REVIEW: Carnosine as a Regulator of Soluble Guanylate Cyclase
I. S. Severina*, O. G. Bussygina, and N. V. Pyatakova
Institute of Biomedical Chemistry, Russian Academy of Medical Sciences, ul. Pogodinskaya 10, Moscow, 119832 Russia; fax: (095) 245-0857* To whom correspondence should be addressed.
Received November 29, 1999
The molecular mechanism of the participation of carnosine in the functioning of soluble guanylate cyclase is discussed. It is shown that carnosine inhibits the activation of soluble guanylate cyclase by sodium nitroprusside and a derivative of furoxan--1,2,5-oxadiazolo-trioxide (an NO donor). However, carnosine has no effect on stimulation of the enzyme by a structural analog of the latter compound, a furazan derivative (1,2,5-oxadiazolo-dioxide) that is not an NO donor; nor was carnosine involved in the enzyme activation by protoporphyrin IX, whose stimulatory effect is not associated with the guanylate cyclase heme. The inhibition by carnosine of guanylate cyclase activation by an NO donor is due to the interaction of carnosine with heme iron with subsequent formation of a chelate complex. It was first demonstrated that carnosine is a selective inhibitor of NO-dependent activation of guanylate cyclase and may be used for suppression of activity of the intracellular signaling system NO-soluble guanylate cyclase-cGMP, whose sharp increase is observed in malignant tumors, sepsis, septic shock, asthma, and migraine.
KEY WORDS: soluble guanylate cyclase, nitric oxide, carnosine, sodium nitroprusside, protoporphyrin IX
Carnosine (beta-alanyl-L-histidine) was isolated by V. S. Gulewitsch as a component of compounds extracted from muscle tissue at the beginning of the XXth century [1].
It has been established that this natural dipeptide performs important biological functions; in particular, it exhibits antioxidative properties [2, 3] directed at suppression of free-radical reactions. Study of the antioxidative action of carnosine has shown that its effects are realized not only owing to binding of lipid oxidation products in the course of free-radical reactions, but also through interaction with active oxygen species [4, 5]. Carnosine may also serve as a scavenger of peroxyl [2] and hydroxyl [2] radicals, for singlet oxygen [2] and superoxide anion oxygen [2], and can neutralize hypochlorite anion by forming with it stable chloramine complexes [2]. The antioxidative properties of carnosine provide its successful application for treatment of cataract, superficial burns of epiderma, wound healing, i.e., for various inflammatory processes developing on the background of cellular membrane damage [2, 6, 7]. The exact molecular mechanism of carnosine action and its biological significance are not yet clear.
Soluble guanylate cyclase (EC 4.6.1.2) catalyzing the biosynthesis of cyclic 3´,5´-guanosine monophosphate (cGMP) is directly connected with lipid peroxidation and with free-radical reactions. It is known that soluble guanylate cyclase activity is stimulated by unsaturated fatty acids [8], by products of their peroxidation [9], and by free radicals, including hydroxyl radicals [10]. Earlier, we demonstrated the stimulation of soluble guanylate cyclase by a stable free radical and cumole hydroperoxide [11]. Acute disturbances in platelet guanylate cyclase activity have been revealed in experimental pathologies such as allergic heart lesion in rabbit [12] and acute ischemia in rats [13]. Both these pathologies are characterized by intensification of free-radical reactions. It is very likely that free radicals are endogenous regulators of soluble guanylate cyclase. The mechanism of their action is unknown, and the role of antioxidants in the functioning of guanylate cyclase is not yet clear. Studies in this area have acquired especial significance after identification of yet another free radical molecule--nitric oxide (NO)--one of the key endogenous regulators of various physiological processes. Endogenous NO, formed from L-arginine by L-arginine-NO-synthase [14] and identical to the endothelial derived relaxation factor (EDRF), is also a neurotransmitter, a cytotoxic agent [16], and a powerful factor of hemostasis. NO inhibits platelet aggregation [17] and is presently considered as an endogenous vasodilator. Many physiological functions of NO, particularly its vasodilatory and anti-aggregatory effects are directly associated with soluble guanylate cyclase activation and cGMP accumulation. Activation of guanylate cyclase by nitric oxide is due to the interaction of NO with the guanylate cyclase heme iron. It is known that the immediate heme precursor in vivo is protoporphyrin IX [18], which proved to be a potent activator of the enzyme. Introduction of iron into the porphyrin ring yields ferroprotoporphyrin IX (or heme), which acts as an inhibitor of the enzyme [19]. This reaction is considered as very important in the endogenous regulation of soluble guanylate cyclase. Following the interaction of NO with the heme iron, the iron protrudes from the plane of the porphyrin ring; as a result, the structure of the nitrosyl-heme complex formed becomes similar to that of protoporphyrin IX--one of the potent enzyme activators [18]. Thus, it appears that the nitrosyl-heme complex is the true activator of the enzyme [20]. It is known that the therapeutic effect of commonly used vasodilators (such as nitroglycerin) is closely related to the interaction of NO released in the course of their biotransformation with guanylate cyclase heme (according to above mechanism), enzyme activation, and accumulation of cGMP.
As mentioned above, carnosine is able to bind to the products and inhibitors of free-radical reactions [2]. No evidence has been presented that carnosine can interact with NO. However, it is known that carnosine is able to form chelate complexes with ions of bivalent metals (Cu, Co, Zn, Mn) [21, 22] including bivalent iron [23]. The antioxidative properties of carnosine and its ability to form chelate complexes may modify the functioning of guanylate cyclase.
The influence of carnosine on the activity of human platelet guanylate cyclase and on sodium nitroprusside-induced enzyme activation is shown in Table 1. One can see that in experiments with 105,000g supernatant, obtained from sonicated platelets suspension, carnosine (at a final concentration of 1 mM) had no effect on the guanylate cyclase activity (with Mg2+); however, at the concentration 5 mM, carnosine lowered the basal value by ~22%. In these experiments, carnosine was preincubated with guanylate cyclase for 10 min at 0°C before substrate addition. Increasing the preincubation time (up to 40 min) enhanced the inhibitory effect of carnosine to 35%. Carnosine also suppresses the ability of guanylate cyclase to be activated by sodium nitroprusside (Table 1). At the carnosine concentration 1 mM, the nitroprusside-induced activation was lowered 3-fold, while at 5 mM carnosine, 5-fold.
Table 1. Effect of carnosine on the activity
of human platelet guanylate cyclase and on the activation of the enzyme
by sodium nitroprusside (SNP, 100 µM) and protoporphyrin IX
(PTP IX, 5 µM)
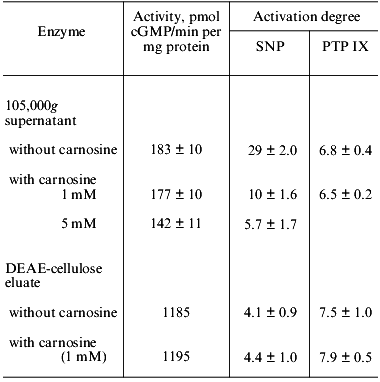
Note: Data represent the mean ± SD of four-five independent
experiments.
The intensity of the inhibitory action of carnosine on the sodium nitroprusside-induced guanylate cyclase activation was dependent not only on the carnosine concentration in the sample, but also on the order of addition of the components. We have shown that if guanylate cyclase was first preincubated with sodium nitroprusside (10 min at 0°C) and then 1 mM carnosine was added, the inhibition was 51 ± 4%. If guanylate cyclase was preincubated with 1 mM carnosine (10 min at 0°C) with subsequent addition of sodium nitroprusside, the inhibition increased to 74 ± 5%. These results are suggestive of a competition between carnosine and sodium nitroprusside for the same binding site on the enzyme molecule.
Since the action of sodium nitroprusside is due to the interaction of the NO group with the guanylate cyclase heme iron, we studied the influence of carnosine on sodium nitroprusside-induced guanylate cyclase activation upon heme deficiency of the enzyme. Although the nature of the bond between the protein and heme in guanylate cyclase is not yet fully clarified, it is known that this bond is labile. The heme can dissociate from the protein on a decrease in pH to 5.0, on storage, and in the course of some purification procedures (ion-exchange chromatography), thus causing a certain degree of heme deficiency of guanylate cyclase [20].
Earlier [24], we have shown that the 7-8-fold purified human platelet guanylate cyclases (with Mg2+) lost its ability to be activated by sodium nitroprusside by 70-80%. The absorbance spectra of such an enzyme preparation was lacking the maximum at 415-420 nm (Soret band) that was present in the initial 105,000g supernatant. In other words, the partially purified guanylate cyclase preparation was heme deficient. The degree of activation of such a preparation with sodium nitroprusside was lowered by 86% (from 29 to 4.1) (Table 1). The heme deficiency of the enzyme was further supported by the loss of the absorbance maximum at 410-415 nm (Soret band). From Table 1 one can also see that carnosine (at 1 mM or at even lower concentration, to 0.1 mM) does not affect the basal activity of the preparation and its ability to be activated by sodium nitroprusside. It appears therefore that carnosine does not inhibit the sodium nitroprusside-induced activation of the heme-deficient guanylate cyclase.
We [25] and others [18] have shown that the stimulatory effect of protoporphyrin IX (immediate heme precursor) is not associated with the heme. Therefore, we compared the influence of carnosine on the sodium nitroprusside-induced and the protoporphyrin IX-induced guanylate cyclase stimulation. The optimal concentration of protoporphyrin IX (with both 105,000g supernatant and partially purified heme-deficient guanylate cyclase preparation) is the same, 5 µM. In experiments with 105,000g supernatant, addition of carnosine (1 mM) lowered the stimulatory effect of sodium nitroprusside by ~66% but had no effect on the protoporphyrin IX-induced enzyme activation. In experiments with partially purified heme-deficient preparation, the activation of the enzyme by sodium nitroprusside was lowered, and this level remained unchanged upon carnosine addition (1 mM) (Table 1). The intensity of the activating effect of protoporphyrin IX on the heme-deficient enzyme preparation remained the same as in experiments with supernatant and the activation degree did not change upon carnosine addition (1 mM) (Table 1).
The tightness of heme binding to guanylate cyclase varies depending on the source of the enzyme [26, 27]. The literature contains no indication of the possible existence in tissues of soluble guanylate cyclase in the heme-deficient form. We were the first to demonstrate that rat platelet guanylate cyclase could not be activated by sodium nitroprusside [11]. More detailed investigation of this phenomenon showed that, contrary to the generally accepted notion, heme is not a constituent part of the rat platelet guanylate cyclase [28]. Therefore, rat platelets cannot be used as a model to study the effect of NO and NO-generating compounds on platelet guanylate cyclase.
In this context, of special interest is that with some human platelet 105,000g supernatants the activation of guanylate cyclase by sodium nitroprusside was only 4.5-fold (instead of the usual 25-fold); this activating effect did not change after DEAE chromatography.
Based on this and some other facts, we have concluded that guanylate cyclase present in such supernatants is initially heme deficient. In this case, carnosine must not inhibit sodium nitroprusside-induced guanylate cyclase activation. We compared the ability of such supernatant (conventionally designated as one “containing the heme-deficient guanylate cyclase”) and the ability of the preparation obtained after DEAE-cellulose chromatography to be activated by sodium nitroprusside and by protoporphyrin IX. Concurrently, we investigated the influence of carnosine on these processes. The results are presented in Table 2. One can see that with sharply reduced ability of guanylate cyclase from 105,000g supernatant to be activated by sodium nitroprusside (6-fold as compared to 29-fold, Tables 1 and 2), the extent of enzyme activation by protoporphyrin IX did not differ significantly from that obtained earlier (the corresponding values are 5.1 and 6.8, Tables 1 and 2). Carnosine addition (1 mM) lowered the sodium nitroprusside-induced stimulation only insignificantly (by 20%) and had no effect on the enzyme activation by protoporphyrin IX (Table 2). Following the DEAE-chromatography of such supernatant, the guanylate cyclase preparation obtained did not differ significantly from the initial supernatant (Table 2) with regard to its ability to be activated by sodium nitroprusside and by protoporphyrin IX. These stimulatory effects did not change upon addition of 1 mM carnosine (Table 2). Thus, the unexpectedly low extent of sodium nitroprusside-induced guanylate cyclase activation in the series of experiments with 105,000g supernatant, along with the lack of the inhibitory effect of carnosine on this activation (Table 2) suggests that in this case the stimulation of guanylate cyclase is not associated with its heme.
Table 2. Effect of carnosine (1 mM) on
the activation degree of heme-deficient human platelet guanylate
cyclase by sodium nitroprusside (SNP, 100 µM) and by
protoporphyrin IX (PTP IX, 5 µM)
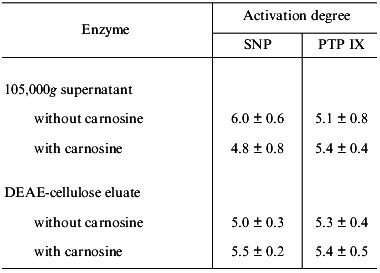
Note: Acivation degree was determined with respect to the basal
guanylate cyclase activity taken as 1. Data represent means + SD
of three independent experiments.
It is known that one distinctive feature of soluble guanylate cyclase is the presence of labile sulfhydryl groups on its surface which are easily oxidized by various endogenous and exogenous oxidants. Oxidation of these labile SH-groups stimulates the enzyme activity [29], while prolonged action of oxidants inhibits the enzyme [30]. We believe that the activating action of sodium nitroprusside is a combination of two effects: nonspecific, heme-independent increase in activity (probably caused by oxidation of labile sulfhydryl groups) and stimulation of activity due to formation of nitrosyl-heme complex between the NO group and guanylate cyclase heme. It is this process that is inhibited by carnosine.
Inhibition by carnosine of the sodium nitroprusside-induced enzyme stimulation (Table 1) could possibly be explained by the antioxidative properties of carnosine directed at the NO group formed from sodium nitroprusside. However, the data presented were interpreted as indicating that the effect of carnosine is due to its interaction with guanylate cyclase and is heme-directed. This suggestion is supported by data on the competition between carnosine and sodium nitroprusside for heme binding and on the strengthening of the inhibitory effect of carnosine (from 51 to 74%) if it was initially incubated with the enzyme before sodium nitroprusside addition. In favor of this suggestion is the loss of the inhibitory effect of carnosine in experiments with partially purified heme-deficient preparation of the enzyme (Table 1) and the virtual absence of the carnosine effect in experiments with heme-deficient guanylate cyclase (Table 2). Interaction of carnosine with guanylate cyclase heme may be realized trough the interaction of this dipeptide with the bivalent heme iron; the ability of carnosine to form chelate complexes with bivalent metal ions is well known [21-23]. Although the carnosine Fe2+ complex readily dissociates, it cannot be excluded that a result of interaction of carnosine with heme Fe2+ is that the complex formed is more stable and prevents binding of heme with the NO group. The evidence for a carnosine-heme Fe2+ interaction also comes from the finding that absorption at 428 nm in the reduced hemoglobin spectrum (Soret band) is lowered upon carnosine addition (data not shown).
After identification of EDRF as NO, which proved an endogenous activator of soluble guanylate cyclase and regulator of many physiological processes, a better insight was gained into the understanding of the physiological significance of a new intracellular signaling system: NO-soluble guanylate cyclase-cGMP. It is currently established that this signaling system is widespread in mammalian organs and tissues and plays an important role in a variety of physiological processes [31]. Enhancement of activity of this system in malignancy, sepsis, septic shock, asthma, and migraine motivates the search for inhibitors of NO-dependent guanylate cyclase activation which might serve as a basis for creation of new drugs able to selectively inhibit this activity [31]. Such inhibitors are virtually absent. Known blockers of NO-dependent guanylate cyclase activation, such as methylene blue and LY-83583, are nonspecific [31]. A recently proposed inhibitor of NO-dependent guanylate cyclase activation--ODQ (1-H[1,2,4]oxadiazolo[4,3-alpha]quinoxalin-1-one) [32]--only partially satisfies the necessary requirements [31]. In this context the ability of carnosine to inhibit the sodium nitroprusside-induced guanylate cyclase activation merits special consideration and requires the elucidation of the question as to what extent this ability of carnosine is revealed with respect to other NO donors.
We have investigated the influence of carnosine on the stimulation of human platelet guanylate cyclase by a yet unstudied furoxan derivative condensed with pyridazine di-N-oxide--4,7-dimethyl-1,2,5-oxadiazolo[3,4-d]pyridazine-1,5,6-trioxide (OPTO)--and the corresponding furazan derivative--4,7-dimethyl-1,2,5-oxadiazolo[3,4-d]pyridazine-5,6-dioxide (OPDO). The data presented in Table 3 show that both compounds at 0.01 mM concentration are potent guanylate cyclase activators, whose stimulatory effect exceeds the activation of the enzyme by sodium nitroprusside (0.1 mM). Preliminary incubation of carnosine (1 mM) with guanylate cyclase (10 min at 0°C) with subsequent addition to samples of separately introduced sodium nitroprusside (0.1 mM) and OPTO (0.01 mM) sharply inhibits the guanylate cyclase activation with these compounds (by 74 and 61%, respectively).
Table 3. Effect of carnosine (1 mM) on
human platelet guanylate cyclase (GC) activation by sodium
nitroprusside (SNP),
4,7-dimethyl-1,2,5-oxadiazolo[3,4-d]pyridazine-1,5,6-trioxide (OPTO),
and 4,7-dimethyl-1,2,5-oxadiazolo[3,4-d]pyridazine-5,6-dioxide
(OPDO)
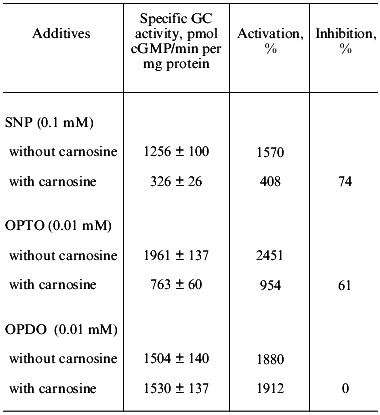
Note: Values of specific GC activity presented are means of
three-four independent experiments ± SD. Basal specific activity
is 80 ± 7 pmol cGMP/min per mg protein.
However, carnosine (1 mM) in the same conditions has no effect on the guanylate cyclase stimulation by the furazan derivative (0.01 mM OPDO). Water solutions of OPTO and OPDO are stable at pH 7.4 in the absence of thiols. Addition of cysteine or glutathione leads, in the case of OPTO, to rapid modification of SH-groups (2.2-3.5 mol per mol of compound) accompanied by generation of nitrite (the product of NO oxidation with air oxygen) (up to 0.48 mol/mol), of S-nitrosothiols (up to 0.17 mol/mol), or hydroxylamine (up to 0.44 mol/mol). OPDO incubation in the presence of thiols does not lead to formation of NO or its forms [33]. In other words, OPTO (but not OPDO) is a thiol-dependent NO donor because it contains the furoxan system. These data are in accord with those reported in the literature [34, 35] indicating the NO-donor properties of furoxan (but not furazan) derivatives. The effect of carnosine on the guanylate cyclase activation by OPTO and OPDO was compared with that of ODQ that was proposed by Moncada et al. [32] as a selective inhibitor of NO-dependent guanylate cyclase activation. We have shown that ODQ, at the optimal concentration in the sample of 3·10-7 M, supresses with equal intensity (by 40 and 47%) the guanylate cyclase activation induced by OPTO and OPDO. Carnosine (1 mM) inhibited only the guanylate cyclase activation induced by OPTO, an NO donor (Table 3). Suppression by ODQ of the 16-fold activation of the enzyme by sodium nitroprusside (0.1 mM) is, in analogous conditions, 75%. These data do not agree with the notion of ODQ as a selective inhibitor of the NO-dependent guanylate cyclase activation because OPDO is not an NO donor. Indeed, at present ODQ is considered as an inhibitor of heme-dependent enzyme activation [36].
The ability of carnosine to interact with the guanylate cyclase heme may be used for assessing the enzyme saturation with heme. The importance of such a test is due to disturbances of endogenous regulation of guanylate cyclase (in case of its heme deficiency) by the parallel lowering of the efficiency of action of organic nitrovasodilators (nitroglycerin, etc.) and by disturbances of vascular tone.
Since the presence or absence of heme in the guanylate cyclase molecule does not significantly influence the enzyme activity [20], we believe that the insignificant inhibition of activity of platelet guanylate cyclase by carnosine (Table 1) is connected with the antioxidative properties of the latter, i.e., its ability to suppress free-radical guanylate cyclase-stimulating reactions in platelets.
The most important result of this study is that carnosine appears to be a selective inhibitor of NO-dependent guanylate cyclase activation. The possibility exists of using this compound as a pharmaceutical drug for treatment of malignant tumors, sepsis, septic shock, asthma, migraine, i.e., pathologies which are associated with increasing activity of the intracellular signaling system: NO-soluble guanylate cyclase-cGMP. It should be stressed that carnosine is not toxic and is convenient for clinical application [2]. The compound is fully metabolized in the human body and is not accumulated in mammalian tissues upon prolonged use. Although carnosine has long been considered as a potential drug, its practical application is delayed, mostly because of insufficient exploration of its biological activity [2]. It may well be that the now-revealed ability of carnosine to selectively inhibit the NO-dependent activation of guanylate cyclase will attract closer attention of biochemists, pharmacologists, and clinical physicians to this compound and enlarge the area of its application as a medical preparation.
This work received financial support from the Russian Foundation for Basic Research.
REFERENCES
1.Gulewitsch, W. S., and Amiradzibi, S. (1900)
Ber. Deutschen Chem. Ges., 33,1902-1903.
2.Boldyrev, A. A. (1998) in Carnosine. Biological
Significance and Possibility of Application in Medicine [in
Russian], Moscow State University, Moscow, pp. 252-269.
3.Boldyrev, A. A., Dupin, A. M., Pindel, E. V., and
Severin, S. E. (1988) Comp. Biochem. Physiol., 89B,
245-250.
4.Dupin, A. M., Bemanadzara, M., Stvolinsky, S. L.,
Boldyrev, A. A., and Severin, S. E. (1987) Biokhimiya,
52, 783-787.
5. Chan, W. K. M, and Decker, E. A. (1994) Crit. Rev. Food Sci.
Nutr., 34, 403-426.
6.Nagai, K., and Suda, T. (1988) Methods Finding
Exp. Clin. Pharmacol., 10, 497-507.
7.Boldyrev, A. A., Dupin, A. M., Babizhaev, M. A.,
and Severin, S. E. (1987) Biochem. Int., 15,
1105-1113.
8.Glass, D. B., Frey, W. H., Carr, D. W., and
Goldberg, N. D. (1977) J. Biol. Chem., 252,
1279-1285.
9.Graff, G., Stephenson, J. B., Glass, D. B., Haddox,
M. K., and Goldberg, N. D. (1978) J. Biol. Chem., 253,
7662-7673.
10.Mittal, C. K., and Murad, F. (1977) Proc.
Natl. Acad. Sci. USA, 74, 4360-4364.
11.Severina, I. S. (1988) Biochem. Int.,
17, 265-278.
12.Miroshnichenko, V. P., Ryaposova, I. K.,
Charakhchyan, I. A., Kryukova, G. V., and Severina, I. S. (1988)
Vopr. Med. Khim., No. 2, 34-36.
13.Bussygina, O. G., and Severina, I. S. (1987)
Vopr. Med. Khim., No. 2, 24-26.
14.Palmer, R. M. J., Ashton, D. S., and Moncada, S.
(1988) Nature, 333, 664-666.
15.Palmer, R. M. J., Ferrige, A. G., and Moncada, S.
(1987) Nature, 327, 524-526.
16.Dawson, T. M., Dawson, V. L., and Snyder, S. H.
(1994) in The Neurobiology of NO and OH (Chiueh, C. C., Gilbert,
D. L., and Colton, C. A., eds.) New York Academy of Sciences, N.
Y.,Vol. 738, pp. 76-86.
17.Busse, R., Luchoff, A., and Bassenge, E. (1987)
Naunyn-Schmiedeberg Arch. Pharmacol., 336, 566-571.
18.Ignarro, L. J., Wood, K. S., and Wolin, M. S.
(1982) Proc. Natl. Acad. Sci. USA, 79, 2870-2873.
19.Ignarro, L. J., Wood, K. S., and Wolin, M. S.
(1984) Adv. Cycl. Nucl. Protein Phosphor. Res., 17,
267-274.
20.Graven, P., and de Rubertis, F. (1983)
Biochim. Biophys. Acta, 745, 310-321.
21.Viola, R. E., Hartzell, C. R., and Villafranca,
J. J. (1979) J. Inorg. Biochem., 10, 281-292.
22.Viola, R. E., Hartzell, C. R., and Villafranca,
J. J. (1979) J. Inorg. Biochem., 10, 293-307.
23.Vladimirov, Y. A. (1996) Proc. Int. Symp.
Natural Antioxidants. Molecular Mechanisms and Helth Effects
(Parcker, L., Traber, M. G., and Xin, W., eds.) AOCS Press,
Champaign, Illinois, pp. 125-144.
24.Bussygina, O. G., and Severina I. S. (1990)
Biokhimiya, 55, 1812-1818.
25.Chirkov, Y.Y., Tyshchuk, I. A., and Severina, I.
S. (1990) Vopr. Med. Khim., No. 4, 57-60.
26.Gerzer, R., Hoffmann, F., and Schultz, G. (1981)
Eur. J. Biochem., 116, 479-486.
27.Tsai, S., Adamik, R. A., Manganiello, V., and
Vaughan, M. (1988) Biochem. J., 215, 447-455.
28.Bussygina, O. G., and Severina, I. S. (1991)
Biokhimiya, 56, 487-493.
29.Braughler, J. M. (1980) Biochim. Biophys.
Acta, 616, 94-104.
30.Braughler, J. M., Mittal, S. K., and Murad, F.
(1997) J. Biol. Chem., 254, 12450-12454.
31.Hobbs, A. J. (1997) TIPS, 18,
484-491.
32.Cellek, S., Kasakov, L., and Moncada, S. (1996)
Brit. J. Pharmacol., 118, 137-140.
33.Kots, A. Ya., Grafov, M. A., Khropov, Y. V.,
Bussygina, O. G., Belushkina, N. N., Gavrilova, N. N., Ukraintsev, K.
E., Ovchinnikov, I. V., Kulikov, A. S., Makhova, N. N., Medvedeva, N.
A., Severina, I. S., and Bulargina, T. V. (1998) Proc. Int. Symp.
“Receptiveness, Intracellular Signalling”, Institute of
Cell Biophysics, Pushchino, pp. 147-149.
34.Feelish, M., Schonafinger, K., and Noack, E.
(1992) Biochem. Pharmacol., 44, 1149-1157.
35.Medana, C., Ermondi, G., Fruttero, R., di Stilo,
A., Ferreti, C., and Gasco, A. (1994) J. Med. Chem., 37,
4412-4416.
36.Schrammel, A., Behrendes, S., Schmidt, K.,
Koesling, D., and Mayer, B. (1996) Mol. Pharmacol., 50,
1-5.