Purification and Characterization of a Soluble Polyphosphatase from Mitochondria of Saccharomyces cerevisiae
L. P. Lichko*, T. V. Kulakovskaya, and I. S. Kulaev
Institute of Biochemistry and Physiology of Microorganisms, Russian Academy of Sciences, Pushchino, Moscow Region, 142292 Russia; fax: (7-095) 923-3602; E-mail: alla@ibpm.serpukhov.su* To whom correspondence should be addressed.
Received July 26, 1999
A polyphosphatase with the specific activity 2.2 U/mg was purified to apparent homogeneity from a soluble preparation of mitochondria of Saccharomyces cerevisiae. The polyphosphatase is a monomeric protein of ~41 kD. The purified enzyme hydrolyzes polyphosphates with an average chain length of 9 to 208 phosphate residues to the same extent, but its activity is ~2-fold higher with tripolyphosphate. ATP, PPi, and p-nitrophenyl phosphate are not substrates of this enzyme. The apparent Km values are 300, 18, and 0.25 µM obtained at hydrolysis of polyphosphates with a chain length of 3, 15, and 188 phosphate residues, respectively. Several divalent cations stimulated the enzyme activity 1.2-27-fold (Mg2+ = Co2+ = Mn2+ > Zn2+). Determination of the protein N-terminal sequence and its comparison with the EMBL data library indicates that the soluble polyphosphatase of mitochondria of S. cerevisiae is not encoded by the gene of the major yeast polyphosphatase PPX1.
KEY WORDS: polyphosphatase, mitochondria, purification, molecular and kinetic properties, Saccharomyces cerevisiae
Abbreviations: polyPn) polyphosphates, where n is the number of phosphate residues in the polymer chain; Me2+) divalent metal cations.
Recent studies on the biochemistry of inorganic polyphosphates confirm
that these biopolymers represent not only a phosphate reserve, but also
are important regulatory molecules [1]. One of the
approaches for elucidating the functions of polyphosphates is to
investigate the enzymes involved in their metabolism.
Exopolyphosphatases (polyphosphate phosphohydrolase, EC 3.6.1.11)
splitting Pi from the chain end play an essential role in
the polyphosphate metabolism in yeast. The level of the polyphosphatase
activity in yeast is 10-20-fold higher than in bacteria [2, 3]. Furthermore, each yeast
compartment possesses its own exopolyphosphatase [4]. The yeast has been found to have at least five
types of polyphosphatases localized in the cytosol and cell envelope,
vacuoles, nuclei, and soluble and membrane-bound preparations of
mitochondria [5-9]. These
enzymes differ in a number of kinetic properties including substrate
specificity. The occurrence of these forms is possibly due to different
origin of cell organelles in the process of evolution and different
functions of polyphosphates, which requires the distinctive ways for
regulating the content of biopolymers in each compartment.
In connection with this, studies of mitochondrial polyphosphatases are of special interest. On one hand, these organelles contain their own polyP pool whose functions are associated with bioenergetic processes [10]. On the other hand, a comparison of their enzyme systems with those of bacteria is important from the point of view of evolutionary biochemistry. Besides, these organelles have two polyphosphatases differing much in their forms: a soluble and a membrane-bound one [9].
Here we report on the purification and N-terminal sequencing of a soluble polyphosphatase from mitochondria of S. cerevisiae.
MATERIALS AND METHODS
Cultivation of the yeast S. cerevisiae VKM Y-1173 (IBFM-366) and isolation of spheroplasts and mitochondria were performed as described earlier [9]. A soluble preparation of mitochondria included both the intermembrane space and the matrix.
All steps of enzyme purification were carried out at 4°C. The soluble preparation of mitochondria in 20 mM Tris-HCl, pH 7.2, containing 4 mM MgSO4 and 1 mM EDTA was loaded onto a column (1.6 × 25 cm) with DEAE-Toyopearl 650M (Toson, Japan) equilibrated in 20 mM Tris-HCl, pH 7.2 (buffer A). The column was washed with 100 ml of buffer A, and polyphosphatase was eluted at a flow rate of 30 ml/h with increasing concentrations of KCl (0-0.3 M) in buffer A. The gradient volume was 200 ml. Fractions with polyphosphatase activity were pooled and dialyzed using an Amicon system (Amicon, USA) (PM-10 membrane) against three volumes of buffer A. The resulting preparation (1-1.5 ml) was subjected to fast-flow protein liquid chromatography (FPLC) on an HR 5/5 Mono-Q column equilibrated with buffer A. The sample was eluted at a flow rate of 0.5 ml/min with a salt gradient of 0-0.25 M KCl in buffer A (gradient volume 25 ml). Protein peaks were collected, and aliquots were assayed for polyphosphatase activity.
To determine molecular mass, proteins of the soluble preparation of mitochondria were chromatographed using columns (1.6 × 90 cm) with Sephacryl S-200 or S-300 (Pharmacia, Sweden) equilibrated with 20 mM Tris-HCl, pH 7.2, containing 0.1% Triton X-100, 1 mM dithiothreitol, 0.1 M NaCl, and 4 mM MgSO4 (buffer B), and then eluted with the same buffer at 15 ml/h. The fraction volume was 3 ml. Sepharose 6B was also used for determination of the molecular mass. It was equilibrated and eluted with buffer B containing 0.02 M NaCl instead of 0.1 M NaCl. The following marker proteins were used: ferritin (440 kD), catalase (232 kD), beta-amylase (200 kD), aldolase (158 kD), alcohol dehydrogenase (150 kD), bovine serum albumin (67 kD), ovalbumin (43 kD), and chymotrypsinogen A (25 kD) (Pharmacia, Sweden; Serva, Germany).
To identify the purified polyphosphatase, the N-terminal sequence was analyzed according to the Edman procedure with an Applied Biosystems 477A sequencer (USA). Phenylthiohydantoin amino acids were detected using an on-line analyzer (Applied Biosystems, Model 120A, USA). Prior to sequencing, the protein bands obtained by electrophoresis in 12% polyacrylamide gel with SDS were electroblotted onto a PVDF membrane (Sigma, USA) using 50 mM sodium-borate buffer, pH 8.0, with 20% methanol and 0.2% mercaptoethanol. After staining the membrane with 0.1% Coomassie Brilliant Blue R-250 in methanol-water-acetic acid (40:59:1 v/v) for 1 min, destaining in methanol-water (50:50 v/v), and drying, the bands of interest were cut off and subjected to the reaction cartridge of the sequencer.
All the other methods, including enzyme assays and SDS-PAGE were described in our recent paper [9].
RESULTS AND DISCUSSION
Polyphosphatase purification. The soluble preparation of mitochondria (~15 ml) including both the intermembrane space and matrix was applied onto a column with DEAE-Toyopearl 650M and chromatographed as indicated in “Materials and Methods”. The results of a standard chromatographic resolution of proteins are depicted in Fig. 1. About 70-85% of pyrophosphatase and ~5% of poly- and tripolyphosphatase activities were not bound to the carrier. Further separation of the pyrophosphatase activity from poly- and tripolyphosphatase activities was made with a KCl gradient (Fig. 1).
The fractions with polyphosphatase activity were pooled, desalted, and subjected to FPLC on Mono-Q. Almost all polyphosphatase activity was found in one protein peak after FPLC (Fig. 2). The resulting enzyme preparation had a specific activity of 2.2 U/mg protein and 12% yield relative to the soluble preparation of mitochondria (Table 1). We did not evaluate the purification degree with relation to cell homogenate because the latter contained large amounts of other polyphosphatases [4]. The enzyme rapidly lost its activity during chromatographic steps but was rather stable when dialyzed on a PM-10 membrane against 20 mM Tris-HCl, pH 7.2. Its activity decreased by only 5-20% in this case. Addition of 1 mM dithiothreitol to the buffer caused an almost 3-fold decrease of enzyme activity. The recovery of enzyme activity during chromatography improved significantly when KCl was used instead NaCl.Fig. 1. Chromatography of a soluble preparation of mitochondria on DEAE-Toyopearl 650M: 1) protein; 2) pyrophosphatase; 3) tripolyphosphatase; 4) polyphosphatase. Elution with a KCl gradient.
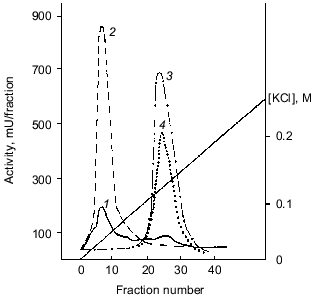
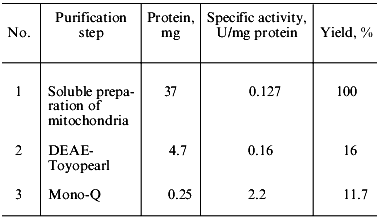
Table 1. Purification of soluble polyphosphatase from mitochondria of S. cerevisiaeFig. 2. FPLC of pooled polyphosphatase fractions from DEAE-Toyopearl (Fig. 1) on Mono-Q. 1-11) Protein peaks at 280 nm. Numbers above the protein peaks represent the total activities (mU/fraction).
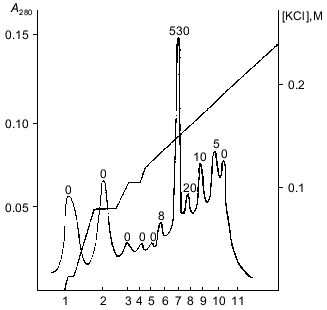
The specific activity of the polyphosphatase purified from yeast mitochondria was close to that of purified polyphosphatases from prokaryotes, 2 U/mg protein for Acinetobacter johnsonii [2] and 22 U/mg protein for Escherichia coli [3], and considerably lower than of other purified polyphosphatases from S. cerevisiae. Thus, polyphosphatases purified from cell homogenates of S. cerevisiae had specific activities 202 [11] and 890 U/mg of protein [12] at 37°C. The specific activities of purified polyphosphatases from different cell compartments of the S. cerevisiae strain used in this work were as follows: 220, 420, and 60 U/mg of protein for the cell envelope, cytosol, and vacuoles, respectively [5-7].
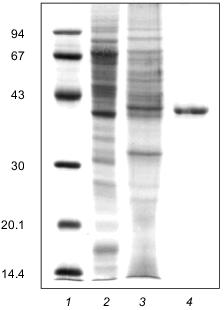
Molecular mass. For estimating the molecular mass of native enzyme, three types of resins were used: Sephacryl S-200 and S-300 and Sepharose 6B. Polyphosphatase was eluted from all three columns at a volume corresponding to a molecular mass of 42 ±_6 kD. Electrophoresis of the polyphosphatase preparation in 12% SDS-polyacrylamide gel showed one main band with a molecular mass of 41 ±_2 kD (Fig. 3). It is noteworthy that in the course of purification the amount of the polypeptide of .41 kD was enriched (Fig. 3). Electrophoresis of the protein fractions after FPLC in 12% SDS-polyacrylamide gel revealed a simultaneous enrichment of this protein and increase of the polyphosphatase activity (Fig. 4). The results of gel filtration and electrophoresis under denaturing conditions indicate that native enzyme is a monomer.
Fig. 3. Electrophoresis of the polyphosphatase preparation in 12% SDS-polyacrylamide gel at different stages of purification: 1) protein standards (phosphorylase b, BSA, ovalbumin, carbonic anhydrase, trypsin inhibitor, lactalbumin) (molecular masses in kD are shown on the left); 2) soluble preparation of mitochondria (30 µg protein); 3) DEAE-Toyopearl (25 µg protein); 4) Mono-Q (7 µg protein). Proteins after electrophoretic resolution were stained with Coomassie Brilliant Blue.
By their molecular mass (.40 kD), yeast polyphosphatases from cell homogenate of S. cerevisiae [12] and the cell envelope and cytosol [5, 6] are close to the tested mitochondrial polyphosphatase. Some other monomeric polyphosphatases from eukaryotic cells are known as well. The molecular mass of polyphosphatases from cell homogenate of Endomyces magnusii [13] and Neurospora crassa [14] were estimated as 48 and 50 kD, respectively. Two polyphosphatases of E. coli were found to be dimers with a molecular mass of native enzyme of 100 and 120 kD [3, 15]. The molecular mass of native polyphosphatase from A. johnsonii was 55 kD and SDS-PAGE revealed only one subunit of .43 kD [2].Fig. 4. Electrophoresis of protein peaks after FPLC (Fig. 2) in 12% SDS-polyacrylamide gel. Protein fractions (100 µl) were precipitated with deoxycholate and TCA. Pellets were resuspended in 80 µl of the sample buffer and incubated 5 min at 100°C. Proteins after electrophoretic resolution were stained with Coomassie Brilliant Blue. Numbers above the lanes represent the protein peaks after FPLC; total activities of polyphosphatase (mU/fraction) are indicated under the lanes. MP, marker proteins (phosphorylase b, BSA, ovalbumin, carbonic anhydrase, trypsin inhibitor, lactalbumin) (molecular masses in kD are shown on the right).

Substrate specificity. The purified soluble polyphosphatase of mitochondria hydrolyzed polyP with different chain length, including tripolyphosphate (Table 2). Polyphosphatase activity was nearly the same when polyP with average chain length of 9 to 208 phosphate residues were used, but it was by 1.9 times higher with tripolyphosphate than with polyP15 (Table 2). The mitochondrial polyphosphatase was inactive with ATP, PPi, and p-nitrophenyl phosphate.
Table 2. Influence of chain length on polyP
hydrolysis by soluble polyphosphatase from mitochondria of S.
cerevisiae
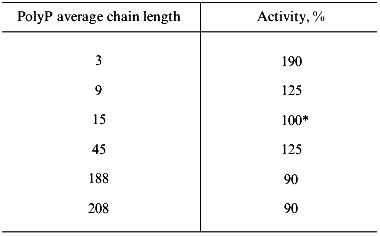
*100% corresponds to a specific activity of 2 U/mg
protein.
The apparent Km values in the presence of 2.5 mM Mg2+ were 300 µM for polyP3 and 18 and 0.25 µM for polyP15 and polyP188, respectively. While substrate specificity of the purified enzyme and soluble preparation of mitochondria was similar [9], the affinity of purified enzyme to polyP somewhat decreased. Earlier we reported that the soluble preparation of mitochondria had the following apparent Km values: 150, 4, and 0.06 µM for polyP with an average chain length of 3, 15, and 188 phosphate residues, respectively [9]. This may be is due to removal of some components required for the display of polyphosphatase activity during purification. Notwithstanding this, the purified soluble polyphosphatase of mitochondria has the highest affinity to polyP among the reported yeast polyphosphatases.
pH optimum. The purified soluble polyphosphatase of mitochondria was most active at neutral pH, which coincided with a pH-dependence profile of crude enzyme [9]. The mitochondrial polyphosphatase was similar in its pH optimum to polyphosphatases from other compartments of the same yeast strain [5-8] as well as to most known microbial polyphosphatases [14, 16, 17].
Effect of some Me2+. The soluble polyphosphatase of mitochondria exhibited low activity in the absence of divalent cations. Addition of Me2+ to the enzyme preparation resulted in considerable stimulation of the activity, which depended on the nature and concentration of Me2+ (Table 3). Based on a degree of stimulation of the polyphosphatase activity of the purified enzyme, tested Me2+ were arranged in the following order: Mg2+ = Mn2+ = Co2+ > Zn2+, which was fairly similar to the data obtained for crude mitochondrial polyphosphatase [9]. The activating effect of Me2+ on the purified enzyme was .6-fold higher than in the case of crude polyphosphatase, which indicates possible loss of divalent cations presumably incorporated into the protein structure during purification.
Table 3. Influence of divalent metal cations
on the soluble polyphosphatase activity of mitochondria from S.
cerevisiae
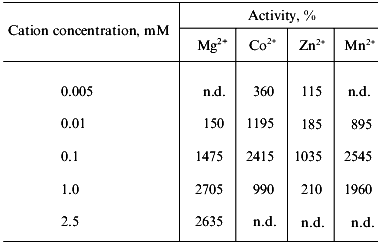
Note: Activity in the absence of divalent metals taken as 100% was 0.13
U/mg protein; n.d., not determined.
By the effect of Me2+, most similarity was found between the soluble polyphosphatase of mitochondria, the cytosol polyphosphatase of the same strain of S. cerevisiae [6], and purified polyphosphatase from cell homogenate of S. cerevisiae [12]. In the last case, however, the enzyme stimulation was lower than in the case of the polyphosphatase under study.
Effect of some specific reagents. The purified mitochondrial polyphosphatase was almost insensitive to 10 mM sodium azide, 1 mM ammonium molybdate, and 1 mM sodium orthovanadate. Sodium fluoride, a known inhibitor of pyrophosphatases, at a concentration of 1 mM, decreased the tripolyphosphatase activity of the enzyme by only 10% and had no effect on the polyphosphatase activity (Table 4).
Table 4. Influence of some reagents on the
poly- and tripolyphosphatase activities of a soluble preparation of
exopolyphosphatase purified from mitochondria of S.
cerevisiae
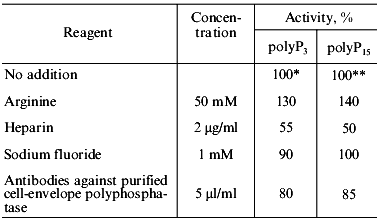
*,**Specific activities taken as 100% were 3.4
and 2.1 U/mg protein, correspondingly.
Heparin, an effective inhibitor of polyphosphatases from all yeast cell compartments studied earlier, suppressed the polyphosphatase activity of the purified enzyme by 50% (Table 4) compared with complete inhibition in the isolated yeast mitochondria [8]. There was some stimulation of purified enzyme by 50 mM arginine: 30% and 40% in the case of the tripolyphosphatase and polyphosphatase activity, respectively (Table 4).
Antibodies against the purified cell-envelope polyphosphatase of S. cerevisiae decreased the activity of the purified mitochondrial polyphosphatase to a lesser extent (15%, Table 4) compared with crude enzyme (30%) [9]. The above antibodies inhibited polyphosphatases of the cell envelope and cytosol more effectively (by 80-90%). Thus, despite of the similar molecular mass, the soluble polyphosphatase of mitochondria differs in its immune properties from that of the cell envelope and cytosol.
N-Terminal sequence. The amino-terminal sequence of a major protein with a molecular mass of .41 kD found in the preparation of polyphosphatase purified from the soluble preparation of mitochondria of S. cerevisiae was as follows: Met-Leu-Ala-Glu-Lys-Thr-Arg-Ser-Ile-Ile. The only yeast gene encoding this sequence, YHB1, was extracted from the EMBL data library [18, 19]. This gene corresponds to a protein of 399 amino acids and a molecular mass of 44,647 daltons, which belongs to a large family of yeast flavohemoproteins [18-20]. According to the EMBL data library, none of the other members of this family has the sequence indicated. The function of the protein encoded by the YHB1 gene remains unknown. But it is known from study of the corresponding mutants that they are associated with yeast cell defense reactions against various stresses [19].
An important role of polyphosphatases in cell survival under stresses was demonstrated for bacteria [15]. One of polyphosphatases of E. coli hydrolyzes guanosine pentaphosphate to guanosine tetraphosphate [15]. Guanosine tetraphosphate, the so-called stringent factor, is a regulator of ribosomal functioning and biogenesis in response to amino acid starvation in bacteria. Although the polyphosphatase in question differs greatly from the tested mitochondrial polyphosphatase in molecular mass and some other properties, a detailed analysis of the corresponding sequences may be useful.
To date, the primary structure of polyphosphatases has been demonstrated only for S. cerevisiae and E. coli in Kornberg's laboratory. The amino acid sequence of polyphosphatase purified from the cell homogenate of S. cerevisiae was determined by sequencing a PPX1 gene isolated from the yeast gene library and encoding a protein with a molecular mass of .43 kD [21]. Two genes, ppx and gppA, encoding the subunits of two different exopolyphosphatases with a molecular mass of 58 and 50 kD, respectively, were detected in E. coli [3, 15]. The second polyphosphatase was purified from the ppx mutant and identified as guanosine pentaphosphate phosphohydrolase (EC 3.6.1.40) [15]. When the amino acid sequence of the mitochondrial polyphosphatase was compared with those recognized for S. cerevisiae and E. coli [3, 15, 21] with the help of a program described earlier [22], there was apparent lack of significant similarity.
Comparison of the primary structure of polyphosphatases of E. coli with that of the tested polyphosphatase from yeast mitochondria is especially interesting in the context of evolutionary origin of mitochondria from ancient bacteria. Lack of similarity between the primary structure of the soluble polyphosphatase from yeast mitochondria and that of the polyphosphatase from E. coli encoded by the ppx gene is supported by differences in the main properties of these enzymes: structure (the yeast mitochondrial enzyme is a monomer, while the enzyme of E. coli is a dimer), substrate specificity (the mitochondrial enzyme hydrolyzes polyP3, while the enzyme of E. coli fails to do so), and relationship to K+ (the bacterial polyphosphatase is active only in the presence of K+ in contrast to the mitochondrial enzyme of S. cerevisiae). We suggest that a polyphosphatase of bacterial type has not been conserved in yeast mitochondria in the evolutionary process.
The program described in [22] did not allow us to detect any similar sites in the PPX1, ppx, and gppA genes, which has already been intimated earlier [21]. Thus, the regions responsible for substrate binding in the molecules of these enzymes, which could possess a similar primary structure, have not been found yet. A more detailed analysis of the above sequences will permit probably reveal the corresponding regions. However, the mitochondrial polyphosphatase and polyphosphatases encoded by the respective genes differ in substrate specificity and, possibly, the mechanism of action. Therefore, the structure of their active centers might have some special features.
The data show that the soluble polyphosphatase of mitochondria is not encoded by the PPX1 gene and confirms our view that exopolyphosphatases of different yeast compartments that vary in their properties are the products of different genes [4]. Among yeast polyphosphatases only enzymes of the cell envelope and cytosol [6, 7] are obviously the products of the PPX1 gene, since their main properties coincide with those of a polyphosphatase purified from cell homogenate of S. cerevisiae that is encoded by this gene [12, 21].
Identification of the genes encoding other polyphosphatases of the yeast cell is a necessary stage for studying the functions of these enzymes. We showed earlier that yeast mitochondria possess both a soluble polyphosphatase and a membrane-bound one, which is unique in its properties [9] and most likely is encoded by a different gene. The aim of our future work will be purification and sequencing of the membrane-bound polyphosphatase from yeast mitochondria.
The authors are grateful to N. Andreeva, T. Ivashina, A. Karpov, T. Muranova, V. Petrov, C. Slayman, A. Tsiomenko, and T. Zhuravleva, all of whom contributed to our investigation.
This work was supported by grants of the Russian Foundation for Basic Research Nos. 96-15-98024 and 99-04-48246 and the INCO-COPERNICUS-PL971185 grant.
REFERENCES
1. Kulaev, I. S. (1979) Biochemistry of Inorganic
Polyphosphates, J. Wiley & Sons, Chichester-New York.
2. Bonting, C. F. C., Kortstee, G. J. J., and
Zehnder, J. B. (1993) Antonie Leeuwenhoek, J. Microbiol.
Serol.,64, 75-81.
3. Akiyama, M., Crooke, E., and Kornberg, A. (1993)
J. Biol. Chem.,268, 633-639.
4. Kulaev, I. S., Kulakovskaya, T. V., Andreeva, N.
A., and Lichko, L. P. (1997) J. Evol. Biokhim. Fiziol. (St.
Petersburg), 33, 74-82 (Russ.).
5. Andreeva, N. A., and Okorokov, L. A. (1993)
Yeast, 9, 127-139.
6. Andreeva, N. A., Kulakovskaya, T. V., and Kulaev,
I. S. (1996) Biochemistry (Moscow), 61, 1213-1220.
7. Andreeva, N. A., Kulakovskaya, T. V., and Kulaev,
I. S. (1998) FEBS Lett., 429, 194-196.
8. Lichko, L. P., Kulakovskaya, T. V., and Kulaev, I.
S. (1996) Biochemistry (Moscow), 61, 499-506 (Russ.).
9. Lichko, L. P., Kulakovskaya, T. V., and Kulaev, I.
S. (1997) Biochemistry (Moscow), 62, 1146-1151.
10. Beauvoit, B., Rigoulet, M., Guerin, B., and
Canioni, P. (1989) FEBS Lett., 252, 17-22.
11. Lorenz, B., Muller, W. F. G., Kulaev, I. S., and
Schröder, H. C. (1994) J. Biol. Chem., 269,
22198-22204.
12. Wurst, H., and Kornberg, A. (1994) J. Biol.
Chem., 269, 10996-11001.
13. Afanasieva, T. P., and Kulaev, I. S. (1973)
Biochim. Biophys. Acta, 321, 336-347.
14. Umnov, A. M., Umnova, N. S., and Kulaev, I. S.
(1975) Mol. Biol. (Moscow), 9, 594-601.
15. Keasling, J. D., Bertsch, L. R., and Kornberg,
A. (1993) Proc. Natl. Acad. Sci. USA, 90, 7029-7033.
16. Muhammed, A., Rodgers, A., and Hughes, D. E.
(1959) J. Gen. Microbiol.,20, 482-495.
17. Felter, S., Dirheimer, G., and Ebel, J. P.
(1970) Bull. Soc. Chim. Biol.,52, 433-446.
18. Van der Aart, Q. J. M., Kleine, K., and
Steensma, H. Y. (1996) Yeast, 12, 385-390.
19. Buisson, N., and Labbe-Bois, R. (1998) J.
Biol. Chem.,273,9527-9533.
20. Zhu, H., and Riggs, A. F. (1992) Proc. Natl.
Acad. Sci. USA, 89, 5015-5019.
21. Wurst, H., Shiba, T., and Kornberg, A. (1995)
J. Bacteriol., 177, 898-906.
22. Thompson, J. D., Higgins, D. G., and Gibson, T.
J. (1994) Nucleic Acids Res.,22, 4673-4680.