Mechanism of Ca2+-Induced Inhibition of Escherichia coli Inorganic Pyrophosphatase
S. M. Avaeva1*, N. N. Vorobyeva2, S. A. Kurilova1, T. I. Nazarova1, K. M. Polyakov3, E. V. Rodina1, and V. R. Samygina3
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-3181; E-mail: avaeva@libro.genebee.msu.su2School of Chemistry, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 932-8846; E-mail: protein@libro.genebee.msu.su
3Shubnikov Institute of Crystallography, Leninskii pr. 59, Moscow, 117333 Russia; fax: (095) 135-1011; E-mail: kostya@ns.crys.ras.ru
* To whom correspondence should be addressed.
Received October 11, 1999
The causes of inhibition of Escherichia coli inorganic pyrophosphatase (PPase) by Ca2+ were investigated. The interactions of several mutant pyrophosphatases with Ca2+ in the absence of substrate were analyzed by equilibrium dialysis. The kinetics of Ca2+ inhibition of hydrolysis of the substrates MgPPi and LaPPi by the native PPase and three mutant enzymes (Asp-42-Asn, Ala, and Glu) were studied. X-Ray data on E. coli PPase complexed with Ca2+ or CaPPi solved at atomic resolution were analyzed. It was shown that, in the course of the catalytic reaction, Ca2+ replaces Mg2+ at the M2 site, which shows higher affinity for Ca2+ than for Mg2+. Different properties of these cations account for active site deformation. Our findings indicate that the filling of the M2 site with Ca2+ is sufficient for PPase inhibition. This fact proves that Ca2+ is incapable of properly activating the H2O molecule for nucleophilic attack on PPi. It was also demonstrated that Ca2+, as a constituent of the non-hydrolyzable substrate analog CaPPi, competes with MgPPi at the M3 binding site. As a result, Ca2+ is a powerful inhibitor of all known PPases. Other possible reasons for the inhibitory effect of Ca2+ on the enzyme activity are also considered.
KEY WORDS: inorganic pyrophosphatase, equilibrium dialysis, Ca2+, inhibition, complex, spatial structure
Soluble inorganic pyrophosphatases catalyze reversible hydrolysis of pyrophosphate (PPi) to two orthophosphate molecules (Pi) and are the key factors controlling PPi intracellular level. Pyrophosphate is a product of more than 120 enzymatic reactions. Catalyzing pyrophosphate hydrolysis, pyrophosphatases ensure the synthesis of all vital cellular biopolymers.
Similar to many enzymes involved in phosphate metabolism, PPases are metal-dependent enzymes. Three or four bivalent metal ions are necessary for their catalytic activity; among them, Mg2+ is the most powerful effector.
The development of X-ray analysis and PPase synthesis by genetic engineering provided the basis for recent progress in determination of the three-dimensional structures of PPases from Escherichia coli, Saccharomyces cerevisiae, Thermus thermophilus, and Sulfolobus acidocaldarius. The crystal structures of the apoenzymes and their complexes, including those with activator metal ions, are presently known [1-5]. These data show that, despite considerable differences in amino acid sequences and subunit structure, PPases from various organisms share similarity in the active site structure and probably in the mechanism of catalysis.
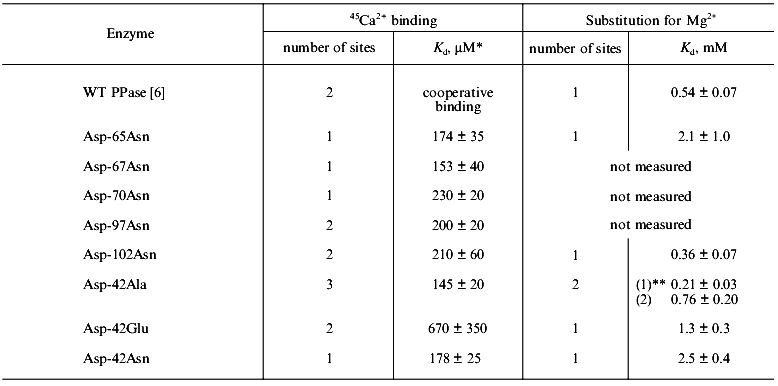
Structure determination of E. coli PPase complexes with Mn2+ [1] and Mg2+ [2, 4] and of S. cerevisiae PPase complexes with Mn2+ and Pi [3, 5] made it possible to locate the sites responsible for binding of activator metal ions. Figure 1 shows the position of four Mn2+ ions at the active site of the PPase subunit [3]. Two sites, M1 and M2, are filled in the absence of substrate; Mg2+ and Mn2+ bind to these sites similarly [6]. The affinities of M1 and M2 for activator metal ions differ more than tenfold. Thus, for E. coli PPase at pH 7.5, Kd (Mg2+) is 50 µM at site M1 and 1.4 mM at site M2 [7]. Two other sites, M3 and M4 (Fig. 1), are occupied with metal cations in enzyme complexes with the substrate or products of the catalytic reaction.
In addition to Mg2+ and Mn2+, only Zn2+ and Co2+ can stimulate pyrophosphate hydrolysis catalyzed by PPases [8]. Ca2+, conversely, is an efficient inhibitor of magnesium pyrophosphate hydrolysis. The enzyme interactions with Ca2+, and inhibition kinetics are best studied for S. cerevisiae (baker's yeast) PPase [8-11]. In this instance, inhibition by Ca2+ was shown to be due to competition with Mg2+ for binding to two binding sites on the enzyme and to a third site on the substrate [10, 12]. In 1983, the structure of yeast PPase complexed with calcium pyrophosphate was reported [13]; however, low resolution (5 Å) allowed the authors to draw only preliminary conclusions concerning Ca2+ and CaPPi binding at the active site.
For E. coli PPase, it was demonstrated that Ca2+-induced inhibition is also connected with the replacement of Mg2+ from the substrate and from at least one site on the enzyme [14]. Similar results were obtained for all other PPases [15-19]. This suggests that the causes and mode of Ca2+ inhibition are common to all PPases. A strong inhibitory effect of Ca2+ on PPases may contribute to activity-regulation mechanisms of these constitutive enzymes in vivo.
Numerous recent studies have shown that Ca2+ is the most common regulator used by living cells from the earliest evolutionary stages [20-22]. The unique properties of Ca2+ account for this phenomenon: it is widely spread in nature and capable of rapid and tight binding to proteins; in addition, cells possess a specific system providing quick removal of excess Ca2+. In eucaryotes, Ca2+ is involved in regulation of dozens of processes, such as mitosis, muscle contraction, proliferation and differentiation in different types of cells, hormone secretion, etc; it also plays an integrating role controlling coordinated function of different compartments, cells, and organs [22]. Although the utilization of Ca2+ in procaryotic cells is not so diverse, it has been shown that Ca2+ participates in regulation of such processes as metabolic shift toward the use of a variant food substrate, directional cell movement, etc. [23]. The intracellular Ca2+ concentration is normally 10-7-10-6 M [19, 20]. Elevated Ca2+ concentrations (up to 10-5 M) serve as a signal to the cell. At such concentrations, Ca2+ produces a strong effect on PPases and other enzymes of phosphate metabolism (kinases, ATPases, and phosphatases [21]). In all probability, this effect forms the basis for the regulatory role of Ca2+ in procaryotic cells.
In this work, we investigated the interaction of Ca2+ with E. coli PPase and some of its mutants both in the absence of substrate and during the catalytic reaction. Such an approach is helpful for the comparison of Ca2+ binding sites with those responsible for binding activator metal ions, Mg2+ and Mn2+, already located in the PPase three-dimensional structures (Fig. 1).
In collaboration with the staff of the Institute of Crystallography of the Russian Academy of Sciences, we examined the structure of two E. coli PPase complexes with Ca2+ and calcium pyrophosphate. Their recently solved three-dimensional structures will soon be published. X-Ray data proved useful for understanding the results and, therefore, are used in the discussion.
MATERIALS AND METHODS
The recombinant E. coli inorganic pyrophosphatase and mutant enzymes were obtained as described previously [24]. Enzyme suspensions were stored in ammonium sulfate (90% saturation) and desalted on a Sephadex G-50 (fine) column equilibrated with 50 mM Tris-HCl, pH 7.5, before use.
The concentration of pyrophosphatase solutions was determined spectrophotometrically using the absorption value A280 nm0.1% of 1.18 [25].
Equilibrium dialysis of inorganic pyrophosphatase and mutants with 45CaCl2, and equilibrium dialysis to substitute Ca2+ for Mg2+ were conducted as described [6].
In this work, we used carrier-free 45CaCl2 (Amersham, England) with radioactivity of 0.5-1.0 mCi/ml; Tris, Hepes, Na4P2O7, and CaCl2 were from Sigma (USA); MgCl2, LaCl3, and methyl green were from Fluka (Switzerland). In all experiments, titrated MgCl2 and CaCl2 solutions were used; all solutions were prepared using twice-distilled water.
Kinetic measurements. All the measurements were performed in 0.1 M Tris-HCl buffer, pH 7.5. To study MgPPi hydrolysis, Mg2+ and Ca2+ concentrations were varied from 0.05 to 2 mM and from 5 to 1500 µM, respectively; MgPPi concentration was changed from 10 to 120 µM. In the investigations of LaPPi hydrolysis, Mg2+ concentration was 0.6 mM, Ca2+ concentration was varied from 5 to 250 µM, and LaPPi concentration was changed from 20 to 200 µM. LaPPi was synthesized just before hydrolysis by mixing equimolar amounts of La3+ and PPi; this excluded CaPPi formation in the reaction mixture. The reaction was initiated by simultaneous addition of Mg2+ and the enzyme.
To calculate concentrations, the following Kd values obtained by recalculation of pH-independent values for pH 7.5 were used: 1.29 µM, 2.42 mM, and 126 µM for MgPPi, Mg2PPi, and CaPPi, respectively [26]. The concentration of Ca2PPi complex was ignored.
Estimation of inhibition parameters. MgPPi hydrolysis. The following scheme for Ca2+-induced inhibition of MgPPi hydrolysis was suggested:
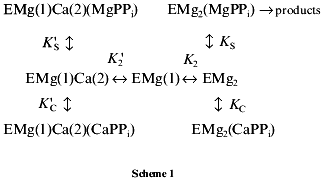
This scheme assumes that MgPPi and CaPPi can bind both to EMg2 and EMgCa. The location of metal cations on the enzyme molecule is shown by figures in parentheses. Thus, EMg(1)Ca(2) is an enzyme subunit with Mg2+ at the M1 site and Ca2+ at the M2 site.
In Scheme 1, the relationship between 1/v and [CaPPi] (Dixon plot) is as follows:
Coefficients of Eq. (1), a, b, and c, are determined from the following equations:
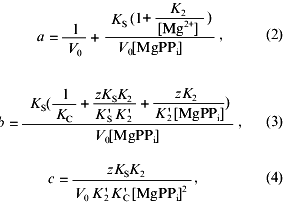
where V0 is the maximum rate of MgPPi hydrolysis in the absence of inhibitor at saturating substrate concentration; K2, K´2, KS, K´S, KC, and K´C are the constants of the equilibria shown on the Scheme 1; z is the ratio of the dissociation constants of pyrophosphates of calcium and magnesium:
The ratio of the concentrations of Ca2+ and CaPPi at the fixed concentrations of [Mg2+] and [MgPPi] is determined by the equation:
Nonlinear regression analysis of the data using Eqs. (1)-(5) allowed us to calculate the parameters V0, K´2/K2, KS, K´S, and K´C. Using K2 values from 0.5 to 2 mM, K´2 was estimated. The KC and K´S parameters were determined interdependently. The calculations show that, in a wide range of K´S values, KC varies not more than 3-5-fold and reach saturation. In Table 2, the limit KC and corresponding K´S values for the native PPase and Asp-42-Asn variant are given. For Asp-42-Glu and Asp-42-Ala PPases, only the lower K´C limit could be determined.
LaPPi hydrolysis. Scheme 2 was suggested to describe inhibition of LaPPi hydrolysis by Ca2+:
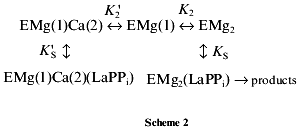
According to this scheme, Ca2+ inhibits LaPPi hydrolysis by competition with Mg2+ for the M2 binding site. In the absence of Mg2+, PPase does not hydrolyze LaPPi, i.e., the form ECa2(LaPPi) is inactive.
It follows from Scheme 2 that under experimental conditions, the dependence of 1/v on Ca2+ concentration is linear:
The coefficients of Eq. (6) are determined from the following equations:
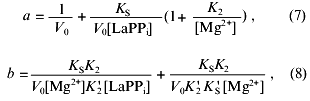
where V0 is the maximal rate of the LaPPi hydrolysis in the absence of the inhibitor at the saturating substrate concentration; KS, K´S, K2, and K´2 are the constants of the equilibria shown on Scheme 2.
Under experimental conditions, the straight lines (1/v, [Ca2+]) at different [LaPPi] are virtually parallel (inhibition is formally assigned to the mixed type), so that:
RESULTS
Ca2+ binding in the absence of substrate. In our previous work [6], using equilibrium dialysis we showed that E. coli PPase subunit binds two Ca2+ cations in the absence of substrate. The form of the curve in Scatchard plot indicates cooperativity of binding (probably between analogous binding sites in different hexamer subunits). One of two Ca2+ cations associated with PPase subunit may be replaced by Mg2+. Kd for Mg2+ at this site is 0.54 ± 0.07 mM (Table 1); this corresponds to the low-affinity Mg2+-binding site (M2 in Fig. 1).
Table 1. Equilibrium dialysis of the native
(WT) and mutant PPases with 45Ca2+ and
Mg2+
*Kd (Ca2+) values for the
high-affinity site are presented, since only this site is saturated
under experimental conditions. Kd (Ca2+)
for other sites, if any, is estimated with large relative errors and is
not shown.
**Dissociation constants for the two sites are given.
Earlier, using fluorometric titration, it was shown [8] that a high-affinity Mg2+-binding site (of two sites filled in the absence of substrate) coincides with a low-affinity Ca2+-binding site and vice versa.Fig. 1. Presumed transition state at the active site of yeast (S. cerevisiae) PPase subunit complexed with Mn2+ and Pi [3]. Designations: Ob, bridge-type oxygen atom from pyrophosphate; Ow, attacking water molecule. The positions of corresponding residues in E. coli PPase are given in parentheses. Metal ions are shown by black circles. Metal-binding sites are numbered from M1 to M4; the phosphate-binding sites are denoted by PA and PL.
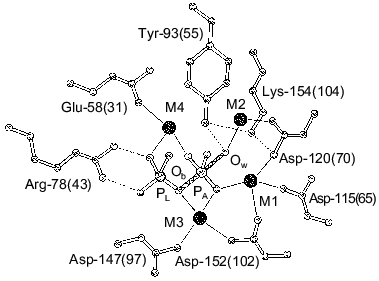
A similar approach is inapplicable to E. coli PPase. To elucidate whether this inversion also takes place in E. coli PPase, we analyzed the mutant PPases with substitutions of active site groups responsible for Mg2+ binding. Equilibrium dialysis with 45Ca2+ was conducted with the mutant variants Asp-65-Asn, Asp-67-Asn, Asp-70-Asn, Asp-97-Asn, Asp-102-Asn, Asp-42-Asn, Asp-42-Glu, and Asp-42-Ala, and for some of them, equilibrium dialysis to substitute 45Ca2+ by Mg2+ was performed. The results are shown in Fig. 2 and summarized in Table 1.
The data demonstrate that, of all mutant PPases, the variants Asp-97-Asn, Asp-102-Asn, and Asp-42-Glu retain the ability of the native enzyme (WT) to bind two Ca2+ cations per subunit. Under experimental conditions, Asp-65-Asn, Asp-67-Asn, Asp-70-Asn, and Asp-42-Asn PPases bind only one Ca2+, while Asp-42-Ala PPase binds three Ca2+ ions.Fig. 2. Equilibrium dialysis of mutant PPases Asp-65-Asn (a) and Asp-102-Asn (b) against 45Ca2+ (double-reciprocal plot). The results of equilibrium dialysis of these enzymes to substitute 45Ca2+ by Mg2+ are given in insets.
Analysis of Ca2+ substitution for Mg2+ shows that all PPases (Asp-65-Asn, Asp-102-Asn, Asp-42-Asn, Asp-42-Glu, and Asp-42-Ala) are similar to WT PPase in their ability to replace one Ca2+ by Mg2+. The dissociation constant for Mg2+ at this site varies from 0.36 mM for Asp-102-Asn PPase to 2.47 mM for Asp-42-Asn PPase; this corresponds to the low-affinity binding site (M2 in Fig. 1).
The data are consistent with the hypothesis that two Ca2+-binding sites with different affinity for Ca2+ are present at the active site of the PPase subunit. It is difficult to estimate Ca2+-binding parameters in WT PPase due to cooperative interactions [6]. In mutant PPases, these sites are filled independently; this makes it possible to determine the dissociation constant for the high-affinity site, which varies from 0.17 mM for Asp-65-Asn PPase to 0.67 mM for the Asp-42-Glu mutant enzyme (Table 1). The dissociation constant for the second Ca2+-binding site cannot be estimated, since under experimental conditions (0.6 mM Ca2+), it is not yet filled.
The mutant Asp-42-Ala PPase has a third Ca2+-binding site, in which the metal may be substituted by Mg2+ with Kd = 212 µM.
Inhibition kinetics of MgPPi hydrolysis by Ca2+. As it follows from equilibrium dialysis [6], spectrophotometric titration, and E. coli PPase protection from proteolysis [7], Ca2+ has at least two binding sites on the enzyme. These sites differ in affinity for Ca2+: Kd(1) measured by different procedures varies from 19 to 68 µM, and Kd(2) is 2.5 mM at pH 7.5 [7]. It was suggested that Ca2+ binding to one of these sites is involved in inhibition of MgPPi hydrolysis; one more calcium ion competes with Mg2+ for substrate binding [14]. It still remains unclear the filling of which sites leads to inhibition of MgPPi hydrolysis.
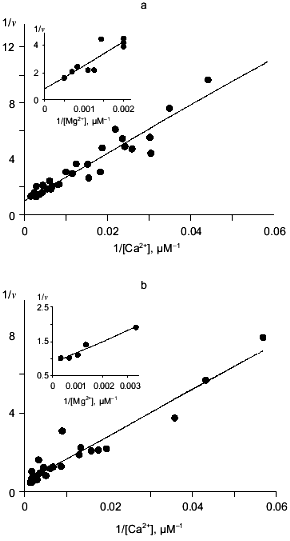
In this work, we explored Ca2+ inhibition kinetics of MgPPi hydrolysis for WT PPase in relation to Mg2+, Ca2+, and MgPPi concentrations. In Fig. 3a, the relationship between hydrolysis rate and free Mg2+ concentration at several constant Ca2+ concentrations is shown. The linear dependences in double-reciprocal plots show that, in a given range of Mg2+ and Ca2+ concentrations, Ca2+ competes with Mg2+ at only one site. The dissociation constant Kd(Mg2+) 0.53 mM calculated at 20 µM Ca2+ concentration is in good agreement with the value obtained by equilibrium dialysis to substitute Ca2+ for Mg2+ (0.54 mM, see Table 1). This value corresponds to the low-affinity Mg2+-binding site (M2 in Fig. 1). The M1 site (high-affinity Mg2+-binding site, Kd(Mg2+) 50 µM at pH 7.5 [7]) is filled under experimental conditions. Hence, we showed that Ca2+ replaces Mg2+ from the M2 site, whose affinity for Ca2+ is considerably higher than for Mg2+ (Kd(Ca2+) determined from the secondary plot is 65 µM; Fig. 3a).
In the absence of Mg2+, E. coli PPase does not hydrolyze pyrophosphate even at excessive Ca2+ concentrations (up to 10 mM). This means that Ca2+ cannot act as an activator, although it is capable of binding to the “activating” M2 site.Fig. 3. a) Dependence of MgPPi hydrolysis rate (v) on Mg2+ concentration at constant concentrations of free Ca2+ (20 (1), 50 (2), and 100 µM (3)) for WT PPase at pH 7.5 and [MgPPi] of 40 µM. A secondary dependence of the slope of lines on [Ca2+] is given in insets. Here and below the rate of hydrolysis is expressed in international units. b) Dependence of the rate of MgPPi hydrolysis by WT PPase on CaPPi concentration. Coordinates are taken from [21] (pH 7.5, [Mg2+] is 0.6 mM, [MgPPi] is 20 µM). c) Inhibition of MgPPi hydrolysis catalyzed by WT PPase by Ca2+ (Dixon plot). Concentration of MgPPi: 10 (1), 20 (2), 40 (3), and 80 µM (4). Calculated Hill coefficients: 0.63 (1), 0.87 (2), 1.16 (3), and 1.58 (4). A secondary dependence of the initial slope of the curves on 1/[MgPPi] is shown in the inset.
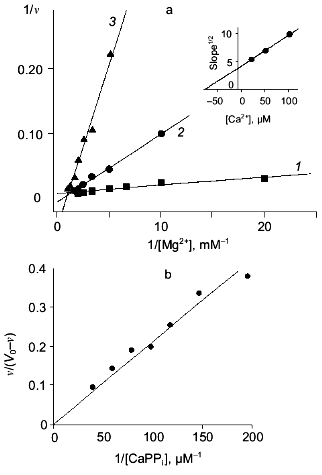
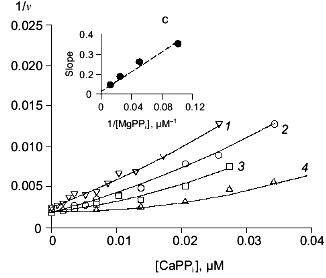
Figure 3, band c, shows the relationship between MgPPi hydrolysis rate and Ca2+ concentration in the presence of 0.6 mM Mg2+. The dependence in (1/[Ca2+], v/(V0 - v)) coordinates (Fig. 3b) allows one to evaluate the degree of inhibition [27]. The fact that the straight line passes through the origin indicates that, in the presence of Mg2+, PPase does not hydrolyze CaPPi at a detectable rate.
Figure 3c shows Dixon relationships between hydrolysis rate and inhibitor concentration ([I], 1/v). For convenience, to calculate kinetic parameters, CaPPi concentrations proportional to [Ca2+] at each constant [MgPPi] were plotted on the abscissa instead of free Ca2+ concentrations. Figure 3c shows a clear-cut nonlinear dependence of 1/v on [I]. This indicates the involvement of several Ca2+ ions in inhibition, which is confirmed by determination of Hill coefficients (given in Fig. 3 legend). Considerable deviation from the straight line indicates the presence of the term [CaPPi]2 in the expression for 1/v. This means that, under experimental conditions, the forms with two calcium ions (EMgCaCaPPi) participate in inhibition. Based on this finding and the above-mentioned consideration that at 0.6 mM Mg2+, the M1 site is completely filled with Mg2+, an inhibition scheme has been suggested (Scheme 1). It assumes that both MgPPi and CaPPi can bind to EMg2 and EMgCa. In this scheme, the equilibria for binding of free pyrophosphate to EMg2 and EMg(1)Ca(2) are omitted. The calculations show that the maximum [PPi]free value under experimental conditions does not exceed 0.2 µM, while Kd of the PPi complex with any enzyme form is above 100 µM [8]. The combination of Eqs. (1)-(5) gives the equation for the hydrolysis rate (1a) in Dixon coordinates as follows:
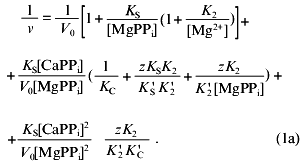
In “Materials and Methods”, we showed that Eq. (1a) allows one to evaluate the parameters of Scheme 1. The calculated values are summarized in Table 2. Analysis of these parameters leads to several intriguing conclusions.
Table 2. Ca2+ inhibition
parameters of MgPPi hydrolysis by native and mutant PPases
(calculated according to Scheme 1 at pH 7.5 and
[Mg2+] of 0.6 mM)
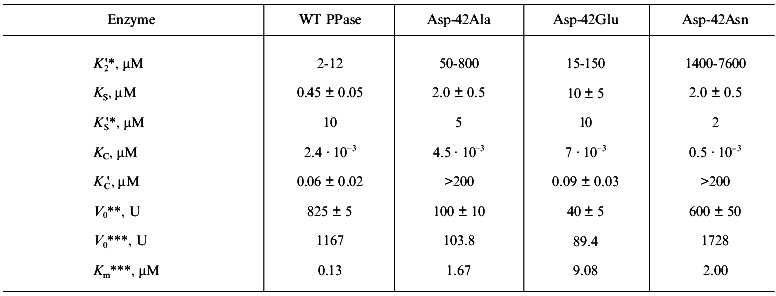
*Estimated parameters determined interdependently are given (for
details see “Materials and Methods”). To estimate
K´2, K2 values from 0.5 to 2
mM were used.
**Calculated according to Scheme 1.
***pH-Independent values obtained from kinetic studies at
[Mg2+] of 5 mM.
1. At the M2 site, as we expected, PPase affinity for Ca2+ is higher than for Mg2+ (K´2 /K2 = 5·10-3). To estimate K´2 given in Table 1, K2 values from 0.53 mM (obtained in the previous kinetic experiment for [Ca2+] of 20 µM, Fig. 3a) to 2.0 mM (determined by UV-spectroscopy [28]) were used. It should be noted that the affinity of this site for metal ions increases in the presence of substrate [29]. This may affect K2 and K´2 values measured by kinetic methods.
2. The K´S value of 10 µM is much higher than KS = 0.45 µM. This is unexpected, since it is assumed that MgPPi affinity for Mg2+- or Ca2+- activated enzyme is virtually the same [8, 10]. The same holds for KC and K´C parameters. Estimation of KC shows good agreement with equilibrium dialysis data for yeast PPase, according to which KCaPPi = .·10-9 M [8]. However, CaPPi affinity for EMg(1)Ca(2) (K´C) is significantly lower than the expected value. The reason for this discrepancy is unknown; however, it indicates different effects of Mg2+ and Ca2+ on the active site conformation.
3. The comparison of KC and KS, as well as K´C and K´S, indicates that the affinity of various PPases for Ca2+ is several orders of magnitude higher than for Mg2+.
On the whole, the experimental data are well described by Scheme 1. Our results show that Ca2+ competes with Mg2+ for the M2 site (low-affinity for Mg2+ and high-affinity for Ca2+), while CaPPi competes with MgPPi. All dissociation constants including K´2 do not exceed 10 µM; this accounts for very high effective Ca2+ inhibition constants for all known PPases [15-19].
Kinetics of Ca2+ inhibition of LaPPi hydrolysis. The binding constants of pyrophosphate complexes with such cations as Cr3+ and La3+ are very high (.1017 M-1 [30]); therefore, we can postulate that Cr3+ (La3+) as substrate constituents cannot be replaced either by Mg2+ or by Ca2+. (The corresponding dissociation constants for MgPPi and CaPPi are given in “Materials and Methods”). In the presence of Mg2+, CrPPi is hydrolyzed by E. coli PPase, although much more less effectively than MgPPi. The inhibition of CrPPi hydrolysis by Ca2+ was earlier used to show that one of the inhibitory Ca2+ ions is a substrate constituent [7].
In this work, we investigated Ca2+ inhibition of LaPPi hydrolysis. Under the experimental conditions, free pyrophosphate was absent from the reaction mixture; therefore, CaPPi was not formed. This allowed us to eliminate the “substrate” Ca2+-binding site and to calculate the inhibition parameters using a simplified model.
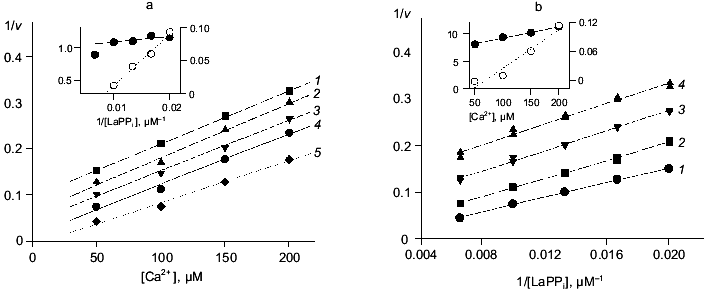
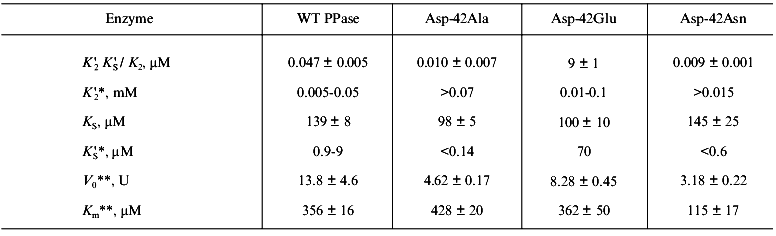
Figure 4 shows the results of this part of the work. The relationships between hydrolysis rate and concentrations of Ca2+ ((a) Dixon plot) and substrate ((b) Lineweaver-Burk plot) are shown. As we expected, the dependences are linear; this means that one Ca2+ ion is sufficient for PPase inhibition. In (1/[Ca2+], v/(V0 - v)) coordinates, all straight lines pass through the origin; this shows that similar to MgPPi inhibition, LaPPi inhibition is complete (not shown). The kinetic parameters of LaPPi hydrolysis at pH 7.5 and [Mg2+] of 0.6 mM in the absence of Ca2+ are V0 = 13.6 U, Km = 356 µM (Table 3).
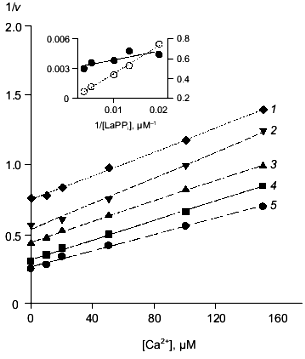
Table 3. Parameters of Ca2+ inhibition of LaPPi hydrolysis by native and mutant PPases (calculated according to Scheme 2 at pH 7.5 and [Mg2+] of 0.6 mM)Fig. 4. a) Dependence of the rate of LaPPi hydrolysis by WT PPase on Ca2+ concentration (Dixon plot). LaPPi concentration: 50 (1), 60 (2), 75 (3), 100 (4), and 200 (5) µM. b) Dependence of the rate of LaPPi hydrolysis by WT PPase on LaPPi concentration (Lineweaver-Burk plot). Ca2+ concentration: 50 (1), 100 (2), 150 (3), and 200 µM (4). Secondary plots for estimating kinetic parameters are shown in insets (the initial slope of the curves is marked with filled circles, left ordinate; open circles show the intercept on the 1/v ordinate, right ordinate).
*Estimated parameters are presented (for details see “Materials and Methods”). To calculate K´2, K2 values from 0.5 to 2 mM were used.
**V0 and Km were determined from kinetic studies at pH 7.5 and [Mg2+] of 0.6 mM.
The experimental data are most adequately described by Scheme 2. According to this scheme, Ca2+ inhibits LaPPi hydrolysis through competition with Mg2+ for the M2 site. In the absence of Mg2+, PPase does not hydrolyze LaPPi, namely, the form ECa2(LaPPi) is inactive. Scheme 2 does not take into consideration the formation of this enzyme form, since as mentioned above, at [Ca2+] < 200 µM, only one calcium cation is involved in inhibition.
The experimental design was such that all pyrophosphate added to the reaction mixture was in the LaPPi form. Therefore, Scheme 2 is a simplified variant of Scheme 1. The combination of Eqs. (6)-(8) gives Eq. (6a) for the rate of LaPPi hydrolysis in Dixon coordinates:
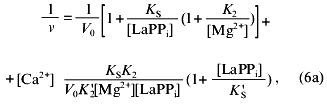
where V0 is the maximum rate of LaPPi hydrolysis in the absence of inhibitor at substrate saturation; other parameters are taken from Scheme 1.
Estimation of inhibition parameters using Scheme 2, gives KS, K´S, and K´2 /KS values. These results together with LaPPi hydrolysis parameters in the absence of Ca2+ (V0 and Km) are summarized in Table 3.
The comparison of MgPPi and LaPPi hydrolysis parameters leads to the following conclusions.
1. The maximum rate of LaPPi hydrolysis by the native PPase is two orders of magnitude lower than that of MgPPi. This finding differs from the literature data on hydrolysis of pyrophosphates of lanthanum and other rare-earth elements by yeast PPase [30]. In all likelihood, the active site of E. coli PPase is more specific with respect to the substrate metal ion than the active site of the yeast enzyme.
2. The affinity of LaPPi for EMg2 is by a factor of 300 weaker than the affinity of MgPPi for EMg2. Similar results were obtained with yeast PPase [30]. However, the dissociation constants of LaPPi and MgPPi complexes with EMg(1)Ca(2) are at least of the same order. These data indicate that the nature of metal ion at the M2 site is important for optimum conformation of the substrate-binding site.
3. The comparison of KS and K´S parameters shows that enzyme affinity for CaPPi is much higher than for LaPPi. A similar result was obtained with MgPPi.
4. The Ca2+ affinity for the M2 site determined from LaPPi hydrolysis is somewhat lower than the parameter determined from MgPPi hydrolysis. This probably reflects the effect of substrate on K´2 and shows that the estimate presented in Table 2 is an effective value.
From these results we may conclude that the filling of M2 site with Ca2+ is sufficient for E. coli PPase inhibition. If the metal in this site is responsible for activating the attacking nucleophile, this fact unambiguously indicates that Ca2+ is ineffective in activating the H2O molecule.
Ca2+ inhibition of mutant PPases with substitutions Asp-42-Ala, Glu, and Asn. It is known that, at concentrations above 5 mM, Mg2+ inhibits E. coli PPase. According to present views, inhibition is caused by the filling of the fourth metal-binding site (taking into account the substrate site) in the PPase active site. X-Ray data explains inhibition by the filling of the M4 site [3]. The carboxyl group of Glu-31, two oxygen atoms from the substrate, and three water molecules, one of which is coordinated by the side chain of Asp-42, are the metal ion ligands at this site.
To explore whether Ca2+ inhibition is connected with the filling of this “inhibitory” site, we studied the behavior of mutant pyrophosphatases with Asp-42 substitutions for Asn, Glu, and Ala under Ca2+ inhibition conditions. The conditions for inhibition of MgPPi and LaPPi hydrolyses were the same as for WT PPase. Results of the experiments are shown on the Figs. 5 and 6.
Fig. 5. Dependence of the rate of MgPPi hydrolysis by mutant PPases on CaPPi concentration (Dixon plot). a) Asp-42-Glu PPase. MgPPi concentration: 80 (1) and 120 µM (2). Calculated Hill coefficients: 1.19 (1) and 1.14 (2). b) Asp-42-Asn PPase. MgPPi concentration: 20 (1), 40 (2), and 80 µM (3). Hill coefficient: 0.94 (1), 1.04 (2), and 1.4 (3). Secondary dependences of the initial slope of the curves on 1/[MgPPi] are given in insets.
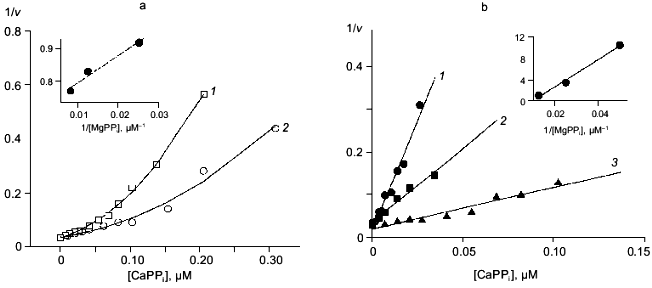
Despite different nature of substitutions, all three mutant PPases retained the essential inhibition characteristics specific to WT enzyme. Therefore, the experimental data were analyzed using Schemes 1 and 2 for inhibition of MgPPi and LaPPi hydrolyses, respectively. Calculated inhibition parameters are given in Tables 2 and 3.Fig. 6. Dependence of the rate of LaPPi hydrolysis by Asp-42-Glu PPase on Ca2+ concentration (Dixon plot). LaPPi concentration: 50 (1), 75 (2), 100 (3), 200 (4), and 300 µM (5). Secondary dependences for estimating kinetic parameters are given in insets (the slope of lines is shown by filled circles, left ordinate; open circles designate the intercept on 1/v axis, right ordinate).
Earlier we studied the kinetics of MgPPi hydrolysis by three mutant PPases, Asp-42-Asn, Glu, and Ala (the paper is submitted for publication). One of two distinctive features of these mutants, as compared to the native enzyme, consists in high Km values (see Table 2). The comparison of parameters in Scheme 1 (Table 2) for the native and all mutant PPases shows that this peculiarity is conserved in all three mutants only for MgPPi binding to EMg2 (KS). The K´S parameters, which characterize MgPPi binding to EMg(1)Ca(2), and KC (CaPPi binding to EMg2) are virtually the same for all four enzymes. This explains why K´S and KS differing by a factor 25 in WT PPase become virtually identical in mutants. This difference shows that, at the active site of the native PPase, there exists an optimum conformation for binding “physiological” ligands, Mg2+ and MgPPi, while the substitution of the Asp-42 residue decreases active site specificity with respect to the nature of the associated cation.
In all four enzymes studied, CaPPi affinity for EMg2 (KC) exceeds MgPPi affinity for EMg2 (KS) by several orders of magnitude. Alternatively, Asp-42-Ala and Asp-42-Asn PPases are characterized by worse K´2 and K´C parameters (Ca2+ affinity for EMg(1) and CaPPi affinity for EMg(1)Ca(2)).
The inhibition parameters of LaPPi hydrolysis, as well as V0 and Km (Table 3), differ in native and mutant pyrophosphatases to an even lesser extent. Thus, the replacement of Mg2+ by La3+ leads to elimination of variation in behavior of different enzymes (WT PPase and mutants). This finding supports our suggestion of the existence of an optimum active site conformation and illustrates an important role of the “substrate” metal ion in generating this conformation.
DISCUSSION
Growth and development of a procaryotic cell mainly depend on the nature of a food substrate and energy resources; H+ (NADH) and organic phosphates (ATP, Pi, and PPi) are the major regulators of metabolic processes in procaryotes [20]. Since Ca2+ affects the enzymes of phosphate metabolism, the changes in intracellular Ca2+ and phosphate concentrations are interrelated [31, 32]. The Ca2+ ions inhibit PPases at a concentration of .10-5 M; this corresponds to Ca2+ maximum level in the cell [20]. In E. coli, an increase in PPi concentration reduces the rate of many processes and finally results in growth inhibition [33]. Accordingly, Ca2+-mediated regulation of pyrophosphatase activity may control PPi concentration during intracellular signal transduction.
Ca2+ binding in the absence of substrate. Our data are consistent with an earlier suggestion that the high-affinity Ca2+-binding site in E. coli PPase (Kd measured by different methods varies from 18 to 68 µM [7]) coincides with the low-affinity Mg2+-binding site (Kd = 1.3 mM [22]; M2 in Fig. 1). Equilibrium dialysis of mutant pyrophosphatases also indicates that the low-affinity Ca2+ binding site (Kd = 2.0 mM [7]) is located in the region of Asp-65, -67, and -70, namely, it probably coincides with the high-affinity Mg2+-binding site (Kd = 50 µM [7]; M1 in Fig. 1). This conclusion supports the results obtained with WT PPase. Consequently, in E. coli PPase, affinity inversion, shown by different methods for yeast PPase, is also observed [8, 11, 12, 28].
The question arises as to why during equilibrium dialysis Mg2+ can replace Ca2+ from the M2 site, while it does not replace Ca2+ from the M1 site, although its affinity for this site is two orders of magnitude higher. This may be due to different properties of Ca2+ and Mg2+. The size of a non-hydrated Ca2+ ion is much greater than that of Mg2+ (their ionic radii are 0.96 and 0.65 Å, respectively [34, 35]). However, a hydrated Ca2+ cation is smaller than a hydrated Mg2+ cation [36]. Metal binding at the M2 site replaces only one solvent molecule in its coordination sphere (by the oxygen atom of Asp-70 carboxyl group). Therefore, the hydration level of the central ion changes insignificantly resulting in easy mutual substitutions at the M2 site, which depend only on [Ca2+]/[Mg2+] ratio. A different picture is observed at the M1 site. During the filling of this site, the metal ion loses three molecules of the solvent and acquires three protein ligands instead (Fig. 1). In this instance, the size of the central ion becomes very important, and the geometry of the M1 site with associated Ca2+ or Mg2+ may differ considerably.
The presence of the third Ca2+-binding site in Asp-42-Ala PPase deserves special attention. The mechanism of binding an additional Ca2+ may be as follows. The Asp-42 residue in the native PPase is hydrogen-bonded to Glu-31 and forms an ion pair with Lys-29. It seems most probable that the substitution of this residue by hydrophobic alanine releases both ligands. The carboxyl groups of Glu-31 and Glu-20 located in this region form a negatively charged cluster capable of Ca2+ binding. Therefore, an additional Ca2+ binding site in Asp-42-Ala PPase is probably located in the same region as the site responsible for binding “inhibitory” Mg2+ ion (M4 in Fig. 1).
Kinetics of Ca2+ inhibition. The kinetic data demonstrate that first, Ca2+ is incapable of activating E. coli PPase even if it is bound at the “activator” M1 and M2 sites; second, CaPPi is not hydrolyzed by E. coli PPase in the absence of Mg2+ and at Mg2+ concentrations below 0.6 mM.
Similar results were obtained with yeast PPase. Of more than 15 metals tested, only 3 were able to activate the enzyme by more than 0.1% compared to Mg2+ [10]. Ca2+ is not among them. Some cations (La3+ and other rare-earth elements) cannot activate the enzyme, although in the presence of a suitable effector, the substrate containing these cations may be hydrolyzed [10, 30]. The available data indicate that the substrate-binding site imposes heavier demands on the nature of the metal ion than the activator site [10]. It was also shown for yeast enzyme that the nature of the substrate metal affects considerably substrate binding and significantly less influences the rate of pyrophosphate hydrolysis [30]. Our findings are in conflict with these data; this is probably another manifestation of the dissimilarity between yeast and E. coli PPases.
Ca2+ also inhibits PPase-catalyzed 18O exchange between phosphate and water [10].
Estimation of the rate constants for the elementary stages of the yeast PPase-catalyzed reaction in the presence of Mn2+, Zn2+, and Co2+ shows that low efficiency of these cations as activators of pyrophosphate hydrolysis is mainly due to a decreased rate of release of hydrolysis products [36]. The inhibition of E. coli PPase by excess Mg2+ is also associated with this phenomenon [37].
Data analysis allowed us to determine the role of Ca2+ at different stages of MgPPi hydrolysis and suggest the possible causes of its inhibitory effect.
1. The release of hydrolysis products and pyrophosphate hydrolysis are the rate-limiting stages for E. coli PPase [36]; therefore, we shall consider them first. The release of pyrophosphate hydrolysis products is probably more complicated in the presence of Ca2+ than of Mg2+. First, as was shown for yeast PPase, this is observed with many cations including Mn2+, Zn2+, and Co2+. They bind stronger to the enzyme active site than Mg2+ that may interfere with the release of the reaction products. At the same time, the maximum rate of pyrophosphate hydrolysis activated by these cations differs insignificantly, as compared to Mg2+, and is even higher with Zn2+ [36]. Second, as we showed in this work, E. coli pyrophosphatases bind CaPPi several orders of magnitude stronger than MgPPi. It probably holds true for calcium and magnesium phosphates. Analysis of the low-molecular-weight compounds of calcium and magnesium demonstrates that Ca2+ exhibits higher affinity for carboxylates than Mg2+ [23]. We may suppose that this is one of the main factors responsible for stronger binding of Ca2+ than of Mg2+ to the PPase molecule. Accordingly, more difficult release of the reaction products may account for Ca2+ inhibition of PPases.
The kinetics demonstrate that CaPPi is not hydrolyzed by PPase. However, we cannot rule out that a single CaPPi hydrolysis act, in which the reaction products remain tightly associated with the enzyme, occurs at the PPase active site in the presence of Mg2+. This makes further hydrolysis of CaPPi impossible. Thus, the role of Ca2+ in the release of the reaction products is still unclear, since these two possibilities are experimentally indistinguishable.
2. Ca2+ can occupy the M1 site. The role of the activator metal at the M1 site consists in conformational rearrangements of the active site for catalytically effective substrate binding. Figure 1 shows that one Mn2+ ligand at the M1 site (Asp-102 carboxyl group) is simultaneously a metal ligand at the M3 “substrate” site. From the above-mentioned considerations it follows that the mode of Ca2+ and Mg2+ binding at the M1 site is different. Even if the ligands for these cations at M1 are similar, their relative position, the distance between them and other active site groups, etc., may vary. This variation must inevitably interfere with substrate binding (MgPPi, CaPPi, or LaPPi). However, this hypothesis is not substantiated by the kinetic data, since under experimental conditions, the M1 site is completely occupied by Mg2+.
3. The substrate metal cation screens pyrophosphate charge, thus providing necessary electrophilicity to the attacked phosphorus atom. The efficiency of the metal ion in this instance is determined by its polarizing effect. The calculated polarizing effect of Ca2+ is lower than that of Mg2+. This suggestion was confirmed by the study of the effect of Ca2+ and Mg2+ on H-O-H angle in liganded water molecules and calculated dehydration energies for Ca2+ and Mg2+ aqua-complexes [38]. However, this problem is more complicated than it may seem. Calcium ions in solution form complexes with a coordination number of 6 or 7 with easy transitions between them. The parameters of Ca2+ binding in these complexes and hence, the polarizing effect of the central ion are different. If we compare Ca2+ and Mg2+ complexes with identical coordination numbers, for example hexahydrates, the effective metal ion charge in these complexes will be 1.57 and 1.18 for Ca2+ and Mg2+, respectively [38]. Accordingly, in some instances, the polarizing effect of Ca2+ may be comparable or even higher than that of Mg2+. Thus, the activating effects of Ca2+ and Mg2+ on pyrophosphate are still incompletely understood.
The data on LaPPi hydrolysis indicate that the nature of metal cation at the M3 site is essential for hydrolysis. The kcat value for LaPPi hydrolysis by WT PPase is two orders of magnitude lower than for MgPPi hydrolysis, despite higher La3+ charge. It follows from this observation that the functions of the substrate metal cation are not limited to shielding of the pyrophosphate charge. Nevertheless, La3+ is capable of substrate activation. In all probability, despite a larger radius as compared to Mg2+ (1.06 Å [35]), La3+ produces a rather strong polarizing effect due to its higher charge.
4. The role of Ca2+ associated at the M2 site is of particular interest. A current model for the mechanism of PPase hydrolytic action suggests that the metal ion at the M2 site functions as a nucleophile activator for the attack on pyrophosphate [3]. Ca2+ and Mg2+ show different efficiency in activating a water molecule. Thus, the pKa of the water molecule included in the coordination sphere of the metal ion is 12.9 for Ca2+ and 11.4 for Mg2+ [34, 35]. This means that under identical conditions, the concentrations of active nucleophile may differ more than tenfold in these two instances. In addition, calcium cation, which differs in size from Mg2+, may destroy the native conformation of the M2 site affecting the position of metal ligands. At the same time, the orientation of the attacking nucleophile is extremely important for SN2 reaction efficiency.
As we showed above, Ca2+ inhibits LaPPi hydrolysis under the conditions when CaPPi is not formed and the concentrations of free Ca2+ correspond to complete filling of the M2 site. This fact clearly indicates that Ca2+ at the M2 site is incapable of nucleophile activation.
Different effects of Ca2+ and Mg2+ on the conformation of the M2 binding site were demonstrated earlier by another example. The fluoride ion is a powerful inhibitor of yeast and E. coli PPases. It was shown for yeast PPase that F- binds to the enzyme or enzyme-substrate complex, and irreversible isomerization of the product occurs. Yeast PPase can bind F- only in the presence of Mg2+, at its concentrations providing complete filling of the M2 site. The resulting enzyme-substrate-magnesium-fluoride complex (E-S-Mg-F) can be isolated by gel-filtration. At the same time, in the presence of Ca2+, the enzyme-substrate complex loses its ability for binding F- [39].
As will be shown below, the X-ray data are consistent with our conclusions on the effect of Ca2+ on the active site conformation.
X-Ray analysis of pyrophosphatase complexed with Ca2+ and CaPPi. The structures of E. coli PPase complexed with Ca2+ and CaPPi have been solved and refined quite recently. The publication of these results is now in progress. They confirm the experimental findings described above, so we present here only a short description of these data.
The structure of E. coli PPase complexed with Ca2+ was solved at 1.1 Å resolution. Each enzyme subunit in the complex contains two metal cations at the active site. Their position is shown in Fig. 7. The carboxyl groups of Asp-65, -70, and -102 and three water molecules are Ca2+ ligands at one site. The Asp-102 carboxyl group is coordinated with Ca2+ in a bidentate manner. The occupancy of this site is 0.8. At the second site, Ca2+ ligands are the Asp-70 carboxyl group and six water molecules; the occupancy of this site is 0.7. Therefore, the positions of calcium ions are similar to those at the M1 and M2 sites in Fig. 1. However, both Ca2+ ions in the structure under consideration have a coordination number of 7, but not of 6, as Mg2+ and Mn2+. This is often observed in proteins and reflects variant properties of these cations.
The structure of the pyrophosphatase-CaPPi complex was solved at 1.2 Å resolution. In this structure, all six subunits are identical. Each subunit contains two Ca2+ ions and CaPPi at the active site (Fig. 8). Metal ions are associated with the sites analogous to M1, M2, and M3. Ca2+ ligands at the M3 site are the carboxyl groups from Asp-97 and Asp-102, two water molecules, and two oxygen atoms from pyrophosphate. It is apparent that Ca2+ binds at the M3 site as a substrate constituent; this fact agrees with the order of filling of Mg2+-binding sites proposed earlier [3].Fig. 7. Active site of E. coli PPase complexed with Ca2+. The side groups of the most important residues are shown. Designations: large circles, metal ions; small circles, water molecules (Ca2+ ligands). Calcium cations are connected with the ligands by dotted lines. Figures 7 and 8 were generated using the MOLSCRIPT program [42].
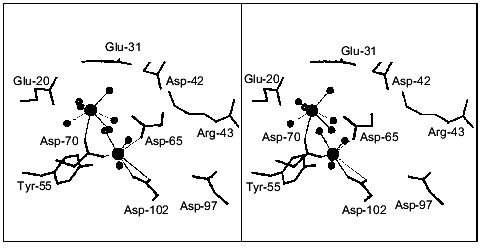
The structural comparison of the M1 and M2 sites of the two PPase complexes reveals several characteristic features. First, the position of Ca2+ at the M1 site differs considerably: in the CaPPi complex, it is shifted by 2.39 Å toward pyrophosphate. At this site, the flexible loops of the polypeptide chain (64-70 and 96-102) with Ca2+ ligands are significantly displaced. The conformation of the M2 site remains virtually unaffected. The position of Ca2+ in these structures differs by 0.8 Å. In the complex with CaPPi, the metal cation at this site has a coordination number of 6, not 7 as in the complex with Ca2+. This results in different arrangement of H2O molecules, the metal ion ligands (the maximum shift Delta is 0.87 Å). Similar to the M1 site, the substrate oxygen atom becomes Ca2+ ligand at M2 instead of one water molecule.Fig. 8. Active site of the E. coli PPase with CaPPi. Metal ions are designated with large filled circles, and water molecules with small filled circles. The bridge-type H2O molecule linking Ca2+ ions is shown as a small gray circle. Pyrophosphate is given in gray color.
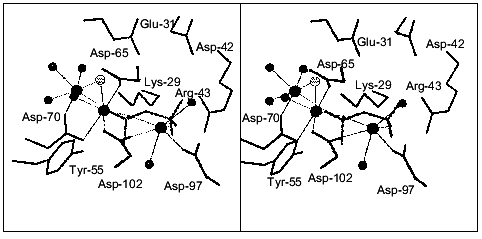
Another interesting structural peculiarity of CaPPi complex is that at the M1 and M2 sites, two metal ions are bound to one more common ligand, a bridge-type water molecule, in addition to the Asp-70 carboxyl group. This molecule is absent from the Ca2+ complex. In all probability, upon substrate binding, one metal ion moves closer to the second ion (3.3 Å), and one of their ligands becomes common to both of them. Similar shifts of Mn2+ ions accompanied by the appearance of a common ligand in the presence of two phosphates were described for yeast PPase [5].
To elucidate the mechanism of catalytic reaction, it is interesting to investigate the arrangement of water molecules relative to CaPPi, which is a non-hydrolyzable analog of the substrate. According to the current concept of PPase catalytic action, two different water molecules are supposed to act as attacking nucleophiles. However, analysis of PPase complexed with Ca2+ and CaPPi demonstrates that neither H2O molecule is located on line to the bridge-type oxygen atom and rather close to phosphorus atoms. Harutyunyan [2, 3] suggested that the attacking water molecule in the PPase complex with the reaction products is replaced by the oxygen atom from one of the phosphates (Ow in Fig. 1). In the complex with non-hydrolyzable CaPPi, the corresponding H2O molecule is replaced by oxygen from pyrophosphate. According to Cooperman's theory [4, 5], the hydroxyl ion generated from the bridge-type water molecule between metal ions at the M1 and M2 sites is an attacking nucleophile. The bridge-type H2O molecule linking two Ca2+ ions (shown in gray in Fig. 8) is 3.48 Å apart from the nearest phosphorus atom, and upon nucleophilic attack, the angle would be 113 instead of 180°. In addition, only one H2O molecule at the active site of PPase complexed with CaPPi is located at a distance, which does not exceed 3.5 Å from the phosphorus atoms; however, it is distant from all three metal cations. The amino group of Lys-148 is its only protein ligand. Thus, neither water molecule can be definitely identified as an attacking nucleophile.
The absence of a specifically oriented water molecule may be connected with different properties of Ca2+ and Mg2+ leading to divergent conformation of the active site. Superimposing the structure of PPase complexed with Ca2+ on the structure of the enzyme-Mg2+ complex [2] shows that the positions of metal ions and their ligands changed. As we expected, Ca2+ and Mg2+ shifted most relative each other at the M1 site (Delta_is 1.37 Å). The picture is different for the M2 site: the metal ions are located similarly (Delta is 0.2 Å), although water molecules are displaced by 0.5-0.6 Å. However, even in the structure of the enzyme complexed with Mg2+, it is impossible to find a H2O molecule located on line with the oxygen atom of calcium pyrophosphate.
Another possible explanation presumes different binding of the substrate at the PPase active site in the presence of various cations. Some kinetic data support this suggestion [36]. Structure investigations of calcium and magnesium pyrophosphates also reveal variation in conformation of these substrates [40]. Superposition of the structures of PPase complexed with CaPPi and yeast PPase complexes with the reaction products (and with Mn2+ as effector [3, 5]) shows that they hardly differ in the mode of substrate binding. Thus, phosphorus atoms are displaced in these structures by 0.99 and 1.38 Å and the straight line P-P shifts by less than 10°. These differences are most likely due to the movement of phosphate groups induced by hydrolysis. Nevertheless, structural analysis of several mutant PPases demonstrates that even relatively small conformational changes may considerably decrease enzymatic activity [41].
We may also suggest the following explanation. The enzymatic reaction of PPi hydrolysis/synthesis represents a sequence of many elementary stages. The available models of PPase catalytic action [3, 5] do not take into consideration the complexity of this process. In particular, both presently available structures of yeast PPase-phosphate complex do not explain the mechanism of PPi synthesis. Two reactions (hydrolysis and synthesis) catalyzed by the enzyme proceed several times during one catalytic act; therefore, it seems reasonable to suggest that after hydrolysis the phosphate groups move relative to each other insignificantly. However, in both instances, the positions of phosphates differed from the expected ones, namely, not a single oxygen atom was in proper orientation for nucleophilic attack on phosphorus. This finding shows that the structural data deals with specific conformation of the active site fixed at an elementary stage following hydrolysis.
The complexity of the processes that occur at the active site during hydrolysis is illustrated by one more observation. As we already noted, in the structure of E. coli PPase complexed with CaPPi, each metal ion at the M1 and M2 sites is associated with only one oxygen from phosphate. Since in this complex hydrolysis did not take place, it means that, upon binding to the active site, the substrate replaces one water molecule from the coordination spheres of each of these ions. If the phosphorus atom of pyrophosphate is attacked by the water molecule included in the coordination sphere of the metal ion, an additional contact of this ion with the phosphate formed should appear. However, in both structures of yeast PPase complexed with hydrolysis products [3, 5], each Mn2+ preserves a contact only with one oxygen atom from phosphate both at the M1 and M2 sites. This may indicate movement of oxygen atoms due to the turn of the phosphate residue after hydrolysis and loss of the contact with Mn2+. A changed position of phosphate was detected upon the comparison of structures of the yeast mutant Arg-78-Lys PPase complexed with phosphate and WT PPase complexed with phosphates and Mn2+ [29]. This fact proves that both structures of PPase complexes with hydrolysis products [3, 5] do not reflect the state of the active site immediately after hydrolysis, but only a subsequent stage of substrate conversion.
In summary, X-ray data are consistent with our hypothesis based on Ca2+ inhibition kinetics of E. coli PPase: inhibition is mainly due to the distortion of the active site conformation resulting in inability of the enzyme to activate and orient a nucleophile for the attack on pyrophosphate.
In this work we analyzed in detail Ca2+-induced inhibition of E. coli PPase. Kinetic studies, equilibrium dialysis of mutant PPases, and X-ray data reveal reasons for the strong inhibitory effect of Ca2+ on PPase. First, in the course of catalysis, Ca2+ replaces Mg2+ from the M2 site, which shows higher affinity for Ca2+ than for Mg2+. This results in inability of the enzyme to acquire the optimum for hydrolysis active site conformation and orientation of the attacking nucleophile. In addition, Ca2+ is incapable of activating thee H2O molecule for the attack. Second, we have shown that the nature of the metal cation at the substrate-binding site and its ability to activate the electrophilic phosphorus atom are important for hydrolysis. Third, it has been suggested that, upon catalysis in the presence of Ca2+, the release of hydrolysis products will be affected. These are the reasons for Ca2+ being a powerful inhibitor of all known PPases.
This work was supported in part by the Russian Foundation for Basic Research (grants No. 97-04-50031 and No. 96-15-97969) and the “Universities of Russia” program (basic investigations, grant No. 5077).
REFERENCES
1.Harutyunyan, E. H., Oganessyan, V. Yu., Oganessyan,
N. N., Terzyan, S. S., Popov, A. N., Rubinskiy, S. V., Vainshtein, B.
K., Nazarova, T. I., Kurilova, S. A., Vorobyeva, N. N., and Avaeva, S.
M. (1996) Kristallografiya, 41, 84-96.
2.Harutyunyan, E. H., Oganessyan, V. Yu., Oganessyan,
N. N., Avaeva, S. M., Nazarova, T. I., Vorobyeva, N. N., Kurilova, S.
A., Huber, R., and Mather, T. (1997) Biochemistry, 36,
7754-7760.
3.Harutyunyan, E. H., Kuranova, I. P., Vainshtein, B.
K., Höhne, W. E., Lamzin, V. S., Dauter, Z., Teplyakov, A. V., and
Wilson, K. S. (1996) Eur. J. Biochem.,239,220-228.
4.Kankare, J., Salminen, T., Lahti, R., Cooperman, B.
S., Baykov, A. A., and Goldman, A. (1996) Biochemistry,
35, 4670-4677.
5.Heikinheimo, P., Lehtonen, J., Baykov, A., Lahti,
R., Cooperman, B. S., and Goldman, A. (1996) Structure,
4, 1491-1508.
6.Avaeva, S. M., Rodina, E. V., Vorobyeva, N. N.,
Kurilova, S. A., Nazarova, T. I., Sklyankina, V. A., Oganessyan, V.
Yu., and Harutyunyan, E. H. (1998) Biochemistry (Moscow),
63, 592-707.
7.Kurilova, S. A., Bogdanova, A. V., Nazarova, T. I.,
and Avaeva, S. M. (1984) Bioorg. Khim., 10,
1153-1160.
8.Ridlington, J. W., and Butler, L. G. (1972) J.
Biol. Chem., 247, 7303-7307.
9.Moe, O. A., and Butler, L. G. (1972) J. Biol.
Chem., 247, 7315-7319.
10.Butler, L. G., and Sperow, J. W. (1977)
Bioinorg. Chem., 7, 141-150.
11.Rapoport, T. A., Höhne, W. E., Heitmann, P.,
and Rapoport, S. (1973) Eur. J. Biochem., 33,
341-347.
12.Braga, E. A., and Avaeva, S. M. (1972) FEBS
Lett., 27, 251-255.
13.Kuranova, I. P., Terzyan, S. S., Voronova, A. A.,
Smirnova, E. A., Vainshtein, B. K., Höhne, W., and Hansen, G.
(1983) Bioorg. Khim., 9, 1611-1619.
14.Kurilova, S. A., Nazarova, T. I., and Avaeva, S.
M. (1983) Bioorg. Khim., 9, 1032-1039.
15.Fraichard, A., Trossat, C., Perotti, E., and
Pugin, A. (1996) Biochimie, 78, 259-266.
16.Volk, S. E., Baykov, A. A., Duzhenko, V. S., and
Avaeva, S. M. (1982) Eur. J. Biochem., 125, 215-220.
17.Maeshima, M. (1991) Eur. J. Biochem.,
196, 11-17.
18.Davidson, A. M., and Halestrap, A. P. (1989)
Biochem. J., 258, 817-821.
19.Uribe, S., Rangel, P., Pardo, J. P., and
Pereira-da-Silva, L. (1993) Eur. J. Biochem., 217,
657-660.
20.Williams, R. J. P. (1998) Biochim. Biophys.
Acta, 1448, 153-165.
21.Santella, L. (1998) Biochem. Biophys. Res.
Commun., 244, 317-324.
22.Berridge, M. J., Bootman, M. D., and Lipp, P.
(1998) Nature, 395, 645-649.
23.Hughes, M. N. (1983) The Inorganic Chemistry
of Biological Processes [Russian translation], Mir, Moscow, pp.
343-355.
24.Oganessyan, V. Yu., Kurilova, S. A., Vorobyeva,
N. N., Nazarova, T. I., Popov, A. N., Lebedev, A. A., Avaeva, S. M.,
and Harutyunyan, E. H. (1994) FEBS Lett., 348,
301-304.
25.Josse, J. (1966) J. Biol. Chem.,
241, 1938-1947.
26.Irani, R. R., and Callis, C. P. (1960) J.
Phys. Chem., 64, 1398-1407.
27.Yoshino, M. (1987) Biochem. J.,
248, 815-820.
28.Avaeva, S. M., Rodina, E. V., Kurilova, S. A.,
Nazarova, T. I., Vorobyeva, N. N., Harutyunyan, E. H., and Oganessyan,
V. Yu. (1995) FEBS Lett., 347, 44-46.
29.Tuominen, V., Heikinheimo, P., Kajander, T.,
Torkkel, T., Hyytiä, T., Käpylä, J., Lahti, R.,
Cooperman, B. S., and Goldman, A. (1998) J. Mol. Biol.,
284, 1565-1580.
30.Ting, S. J., and Dunaway-Mariano, D. (1984)
FEBS Lett., 165, 251-253.
31.Inoue, T., Yamada, T., Furuya, E., and Tagawa, K.
(1989) Biochem. J., 262, 965-970.
32.Griffiths, E. J., and Halestrap, A. P. (1993)
Biochem. J., 290 (Pt. 2), 489-495.
33.Chen, J., Brevet, A., Fromant, M., Leveque, F.,
Schmitte, J.-M., Blanquet, S., and Plateau, P. (1990) J.
Bacter., 172, 5686-5689.
34.Cowan, J. A. (1995) The Biological Chemistry
of Magnesium, VCH Publishers, Inc., New York, pp. 9-11.
35.Spiro, T. G. (1978) in Inorganic
Biochemistry (Eichhorn, G., ed.) [Russian translation], Vol. 1,
Mir, Moscow, pp. 644-650.
36.Welsh, K. M., Jakobyansky, A., Springs, B., and
Cooperman, B. S. (1983) Biochemistry, 22, 2243-2248.
37.Avaeva, S. M., Rodina, E. V., Kurilova, S. A.,
Nazarova, T. I., and Vorobyeva, N. N. (1996) FEBS Lett.,
392, 91-94.
38.Katz, A. K., Glusker, J. P., Beebe, S. A., and
Bock, C. W. (1996) J. Am. Chem. Soc., 118, 5752-5763.
39.Baykov, A. A., Tam-Villosado, J. J., and Avaeva,
S. M. (1979) Biochim. Biophys. Acta, 569, 228-238.
40.McCarthy, W. J., Smith, D. M. A., Adamowicz, L.,
Saint-Martin, H., and Ortega-Blake, I. (1998) J. Am. Chem. Soc.,
120, 6113-6120.
41.Avaeva, S. M., Rodina, E. V., Vorobyeva, N. N.,
Kurilova, S. A., Nazarova, T. I., Sklyankina, V. A., Oganessyan, V.
Yu., Samygina, V. R., and Harutyunyan, E. H. (1998) Biochemistry
(Moscow), 63, 671-684.
42.Kraulis, P. J. (1991) J. Appl.
Crystallogr., 24, 946-950.