REVIEW: Metal Complexes of Carnosine
E. J. Baran
Centro de Quimica Inorganica (CEQUINOR/CONICET,UNLP), Facultad de Ciencias Exactas, Universidad Nacional de La Plata, C. Correo 962, 1900-La Plata, Argentina; fax: (54) 221-4240172/4259485; E-mail: baran@dalton.quimica.unlp.edu.ar
Received November 1, 1999
The ligand properties of carnosine are analyzed. The stoichiometry, stability constants, and structural and spectroscopic characteristics of its coordination compounds with transition and representative metal cations are discussed. Mixed ligand systems containing carnosine are also presented. The biological activity of some of these metallic complexes is briefly considered.
KEY WORDS: carnosine, metal complexes, structures, stability, physicochemical properties, biological activity
Abbreviations: Bipy) alpha,alpha´-dipyridyl; Car) carnosine; CD) circular dichroism; En) ethylenediamine; ENDOR) electron nuclear double resonance; ESR) electron spin resonance; Gly-Gly) glycylglycine; His) histidine; Imid) imidazole; IR) infrared; NADH) reduced nicotinamide adenine dinucleotide; NMR) nuclear magnetic resonance; Ox) oxalate; SOD) superoxide dismutase; UV) ultraviolet; Vinyl-Imid) 1-vinylimidazole.
This review focuses on a very peculiar aspect of the chemistry and
biochemistry of carnosine, namely its ability to act as a ligand in the
generation of coordination complexes with different metallic cations.
This aspect seems to be very important in relation to the general
biochemical behavior of this dipeptide as some of its properties and
biological activities depend on the participation of certain metal
cations [1]. The presence of metal cations is also
important for the stabilization and activation of the enzyme
carnosinase.
As carnosine is the major beta-alanine source in the human body [2], its hydrolysis is of central metabolic importance. In humans, this reaction is catalyzed by two isozymes: tissue (cytosolic) carnosinase (EC 3.4.13.3) and serum carnosinase (EC 3.4.13.20) [2]. The tissue enzyme is a zinc dependent metalloprotein [2-4], and, interestingly, it can be stabilized by other divalent metal cations and the replacement of Zn(II) by other cations also maintains its activity in vitro [4-6]. The apparent efficiency of activation is given by the order Cd(II) > Mn(II) >> Zn(II) > Co(II) [4]. On the other hand, the Mn(II) activated enzyme is inhibited both by cations functioning as activators (Zn(II), Co(II)) and by non-activating ions such as Be(II) and Fe(II). The apparent non-inhibiting capacity of Cu(II) has been explained in terms of the stability of the copper complex of the substrate [4].
The structure of solid carnosine has been reported. Interestingly, its imidazole moiety exists in the N3-H tautomeric form [7], whereas in most of its complexes the N1-H tautomer is present. The “most probable” conformation in solution at pH 7.0 has been deduced from 13C- and 1H-NMR experiments. These experiments showed the predominance of the g-rotamer and folding of the beta-alanyl moiety towards the imidazole ring [8].
In its interactions with metal ions, as shown in Fig. 1, carnosine is a polydentate ligand offering six potential binding sites: the two imidazole nitrogens, one carboxylate and one amino group, and the peptide linkage. As shall be seen from the experimental results discussed below, the type of complexes formed strongly depends on the metal cation, the ligand-to-metal ratios, and the pH of the solution.
Carnosine has three groups that undergo acid-base reactions in the pH range 1-10: the carboxylic acid group, an ammonium group, and the protonated N3 imidazole group. The deprotonation of the amide group becomes important only at pH values in which the hydrolysis of the dipeptide is relevant [9] or in the presence of metal cations, whereas the dissociation of the N1 proton from the imidazole ring is normally not significant if metal ions are absent. The corresponding pK values, reported by different authors, are shown in Table 1 in which pK1, pK2,and pK3 refers respectively to the carboxyl, the N3 imidazole, and the amino groups.Fig. 1. Schematic structure of carnosine.
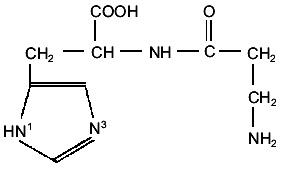
Table 1. pK values for carnosine (at
25°C)

TRANSITION METAL COMPLEXES OF CARNOSINE
All so far investigated complexes of this type belong to metals of the first transition metal series. Copper complexes have been the most investigated ones, probably due to the relatively easy approach offered by magnetic resonance spectroscopic techniques. Interest on zinc complexes has increased in recent years due to its recognized pharmacological activity.
1. Copper complexes. Initial information on the Cu(II)/carnosine interactions was obtained from pH titrations of the ligand in the absence and presence of the cation [11, 14-16]. Dobbie and Kermack [11] proposed for the first time a structure that involved metal binding with participation of the deprotonated amide N-atom. These and other authors agree in that Cu(II) is initially bound to the N1 atom of imidazole and that the terminal NH2 and the deprotonated peptide nitrogen become binding sites as the pH value is increased [14-16]. Except in the early stages of complex formation [16], the possibility of metal binding at the carboxyl group has been completely discarded, presumably because it is impossible to build up strain-free models with the participation of the four potential donor atoms of the dipeptide [17].
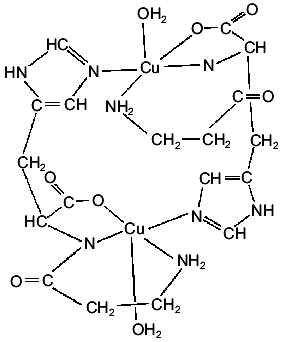
A crystalline Cu(II) complex can be obtained by interaction of Cu(OH)2 and carnosine [17] or by interaction of aqueous solutions of the ligand with copper nitrate and adjusting the pH to 8.0 with KOH [18, 19]. The generated complex is a dimer in which each Cu(II) ion presents coordination number five. As shown in Fig. 2, the four nearest ligand atoms are the terminal amino nitrogen, the amide nitrogen, a carboxylate oxygen of one of the dipeptide molecules, and the N3 nitrogen of the imidazole moiety of the second peptide molecule of the dimer. This means that each carnosine molecule is bonded to two different Cu(II) centers. The fifth ligand on each cation, completing a square-pyramidal coordination, is a water molecule [17].
The magnetic behavior and the somewhat unusual ESR spectrum of this crystalline solid suggest a very weak interdimeric coupling [19]. The ESR results, together with those obtained from the analysis of the electronic spectrum, points to the existence of a dx2-y2 ground state [19]. The IR spectrum of the crystalline complex is also in good agreement with its structural characteristics [19].Fig. 2. Schematic structure of the dimeric Cu(II) complex of carnosine.
The results of the structural analysis opens the question of the validity of the previous solution studies; however, it seems possible that the dimer exists in equilibrium with the monomer in solution, and a great number of studies were performed to clarify this aspect. First, ESR solution studies show no evidence for dimer formation at room temperature [20]. But subsequent low temperature measurements reveal the existence of a thermal equilibrium between monomeric and dimeric forms [21]. Other solution studies using NMR and ESR techniques revealed that in the pH range between 5 and 7, and at high carnosine to Cu(II) ratios, only N-atoms from the imidazole ring are involved in coordination [22]. ESR spectra obtained at 77°K [23, 24] suggested the formation of four copper-imidazole bonds through the N3 atom, a supposition confirmed independently by NMR studies [25]. Interestingly, ESR measurements at pH < 5 point to Cu(II)/carboxylate interactions [23]. When the carnosine/copper ratio approaches 1:1, the four coordination positions of Cu(II) cannot be filled by coordination to a single functional group of carnosine, and formation of the dimeric complex is favored [23]. This was confirmed from ESR measurements of a frozen (77°K) aqueous solution containing equimolecular concentrations of Cu(II) and carnosine at pH 7.2. As the ligand to metal ratio is reduced from 1000:1 to 1:1, there is a gradual transition of the ESR spectrum from the monomer to the dimer. At ratios between 10:1 and 1:1 the spectra are composed of superimposed resonances from both complexes and, thus, this is the range of relative concentrations over which the monomer is replaced by the dimer [23]. Finally, these last mentioned studies have shown that the dimeric structure may also be present in aqueous solutions at room temperature at pH 7.2 when the molar concentrations of carnosine and Cu(II) are equal, indicating that this structure is in fact more stable at ambient temperature than originally supposed [23]. Potentiometric and spectroscopic studies [26] also additionally confirmed this finding, and an FTIR spectroscopic study shows that the dimer is in equilibrium with a monomeric complex in dilute solutions but does not dissociate chemically in saturated solutions, even at elevated temperatures [27].
This last finding is of great biological relevance because it demonstrates that the dimer, although stable at physiological pH and temperature, cannot exist in living organisms because the combination of subsaturated concentrations and competing ligands will shield monomeric species with mixed ligands [27].
On the other hand, comparisons of the Cu(II)/Car with the Cu(II)/Gly-Gly and Cu(II)/Gly-Gly/Imid systems allow a wider insight into the structural characteristics of the copper complex in solution. For the second system, a model in which all the available ligating atoms are involved was finally proposed, with the equatorial positions occupied by one carboxylato O-atom, the (deprotonated) amide and amino N-atoms, and one O-atom from a water molecule, whereas the axial position is occupied by the N3 atom of the imidazole ring [18]. Structural rearrangement to form dimeric units can be easily explained with this model, by the imidazole leaving the axial position and displacing the equatorial water molecule of a neighboring complex. A new solvent water can then fill the appropriate axial binding site [18].
The crystalline complex [Cu(Gly-Gly)(Imid)(H2O)]·1.5 H2O appears as a very good model for each of the subunits which constitute the Cu(II)/Car complex [28]. The coordination sphere around the Cu(II) center is conformed by the amine and peptide N-atoms and one carboxylate O-atom of Gly-Gly, the N3-imidazole atom, and the O-atom of the water molecule. The imidazole ring is approximately coplanar with the peptide ligand, and the Cu(II) ion is displaced from the coordination square in the direction of the axial water molecule [29]. A comparison of the electrochemical behavior of this complex [28] with that of carnosine [19] suggests again the existence of a monomeric Cu(II)/Car species in solution at room temperature, a fact which in this case becomes understandable due to the very low complex concentrations used in these experiments (about 10-3 M) [19, 28].
It must also be mentioned that a great number of papers dealing with the determination of stability constants of the different copper complexes present in solution have been published during the last forty years [9, 11, 13, 14, 16, 26, 30]. Most of the reported results are contradicting or, at least, incomplete. Only with the advent of detailed speciation studies has some clarity emerged. A good and consistent set of values has been reported by Daniele et al. [13]. Their values for the predominant species are shown in Table 2. [H3L]2+ represents the fully protonated ligand, i.e., containing the carboxylate, the imidazole N3, and the terminal amino group protonated. The other used symbols can easily be derived from this one. Values reported for the species [Cu2L2H-2]0, [CuLH]2+, and [CuL]+ are also supported by another, independent study [26].
Table 2. Stability constants in the
Cu(II)/Car system (at 25°C)

The analysis of the data presented in both studies showed that the [Cu2L2H-2]0 species is very relevant, in particular at neutral pH values, whereas the monomeric [CuLH-1]0 species, probably present together with the dimer, is of minor importance. The other monomeric species, [CuL]+ and [CuLH]2+, exist in the acidic-neutral pH region, whereas the homonuclear [Cu2LH-1]2+ species is only significant when an excess of Cu(II) is present in solution. It is also suggested that a second binuclear species, [Cu2LH-2]+, may be formed in such a case [13]. Based on the potentiometric data, complemented with electronic spectroscopy measurements and the help of calculated thermodynamic data, reasonable proposals on the structure of all the species detected in solution could be made [13]. In both [Cu2LH-1]2+ and [CuLH-1]0, coordination involves the participation of the terminal amino group, the deprotonated peptide N-atom, and the N3 imidazole nitrogen, whereas in [CuL]+ only the first two mentioned ligands are involved. For the species [CuLH]2+, the ligand behaves as monodentate, interacting only through the N3 imidazole atom. Finally, for the dimeric [Cu2L2H-2]0 complex, a structure related to that found in the solid state is proposed, i.e., two equivalent Cu(II) ions, each bound to amino, dissociated peptide, and N3 imidazole nitrogens, and to a carboxylate oxygen.
Other physicochemical data for the dimeric complex have been published. Its visible electronic absorption spectrum [13, 26] compares very well with the reflectance spectrum obtained from the solid complex [19]. Detailed analyses of the ultraviolet and circular dichroism spectra, at fixed pH values were also recently reported. Intra-ligand and charge-transfer bands were assigned [31] and gave additional support to previous structural proposals [13]. The electrochemical behavior, investigated by cyclic voltamperometry using a glassy carbon disk as the working electrode, shows that the complex presents a very high redox stability [19]. The electrochemical reduction mechanism was also studied by normal and reverse pulse voltammetry at mercury electrodes. A two-electron reduction occurs through two consecutive steps with an intermediate Cu(I) species stabilized by adsorption at the Hg surface [32].
To conclude this section, it is interesting to note that a Cu(I) complex of carnosine has also been characterized [33]. Potentiometric titration data are compatible with initial formation of a 2:1 Car/Cu(I) complex, in which interaction takes place through the N1 atom of the imidazole ring. At higher pH values a 1:1 species is apparently formed in which carnosine acts as a bidentate ligand through the mentioned imidazole N atom and the N atom of the amino group, generating a 10-membered ring structure [33]. The autooxidation of this complex has also been investigated spectrophotometrically, using stopped flow techniques. Its oxidation rate is unexpectedly slow when compared, for example, with those of the Cu(I) complexes of histidine and histamine [34].
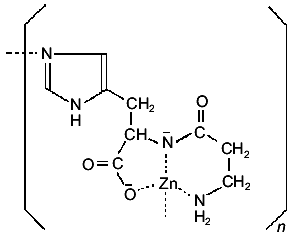
2. Zinc complexes. The Zn(II) complex of carnosine has been intensively investigated in recent years due to its important and interesting pharmacological activity. The solid complex can be prepared either by interaction of carnosine with zinc acetate in methanol solutions, in the presence of sodium methoxide [35], or by direct interaction of the ligand with a Zn(OH)2 suspension in water [36]. Its crystal structure has so far not been reported. But a detailed analysis of the solid, combining IR and NMR techniques, suggested the formation of a complex with a similar environment as that found in the solid Cu(II) complex. However, the IR and far-IR spectra of the two solids differ considerably, indicating that the zinc complex has not the simple dimeric structure of the copper complex, but presents a polymeric structure, as shown in Fig. 3 [35, 36].
Solution studies with the Zn(II)/Car system can only be performed over a very narrow pH range [37, 38]. The extent of complex formation is appreciable only around pH 6, and above pH 7.5 precipitations are observed even in the presence of excess ligand [37]. Logarithms of the stability constants (at 25°C) for the species [ZnLH]2+ and [ZnL]+ were reported to be 11.6 and 4.0, respectively [37, 38]. A slightly lower value (3.86) was reported earlier for the second species [39]. Metal to ligand interactions are surely of the same type as discussed above for the similar Cu(II) complexes, but the stability of the zinc complexes is appreciably lower. Interestingly, and as shown by detailed NMR solution studies with carnosine and other imidazole-containing peptides, zinc complexation reverses the tautomeric preference between protonation of the N-atoms of the imidazole ring as compared to the free ligands, where the N1-H tautomer is predominant [38].Fig. 3. Proposed structure of the Zn(II) complex of carnosine.
3. Cobalt complexes. Complexes of both, Co(II) and Co(III), have been reported. Solution studies with Co(II) have also some pH-range limitations [37]. Stability constants are somewhat lower as those of the respective Zn(II) complexes (logarithms of these constants for [CoLH]2+, [CoL]+, and [CoLH-1]0 are 11.48, 2.85, and -6.10, respectively [37]).
An ESR and NMR investigation of the Co(II)/Car system in aqueous solutions at pH 7.2 shows that two different complexes are formed [40]. At high ligand to metal ratios interaction occurs only through the N3 nitrogen atom of the imidazole moiety, and the number of bonded carnosine molecules (expected to be between 1 and 4) depends on how large is the ligand excess in the solution. When equimolecular concentrations are investigated, chelation takes place through three N-atoms--the terminal amino, the deprotonated peptide group, and the imidazole N1 atom--whereas the free equatorial position is probably occupied by a water molecule. This second complex is very sensitive to oxygen and is unstable toward oxidation to Co(III). It generates a binuclear oxygen carrier if stabilized by an additional carnosine, histidine, or cysteine molecule [40].
A crystalline Co(III) mixed ligand complex of stoichiometry [Co(HCar)(En)(H2O)]Cl2·H2O could be obtained by reaction between carnosine and K[Co(CO3)2(En)] in acidic aqueous solution. The structure, solved by single crystal X-ray diffractometry, showed carnosine coordination through the deprotonated peptide nitrogen, one carboxylic oxygen, and the N1 atom of the imidazole ring. The oxygen atom of the water molecule and the two ethylendiamine N-atoms completes the distorted octahedral arrangement around the metal center [41]. In basic conditions other mixed ligand Co(III) complexes, of compositions [Co(Car)(En)]Cl, Na[Co(Car)(Ox)], and K[Co(Car)(CO3)] could be obtained. In these cases, and as suggested by spectroscopic measurements, carnosine acts as a tetradentate ligand, involving the terminal amino group as an additional donor [42]. The spectroscopic behavior of all these mixed ligand Co(III) complexes has been investigated in detail by means of UV and CD spectroscopy [41, 42].
4. Vanadium complexes. Oxovanadium (IV), VO2+, is probably the most relevant vanadium species present in biological systems [43-47]. Its interaction with carnosine was first investigated by Mulks et al., by means of ENDOR techniques in frozen solutions [48]. These studies suggested a VO2+ coordination through the imidazole N-atoms of four different carnosine molecules, which occupy the equatorial positions around the oxocation and are fully equivalent to each other.
A subsequent investigation of the VO2+/Car system by electron absorption spectroscopy at high ligand-to-metal ratios and at different pH values provided a wider insight into its behavior [49]. In the pH range between 6.0 and 8.5, the interaction of the oxocation with the imidazole group of four different ligand molecules was confirmed, and the results also point to the possible presence of an axially coordinated water molecule, i.e., the stoichiometry of the complex would be [VO(Car)4(H2O)]2+, although the formation of a hydroxo complex of the type [VO(Car)4(OH)]+ cannot be totally excluded [49]. Pure imidazole behaves in a similar manner, as demonstrated also by recent ESR experiments [50]. On the other hand, spectra obtained in the pH range between 2.0 and 4.0, points to coordination through oxygen donors. The spectral characteristics suggest that the carboxylate group of carnosine molecules only occupy a part of the coordination sites, whereas the remaining ones were occupied by water molecules [49]. ESR spectra also suggest some weak interactions through carboxylate groups at pH 2.5-3.0 [51]. In alkaline medium, the spectra suggest an increase of interaction with carboxylate groups, but competition with OH- groups for the coordination sites must also be considered at higher pH values [49].
It is interesting to mention that some other examples of oxovanadium (IV) complexes that can mimic, to a certain extent, the VO2+/Car interactions were recently prepared and characterized. This is the case of a complex with 1-vinylimidazole of stoichiometry [VO(Vinyl-Imid)4Cl]Cl [52] and a histidine complex of the type [VO(His)4]SO4·2H2O [53]. In both cases, interaction to the cation occurs through the N atoms of four imidazole groups.
Recently, it has also been shown that whereas alpha-alanyl-L-histidine interacts strongly with vanadates (V), generating very stable complexes [54, 55], which may be useful mimics for the vanadate/peptide interactions, carnosine does not interact with these anions. This different behavior can probably be related to the stability of the different chelate rings produced by these interactions [54].
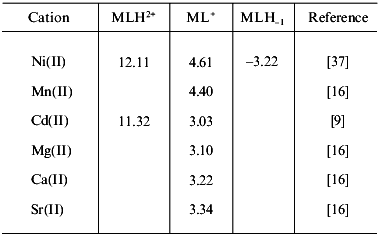
5. Other first row transition metal complexes. For Mn(II) and Ni(II), only a few solution data are available. Stability constants determined in the Ni(II)/carnosine system by Farkas et al. [37] are shown in Table 3. They are consistent with data reported by Brookes and Pettit [30] but differ from those found in older reports [14, 16]. Data reported for the Mn(II) complexes [16] probably also needs revision. One of these values is included in Table 3. Further aspects on the Mn(II)/Car interactions were also investigated by means of ESR spectroscopy during the initial studies with carnosine and carnosinase [6]. NMR studies suggested weak bonding of Mn(II) to carnosine at physiological concentration [56].
Table 3. Logarithms of the stability
constants of different metal-carnosine complexes (at 25°C)
Especially remarkable is the fact that neither Fe(II) nor Fe(III) complexes of carnosine have so far been investigated in detail, although the formation of a weakly bonded Fe(III)/Car complex has been mentioned, suggesting monodentate binding through a single nitrogen atom of the imidazole ring [56]. On the other hand, and although it has been found that carnosine is capable of inhibiting the iron-catalyzed oxidation of phosphatidylcholine liposomes and of lipid peroxides, NMR studies have conclusively shown that carnosine does not form complexes either with Fe(II) or with Fe(III) [57].
For Cr(III), solution data are so far not available. But in this case a crystalline complex containing carnosine has been characterized [58]. It is a hydroxo- and methoxo-bridged dimeric species in which carnosine acts as a tetradentate ligand, in a similar way as in the above described mixed ligand Co(III) complexes, involving chelation by the N-amino, the N-peptide, one O-carboxyl, and the imidazole N1 atoms. This type of complexes are common in the coordination chemistry of Cr(III) [59], and the present structure is stabilized by a pair of hydrogen bonds joining the coordinated N(amino) atom in each half of the dimer and the coordinated O(carboxy) atom in the other [58]. This complex can be prepared by heating trans-dichloro-tetraaqua-chromium(III) chloride with carnosine in methanolic solutions in the presence of NaOH. Dissociation of the dimer occurs after redissolution of the complex in water [58].
OTHER METAL COMPLEXES
Interaction of Ag(I) with carnosine apparently results in the precipitation of a product which was not further characterized [16]. Cd(II) complexes were initially investigated in solution [9, 16]; stability constants are shown in Table 3. Besides these complexes, some evidences exist for the generation of another species of stoichiometry Cd(Car)2 [9]. Recently, a solid Cd(II) complex was also isolated and characterized [60]. Its spectroscopic analysis suggests a dimeric structure similar to that found in the Cu(II) complex but without the coordinated water molecule.
For the alkaline-earth cations, Mg(II), Ca(II), and Sr(II), the formation of very weak 1:1 complexes at high pH values, coordinated through the N1 atom of imidazole and one carboxylate oxygen, was suggested [16]. Reported stability constants are also included in Table 3. A similar type of interaction was also postulated for the Pb(II) and Hg(II) cations [16].
The most recent investigation of the Ca(II) complex was performed by means of NMR techniques at pH 7 and showed the formation of both monomeric and dimeric complexes [8]. The carboxy and the carbonyl oxygens seems to be the binding sites in the monomeric complex, whereas in the dimer intermolecular interactions are promoted by two Ca(II) ions, one of them linking the carbonyl oxygen of one molecule with the carboxy group of the other, and the second ion bonded to the imidazole N1 atom of the first molecule and the carbonyl moiety of the second one [8].
MIXED METAL AND MIXED LIGAND COMPLEXES
Different systems of this type have been investigated and some of them seem to be of particular biological significance, especially mixed ligand complexes, which are often generated in biological fluids [61].
Only a very limited number of mixed metal complexes of carnosine have so far been investigated as they are reduced to the Cu(II)/Zn(II) and Cu(II)/Cd(II) systems [9]. In these cases, the complexes determined to be present in solution are of the types [CuM(Car)H-1]2+ and [CuM(Car)H-2]+ with M = Zn(II) or Cd(II). The logarithms of their stability constants are, respectively, 4.42 and -2.46 for the Zn(II) containing complexes and 4.47 and -3.43 for those containing Cd(II) [9].
As it has been seen in previous sections, a wide number of metal cations can be complexed by carnosine. But a relevant question is if these metal complexes can also be generated in vivo, taking into account the possible competition by other ligands present in biological fluids and tissues and their relative concentrations in these systems. In this context, the finding of Brown and Antholine, mentioned above [40], that the addition of either histidine or cysteine to a solution of the 1:1 Co(II)/Car complex, results in the formation of a more stable Co(II) species, is of special interest.
The formation of mixed ligand complexes of carnosine and a second ligand have been investigated in detail for Co(II), Ni(II), Cu(II), Zn(II), and Cd(II) [37, 62]. These studies reveal that in the systems Cu(II)/Car/Bipy and Cu(II)/Car/His a considerable degree of mixed-ligand complex formation occurs. A speciation study on these systems showed that Bipy is only partially able to suppress the formation of the [Cu2L2H-2]0 dimer, whereas in the presence of histidine the dimer is hardly formed [37]. The same behavior is observed in the presence of cysteine and glutathione [62]. It is evident that the second ligand stabilizes the monomer, by binding to the available coordination position, and this in turn pulls the chemical equilibrium toward the formation of more monomer at the expense of the dimer [62]. In the case of histidine, [Cu(Car)(His)H]+ and [Cu(Car)(His)] species are present in neutral or weakly acidic media, whereas [Cu(Car)(His)H-1]- is formed at comparatively high pH values [37]. On the other hand, interaction of Cu(II) with carnosine in the presence of bovine serum albumin has also been investigated [24, 63]. Serum albumin simultaneously binds the Cu(II) ion and carnosine to separate sites rather than forming a mixed chelate, but carnosine is still capable of competing with serum albumin for subsaturating amounts of copper [24]. It has been suggested that these equilibria may be important in relation to Wilson's disease [24, 62, 63]. Competition studies between Zn(II), carnosine, and serum albumin have also been performed [64].
The Zn(II) and Cd(II) complexes of carnosine do not appear to be affected by addition of histidine, cysteine or alpha,alpha´-dipyridyl [37, 63]. In the case of Ni(II), mixed ligand complexes may be formed with glycine, glycylglycine, and histidine, whereas complex formation is substantially less favored in the case of Co(II), for which, with histidine and in the neutral pH region, the most important species is [Co(Car)(His)]+ [37]. On the other hand, the mechanism by which histidine stabilizes the Co(II)/Car complex towards oxidation was investigated in great detail by means of NMR and vibrational spectroscopic techniques [65]. Histidine stabilizes the Co(II)/Car complex from oxidation by excluding solvent molecules from the equatorial coordination positions. As observed for other cobalt complexes [66], the interaction of the equatorial position with one solvent molecule increases the rate of oxidation of Co(II) to Co(III). Therefore, the addition of an excess of carnosine or histidine reduces the rate of oxidation of cobalt. Apparently, the first formed complex, [O2-Co(II)(Car)2] or [O2-Co(II)(Car)(His)], is slowly transformed to the more stable µ-peroxo-complex [(Car)2-Co(II)-O2-Co(II)-(Car)2] or [(Car)(His)-Co(II)-O2-Co(II)-(Car)(His)] without oxidation of the metal center.
BIOLOGICAL ACTIVITY OF SOME OF THE METAL COMPLEXES
The biological activity, or the interaction with biologically relevant systems, of some of the metallic complexes of carnosine has been investigated. In this final section of the review, brief comments on some of these results are presented.
It has been suggested that carnosine may act as a natural antioxidant in muscle and brain [1, 67], and it has also been shown that the Cu(II)/Car complex possesses SOD-like activity [68], which can also be detected in the Co(II) and Zn(II)/Car systems [69-71]. Notwithstanding, a recent in vitro study with the copper complex shows that this activity is very low [19] in comparison, for example, with that observed in different Cu(II) complexes of amino acids [72] or in Cu(II) salicylates [73]. The copper complex shows also a stereo-specific binding to DNA fibers, keeping its dinuclear structure unchanged [74]. It is also interesting to mention that, although Cu(II) and some of its complexes catalyze the oxidation of NADH by H2O2, L-histidine and carnosine inhibit this effect [75]. Carnosine also inhibits the Cu(II) catalyzed oxidation of ascorbic acid [57].
The Ni(II) complex of carnosine presents a certain SOD and catalase activity, generating OH radicals as final product of both reactions [76]. A detailed study of the SOD-like activity of different Ni(II) complexes revealed only a weak activity in the case of the carnosine complex, whereas in complexes with other tri- or tetrapeptides it is considerably higher [77].
The Zn(II) complex, currently known as Polaprezinc, has a very interesting and varied pharmacological activity. It attenuates gastric mucosal injuries [70, 78, 79], shows a potent anti-ulcer activity, and is also effective against Helicobacter pylori, a causative agent for stomach ulcers. These aspects are described in detail in the chapter by Marsukura and Tanaka in the present volume. It also presents some effects on bone metabolism [80, 81], shows wound healing properties, and appears to be effective in the treatment of hepatitis and hepatopathy. It also shows some antioxidant properties in vitro, presenting SOD-like and OH-radical scavenging activity, quenches singlet oxygen, and inhibits lipid peroxidation [70]. The complex presents also some interesting physiological properties that only very recently began to be explored systematically [64, 82]. The same can be said for the Ca(II) complex, for which a sensitizing action in cardiac and skeletal muscle has been reported [83].
In the case of the Co(II)/Car system, it has been suggested that its probable activity in vivo, and in the form of mixed complexes containing carnosine and a second ligand, may be related in some way to the monitoring of the oxygen tension in kidney. This suggestion is supported by the following facts [40]: a) both, carnosinase and Co(II) are found in relatively high concentrations in kidney, and this organ monitors the oxygen tension of blood; b) when the level of oxygen in the kidney is low, erythropoietin is released and stimulates red cell synthesis in the bone marrow; c) Co(II) bound to carnosine can generate a reversible oxygen carrier. All these findings suggest that the high concentration of the enzyme carnosinase in kidney may be involved with the ability of this organ to monitor oxygen tension. In the absence of carnosinase, carnosine, alone or with the participation of a second ligand, could be expected to chelate Co(II) and subsequently bind oxygen. This in turn would change the apparent concentration of molecular oxygen and the rate of release of erythropoietin. Thus, the high activity of carnosinase in kidney may be necessary to protect this organ from dietary and metabolic sources of carnosine.
The author acknowledges the contributions of colleagues and collaborators whose names appear in the references. Work from this laboratory is supported by the “Consejo Nacional de Investigaciones Cientificas y Tecnicas de la Republica Argentina” (CONICET), the “Comision de Investigaciones Cientificas de la Provincia de Buenos Aires” and, most recently, the “Agencia Nacional de Promocion Cientifica y Tecnologica”. The author is a member of the Research Career from CONICET.
REFERENCES
1.Babizhayev, M. A., Seguin, M. C., Gueyne, J.,
Evstigneeva, R. P., Ageyeva, E. A., and Zheltukhina, G. A. (1994)
Biochem. J., 304, 509-516.
2.Scriver, C. R., and Gibson, K. M. (1995) in The
Metabolic and Molecular Basis of Inherited Disease (Scriver, C. R.,
Beaudet, A. L., Sly, W. S., and Valle, D., eds.) Mc. Graw Hill, New
York, 7th ed., Vol. 1, pp. 1349-1368.
3.Rosenberg, A. (1960) Arch. Biochem.
Biophys., 88, 83-93.
4.Rosenberg, A. (1960) Biochim. Biophys. Acta,
45, 297-310.
5.Rosenberg, A. (1961) Ark. Kemi, 17,
25-40.
6.Rosenberg, A. (1961) Ark. Kemi, 17,
41-50.
7.Itoh, H., Yamane, T., Ashida, T., and Kakudo, M.
(1977) Acta Crystallogr., B33, 2959-2961.
8.Gaggelli, E., and Valensin, G. (1990) J. Chem.
Soc. Perkin Transact., 2, 401-406.
9.Daniele, P. C., Amico, P., and Ostacoli, G. (1982)
Inorg. Chim. Acta, 66, 65-70.
10.Pietta, P. G., and Chersi, A. (1968) Gazz.
Chim. Ital., 98, 1503-1510.
11.Dobbie, H., and Kermack, W. O. (1955) Biochem.
J., 59, 246-257.
12.Agarwal, R. P., and Perrin, D. D. (1975) J.
Chem. Soc. Dalton Transact., 268-272.
13.Daniele, P. G., Prenesti, E., Zelano, V., and
Ostacoli, G. (1993) Spectrochim. Acta, A49,
1299-1306.
14.Martin, B. E., and Edsall, J. T. (1960) J. Am.
Chem. Soc., 82, 1107-1111.
15.Martin, B. E. (1960) J. Am. Chem. Soc.,
82, 6053-6054.
16.Lenz, G. R., and Martell, A. E. (1964)
Biochemistry, 3, 750-753.
17.Freeman, H. C., and Szymanski, J. T. (1967)
Acta Crystallogr., 22, 406-417.
18.Viola, R. E., Hartzell, C. R., and Villafranca,
J. J. (1979) J. Inorg. Biochem., 10, 293-307.
19.Baran, E. J., Parajon-Costa, B. S., Rojo, T.,
Saez-Puche, R., Fernandez, F., Totaro, R. M., Apella, M. C.,
Etcheverry, S. B., and Torre, M. H. (1995) J. Inorg. Biochem.,
58, 279-289.
20.Boas, J. F., Pilbrow, J. R., Hartzell, C. R., and
Smith, T. D. (1969) J. Chem. Soc., A, 572-577.
21.Viola, R. E., Hartzell, C. R., and Villafranca,
J. J. (1976) J. Coord. Chem., 6, 119-121.
22.Viola, R. E., Hartzell, C. R., and Villafranca,
J. J. (1979) J. Inorg. Biochem., 10, 281-292.
23.Brown, C. E., Antholine,W. E., and Froncisz, W.
(1980) J. Chem. Soc. Dalton Transact., 590-596.
24.Brown, C. E. (1981) J. Theor. Biol.,
88, 245-256.
25.Gaggelli, E., Basosi, R., Pogni, R., and
Valensin, G. (1988) J. Inorg. Biochem., 32, 7-12.
26.Sovago, I., Farkas, E., and Gergely, A. (1982)
J. Chem. Soc. Dalton Transact., 2159-2163.
27.Brown, C. E., Vidrine, D. W., Julian, R. L., and
Froncisz, W. (1982) J. Chem. Soc. Dalton Transact.,
2371-2377.
28.Baran, E. J., Parajon-Costa, S. B., Ferrer, E.
G., Lezama, L., and Rojo, T. (1996) J. Inorg. Biochem.,
63, 19-27.
29.Bell, J. D., Freeman, H. C., Wood, A. M., Driver,
R., and Walker, W. R. (1969) Chem. Commun., 1441-1443.
30.Brookes, G., and Pettit, L. H. (1975) J. Chem.
Soc. Dalton Transact., 2112-2117.
31.Daniele, P. G., Prenesti, E., and Ostacoli, G.
(1996) J. Chem. Soc. Dalton Transact., 3269-3275.
32.Bilewicz, R. (1989) J. Electroanal. Chem.
Interfacial Electrochem., 267, 231-241.
33.Kaden, Th., and Zuberbühler, A. (1966)
Helv. Chim. Acta, 49, 2189-2195.
34.Zuberbühler, A. (1970) Helv. Chim.
Acta, 53, 669-675.
35.Matsukura, T., Takahashi, T., Nishimura, Y.,
Ohtani, T., Sawada, M., and Shibata, K. (1990) Chem. Pharm.
Bull., 38, 3140-3146.
36.Förster, M., and Vahrenkamp, H. (1995)
Chem. Ber., 128, 541-550.
37.Farkas, E., Sovago, I., and Gergely, A. (1983)
J. Chem. Soc. Dalton Transact., 1545-1551.
38.Gajda, T., Henry, B., and Delpuech, J. J. (1994)
J. Chem. Soc. Perkin Transact., 2, 157-164.
39.Agarwal, R. P., and Perrin, D. D. (1975) J.
Chem. Soc. Daltons Transact., 1045-1048.
40.Brown, C. E., and Antholine, W. E. (1979)
Biochem. Biophys. Res. Commun., 88, 529-536.
41.Okamoto, K., Yasui, T., Kawaguchi, H., Ama, T.,
and Hidaka, J. (1988) Chem. Lett., 335-338.
42.Ama, T., Kawaguchi, H., Uchijima, M., Kione, N.,
and Yasui, T. (1989) Bull. Chem. Soc. Jpn., 62,
3463-3468.
43.Rehder, D. (1992) Biometals, 5,
3-12.
44.Slebodnick, C., Hamstra, B. J., and Pecoraro, V.
L. (1997) Struct. Bonding, 89, 51-108.
45.Chasteen, D. N. (ed.) (1990) Vanadium in
Biological Systems, Kluwer Academic Publishers, Dordrecht.
46.Baran, E. J. (1997) Bol. Soc. Chil. Quim.,
42, 247-256.
47.Baran, E. J. (2000) J. Inorg. Biochem.,
80, 1-10.
48.Mulks, C. F., Kirste, B., and van Willigen, H.
(1982) J. Am. Chem. Soc., 104, 5906-5911.
49.Ferrer, E. G., Williams, P. A. M., and Baran, E.
J. (1996) Biol. Trace Elem. Res., 55, 79-87.
50.Sanna, D., Micera, G., Strinna Erre, L., Molinu,
M. C., and Garriba, E. (1996) J. Chem. Res. (S), 40-41.
51.Sanna, D., Micera, G., and Molinu, M. G. (1996)
J. Chem. Res. (S), 42-43.
52.Calviou, L. J., Arber, J. M., Collison, D.,
Garner, C. D., and Clegg, W. (1992) J. Chem. Soc. Chem. Commun.,
654-656.
53.Williams, P. A. M., and Baran, E. J. (1997)
Transit. Metal Chem., 22, 589-591.
54.Fritzsche, M., Vergopoulus, V., and Rehder, D.
(1993) Inorg. Chim. Acta, 211, 11-16.
55.Elvingson, K., Fritzsche, M., Rehder, D., and
Pettersson, L. (1994) Acta Chem. Scand., 48, 878-885.
56.Brown, C. E., Margolis, F. L., Williams, T. H.,
Pitcher, R. G., and Elgar, G. (1977) Neurochem. Res., 2,
555-579.
57.Decker, E. A., Crum, A. D., and Calvert, J. T.
(1992) J. Agric. Food Chem., 40, 756-759.
58.Murdoch, Ch. M., Cooper, M. K., Hambley, T. W.,
Hunter, W. N., and Freeman, H. C. (1986) J. Chem. Soc. Chem.
Commun., 1329-1331.
59.Cotton, F. A., and Wilkinson, G. (1980)
Advanced Inorganic Chemistry, 4th. ed., J. Wiley, N. Y., pp.
728-729.
60.Sarkar, A. R., and Sarkar, M. (1997) J. Chem.
Res. (S), 304-305.
61.Sigel, H. (ed.) (1974) Metal Ions in
Biological Systems, Vol. 2, Mixed-Ligand Complexes, Marcel Dekker,
N. Y.
62.Brown, C. E., and Antholine, W. E. (1979) J.
Chem. Phys., 83, 3314-3319.
63.Brown, C. E., and Antholine, W. E. (1980)
Biochem. Biophys. Res. Commun., 92, 470-477.
64.O'Dowd, A., O'Dowd, J. J., and Miller, D. J.
(1996) J. Physiol., 495, 535-543.
65.Brown, C. E., Vidrine, D. W., Czernuszewicz, R.,
and Nakamoto, K. (1982) J. Inorg. Biochem., 17,
247-258.
66.Vogt, L. H., Jr. (1963) Chem. Rev.,
63, 269-277.
67.Kohen, R., Yamamoto, Y., Cundy, K. C., and Ames,
B. N. (1988) Proc. Natl. Acad. Sci. USA, 85,
3175-3179.
68.Kohen, R., Misgav, R., and Ginsburg, I. (1991)
Free Rad. Res. Commun., 12/13, 179-185.
69.Hartman, Z., and Hartman, P. E. (1992) Chem.
Biol. Interact., 84, 153-168.
70.Yoshikawa, T., Naito, Y., Tanigawa, T., Yoneta,
T., and Kondo, M. (1991) Biochim. Biophys. Acta, 1115,
15-22.
71.Gulyaeva, N. V. (1987) Biokhimiya,
52, 1216-1220.
72.Totaro, R. M., Apella, M. C., Torre, M. H.,
Friet, E., Viera, I., Kremer, E., and Baran, E. J. (1993) Acta Farm.
Bonaerense, 12, 73-78.
73.Weser, U., Richter, Ch., Wendel, A., and Younes,
M. (1978) Bioinorg. Chem., 8, 201-213.
74.Chikira, M., and Mizukami, Y. (1991) Chem.
Lett., 189-190.
75.Chan, P. C., and Kesner, L. (1980) Biol. Trace
Elem. Res., 2, 159-174.
76.Cotelle, N., Tremolieres, E., Bernier, J. L.,
Catteau, J. P., and Henichart, J. P. (1992) J. Inorg. Biochem.,
46, 7-15.
77.Ueda, J., Ozawa, N., Miyazaki, M., and Fujiwara,
Y. (1993) Inorg. Chim. Acta, 214, 29-32.
78.Cho, C. H. (1992) Drug Developm. Res.,
27, 61-65.
79.Furuta, S., Toyama, S., Miwa, M., Itabashi, T.,
Sano, H., and Yoneta, T. (1995) Jpn. J. Pharmacol., 67,
271-278.
80.Segawa, Y., Tsuzuike, N., Itokazu, Y., Tagashira,
E., and Yamaguchi, M. (1992) Res. Exp. Med., 192,
317-322.
81.Yamaguchi, M., and Kishi, S. (1994) Biol.
Pharm. Bull., 17, 522-526.
82.O'Dowd, A., O'Dowd, J. J., and Miller, D. J.
(1995) J. Physiol., 483, 112P.
83.Miller, D. J., Lamont, C., and O'Dowd, J. J.
(1993) in Calcium Sensitizing (Allen, D. G., and Lee, J., eds.)
Oxford University Press, Oxford, pp. 117-139.