158.
168.Ikeda, H., and Fujiwara, K. (1993) Biochem.
Biophys. Res. Commun., 191, 675-680.
169.Chen, H., Egan, J. O., and Chiu, J. F. (1997)
Crit. Rev. Eukaryot. Gene Exp., 7, 11-41.
170.Saegusa, M., Ido, A., Nakabayashi, H., Sakai,
M., and Tamaoki, T. (1994) J. Tumor Marker Oncol., 9,
29-34.
171.Tournier-Thurneyssen, I., Feldmann, G., and
Bernuau, D. (1993) Tumour Biol., 14, 201-212.
172.Tournier-Thurneyssen, I., Feldmann, G., and
Bernuau, D. (1992) Biol. Cell., 75, 61-68.
173.Chen, J.-Y., Penco, S., Ostrowski, J.,
Balaguer, P., Pons, M., Starrett, J. E., Reczek, P., Chambon, P., and
Gronemeyer, H. (1995) EMBO J., 14, 1187-1197.
174.Lee, K. C., Crowe, A. J., and Barton, M. C.
(1999) Mol. Cell. Biol., 19, 1279-1288.
175.Pachnis, V., Belayew, A., and Tilghman, S. M.
(1984) Proc. Natl. Acad. Sci. USA, 81, 5523-5527.
176.Blankenhorn, E. P., Duncan, R., Huppi, K., and
Potter, M. (1988) Genetics, 119, 687-691.
177.Vogt, T. F., Solter, D., and Tilghman, S. M.
(1987) Science, 236, 301-303.
178.Jin, D. K., and Feuerman, M. H. (1998) Mamm.
Genome, 9, 256-258.
179.Spear, B. T. (1994) Mol. Cell. Biol.,
14, 6497-6505.
180.Jin, D. K., Vacher, J., and Feuerman, M. H.
(1998) Proc. Natl. Acad. Sci. USA, 95, 8767-8772.
181.Eraiser, T. L., Yazova, A. K., Poltoranina, V.
S., Kuprina, N. I., Karamova, E. R., Shipova, L. Y., Lazarevich, N. L.,
and Abelev, G. I. (1998) Int. J. Cancer, 75, 371-378.
182.McVey, J. H., Michaelides, K., Hansen, L. P.,
Ferguson-Smith, M., Tilghman, S., Krumlauf, R., and Tuddenham, E. G.
(1993) Hum. Mol. Genet., 2, 379-384.
183.Gleiberman, A. S., Kudrjavtseva, E. I.,
Sharovskaya, Yu. U., and Abelev, G. I. (1989) Mol. Biol. Med.,
6, 95-107.