REVIEW: Discrimination between Apoptosis and Necrosis of Neurons under Oxidative Stress
A. A. Boldyrev
International Biotechnological Center and Center of Molecular Medicine, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (7-095) 939-1398; E-mail: aab@genebee.msu.su
Received March 30, 2000
Flow cytometric studies of rat cerebellum neurons are described under conditions inducing cell death. Using a double labeling technique, discrimination between apoptotic and necrotic cell transformations is demonstrated. Histidine containing neuropeptides were found to regulate cell stability, taking part in selection of the preferable way of neuronal death under oxidative stress.
KEY WORDS: neurons, ischemia, reactive oxygen species, glutamate, apoptosis, necrosis, flow cytometry, histidine containing neuropeptides, carnosine, homocarnosine
In the current scientific literature there is no common understanding of the role of reactive oxygen species (ROS) in cell metabolism [1]. Until recently, the useful role of ROS in animals was thought to be restricted to the bactericidal function which superoxide anion of oxygen plays after its generation by NADPH oxidase of the plasma membrane. Very recently, ROS effects were found to be directed toward the cell genome, resulting in protein expression [2]. One could suggest that cellular antioxidant system together with reactions which are illustrated in Table 1 regulates the efficiency of cell metabolism via protein synthesis.
Table 1. Analysis of the participation of
ROS in cellular metabolism [3-9, 11-13]

*Example of participation of ROS in signal transduction mechanism,
when NO forms in one cell and activation of cGMP synthesis occurs
in another.
A dual function of ROS in neuronal function was discussed recently [9, 10]. ROS participate in routine metabolic reactions as indicated by intracellular generation of free radicals in response to activation of glutamate receptors in cultured neurons [4]. The functional role of this signal is still obscure; however, it does not relate to cell death signal and its amplitude is proportional to the neuronal activation [3-5]. It was found that cytoplasmic lipid turnover rather than the mitochondrial respiratory chain is responsible for this ROS generation. In any case, rotenone inhibiting mitochondrial Complex I does not affect the ROS signal, whereas indomethacin, 4-methylpyrazole, and nialamide (cycloxygenase, brain P450, and MAO B inhibitors) suppress ROS production [5, 10].
Studies of the participation of ROS in neuronal function were limited by the absence of appropriate neurochemical methods because neuronal tissue is heterogeneous in its structure and classical biochemical studies provide only integral information not related to specific cellular stimuli. Recently, new approaches have been developed allowing neuronal cells to be obtained in the intact individual state, keeping this intactness long enough for measurements. Using flow cytometry as a tool to analyze cell response provides new information on cell activation. In this review, several experiments will be described which use these techniques.
FLOW CYTOMETRIC CHARACTERIZATION OF NEURONS
Cerebellum of young (10-15-day-old) rats and mice is a suitable source to obtain homogenous suspensions of individual neurons after mild treatment of brain slices with proteolytic enzymes. The method of preparation has been described in detail recently [5, 14, 15]. The neurons obtained are about 10 µm in size and the cell population is rather homogenous. Normally, 85-95% of the cells in suspension are intact, and they remain unbroken for 4-5 h after preparation, which is long enough for careful flow cytometric analysis.
Characterization of individual cells using flow cytometry is widely accepted in relation to blood cells; fluorescence-labeled antibodies can be used to differentiate lymphocytes by the number of specific receptors on their membranes. Preparation of intact neurons in individual state makes possible similar analysis using specific labels for membrane potential, intracellular level of ROS or ionized calcium ions, several parameters being measured in one sample. In such case, each neuron can be characterized by several parameters [16].
This approach is especially important for neuronal studies because it allows estimation of the role in metabolic changes of special cellular receptors. Cerebellum cells, where glutamatergic mechanisms of excitation are widely represented, may be used for separate study of more then 10 different receptors transducing and modifying different signals (kainate receptors, NMDA- and AMPA-receptors, as well as several metabotropic receptors--for each of these more or less specific agonists and antagonists are known).
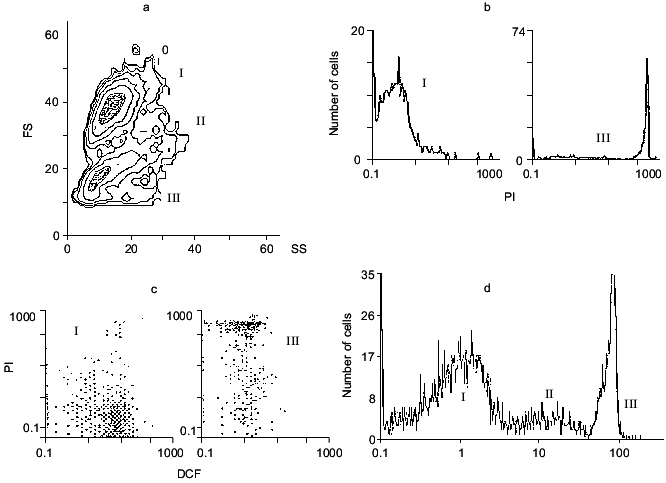
When light scattering of a neuronal suspension is measured, two main populations of the cells are found (Fig. 1a). Their identification using the nucleic acid labeling compound propidium iodide (PI) shows that only a small part of the cells is penetrable for this agent, which accumulates in cells with damaged membrane and interacts with nucleic acid. Thus, the cell population labeled by PI represents dead cells with damaged outer membranes.
The main portion of the cells does not interact with PI, and their fluorescence in the presence of PI is negligible (Fig. 1b). These cells are probably intact. Actually, they efficiently accumulate another dye, 2´,7´-dichlorofluorescein diacetate (DCF-DA), via a membrane bound esterase. In this way the dye is converted into 2´,7´-dichlorofluorescein and accumulated in the intracellular space of intact cells. The dye can interact with hydrogen peroxide, forming 2´,7´-dichlorofluorescin (DCF), whose fluorescent signal can be easily measured. Thus, the total intracellular ROS level can be estimated [11, 17]. One restriction of this method is that the reaction:Fig. 1. Analysis of a neuronal suspension from rat cerebellum using flow cytometry: a) distribution of cells by scattering factor. Ordinate, forward scattering (FS); abscissa, side scattering (SS); b) PI fluorescence of populations I and III; c) DCF and PI fluorescence for populations I and III; d) mean fluorescence of the cells in populations I, II (DCF fluorescence), and III (PI fluorescence).
2´,7´- dichlorofluorescein <--> 2´,7´-dichlorofluorescin,
is accelerated by protons, thus DCF fluorescence will depend on the acidity of the medium. This requires special control of intracellular pH. In the case when activation of specific receptors is studied, a large change of pH inside the cells can be excluded; thus, change in DCF fluorescence reflects mainly the intracellular accumulation of ROS [17, 18].
Preliminary incubation of a neuronal suspension with DCF-DA greatly increases cell fluorescence and differentiates the intact from the damaged cells: the former are labeled with DCF and the latter with PI (Fig. 1c).
There is another, very small population of the cells seen in Fig. 1a (II) in an intermediate position between intact (I) and broken (III) cells. These are not broken because they are rather shrunken, not swollen and not labeled with PI. They accumulate DCF and have higher ROS level (Fig. 1d), so they are living. From this population, apoptotic cells are probably derived, as can be demonstrated using an apoptotic detection kit (see below).
To illustrate a possible advantage of the method, we will describe one set off experimental data. Freshly prepared neurons after loading with DCF-DA are incubated with the agonist of the glutamate receptor, then washed free of its excess and the fluorescence of single neurons (not of the whole suspension) is measured using the flow cytometry technique. From the sample containing (3-5).105 cells, 10,000 cells are usually analyzed. The data can be presented as contour or dot plots (Fig. 1, a and c) or as a mean fluorescence signal characterizing the cell population under analysis (Fig. 1d).
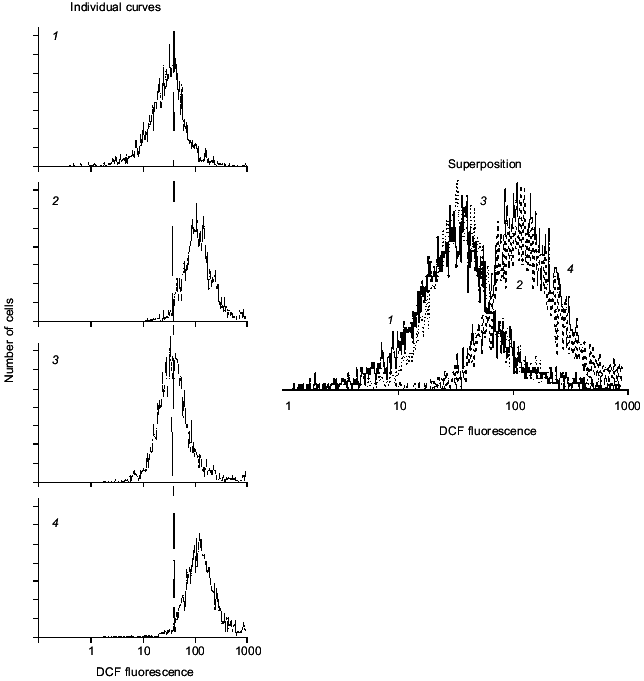
Short-term (10-30 min) incubation of neurons with agonists of glutamate receptors increases intracellular DCF fluorescence, consistent with metabolic activation of the cells, specificity of cellular response being under control of subsequent antagonists. Figure 2 shows that mean DCF fluorescence is increased in response to kainate (KA) and decreased by DNQX (antagonist of KA-receptors) but not by MK-801 (antagonist of NMDA-receptors). In these experiments, short-term activation of glutamate receptors of both KA- and NMDA-subtypes did not result in cell death.
Fig. 2. Cell response to KA and some antagonists. Curves: 1) initial DCF fluorescence; 2) 30 min after exposure of cells to 2 mM KA; 3) the same with 50 µM DNQX (KA antagonist); 4) the same with 50 µM MK-801 (NMDA antagonist).
CELLULAR RESPONSE TO UNFAVORABLE FACTORS
Long-term activation of glutamate receptors taking place during functional disordering of neurons induces cell death. This process is described as the excitotoxic effect of glutamate. This mechanism is involved in several neurodegenerative diseases (Parkinson's and Alzheimer's diseases) as well as in acute disordering of the brain blood supply (stroke) or aging.
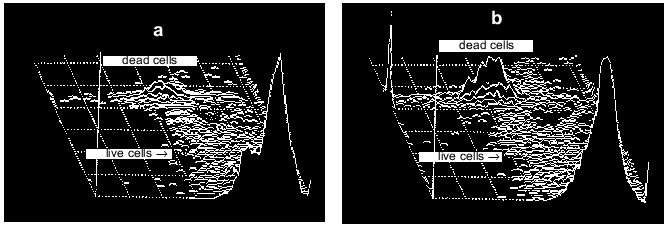
In experiments like those described above, massive cell death can be induced by exposure of cells to millimolar concentrations of glutamate or its analogs KA or NMDA. After such exposure, the number of neurons labeled with PI increases proportionally with a rise in ROS [11, 19]. Disordering of brain blood supply (also resulting in cell death) can be mimicked by exposure of the cells to hypoxic conditions in the absence of glucose in the incubation medium (hypoglycemia) with subsequent re-oxygenation [20]. Under these conditions neither ROS production nor measurable cell death was found after 15 min hypoxia, while both factors were noted after a subsequent 30 min re-oxygenation (Fig. 3). Figure 4 illustrates the cell death after ischemic injury in vitro. A sharp increase in dead cells is seen when ischemic factors are combined with the presence of excitotoxic compounds like NMDA [20].
Fig. 3. Intracellular ROS level under hypoxic conditions (a) with subsequent re-oxygenation (b). a: 1) Control; 2, 3) 5 and 15 min hypoxia, respectively. b: 1) Control; 2) 15 min hypoxia followed by 60 min re-oxygenation. It is seen that mean DCF fluorescence does not shift under hypoxia alone, but does shift to the right demonstrating accumulation of ROS after re-oxygenation.
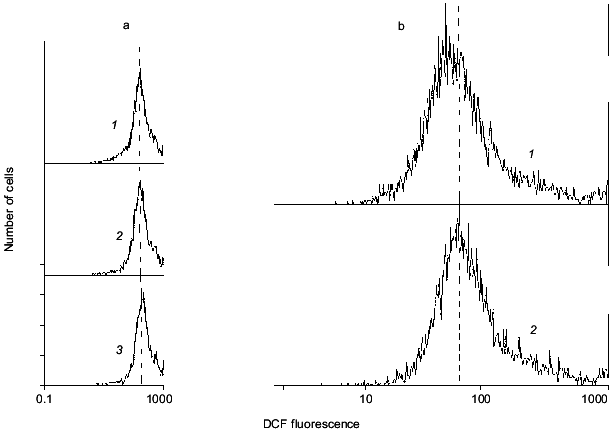
Fig. 4. Distribution of neurons between two populations (live and dead cells) before (a) and after (b) imitation of ischemic attack. Ischemic injury was made by 15 min hypoxia (+ hypoglycemia) followed by 60 min re-oxygenation.
DISCRIMINATION BETWEEN DIFFERENT TYPES OF CELL DEATH (APOPTOSIS VERSUS NECROSIS)
It is important to test the type of cell death under unfavorable conditions. Necrosis is known to differ from apoptosis in principle. The latter is suppressed by protein synthesis inhibitors and requires a cellular pool of energy. This indicates the participation of special genes and the involvement of new proteins in the apoptotic process. Therefore, apoptosis is a programmed cell death, and information directing this program is stored in the cellular genome. Apoptosis is a tool of ontogenic transformation of tissues (for example, during insect development) or a cellular response to unfavorable factors (during neurodegenerative diseases).
From a large body of data describing the apoptotic transformation, it is seen that this phenomenon includes several alternative mechanisms. However, the basic difference between apoptosis and necrosis is that nearly all steps of apoptosis (until formation of the apoptotic bodies and their phagocytosis) the cell membrane remains intact. Decision about the preferable way of cell death follows from estimation of a number of factors, the damaging effect of ROS being one of them. Thus, not only the level of intracellular ROS but also the loss of ability to prevent their damaging action results in the necessity of cellular death.
Choosing of type of cellular death is important for planning the strategy of protection of tissue against oxidative stress. For this and many other reasons, methods for characterizing the earliest steps of cellular death are extremely important. Use of flow cytometry together with specific labels reacting on apoptotic events promises success in these studies.
One of the early events of apoptosis is oxidative disordering of the membrane-cytoskeleton interaction normally provided by cytoskeletal proteins called annexins. As a result of this disordering, diffusional motion of phosphatidylserine in the membrane bilayer is increased. This phospholipid under normal conditions is exclusively concentrated in the inner side of the membrane; after oxidative disordering it undergoes “flip-flop” and appears also in the outer side of the membrane. Therefore, addition to the cell suspension of annexins labeled with fluorescein isothiocyanate (FITC) label those cells that contain phosphatidylserine in the outer side of the membrane. This method is suitable for identifying cells preparing for apoptotic transformation [21-23]. Usually for such experiments FITC-labeled annexin-V is used, which in combination with PI makes it possible to discriminate apoptotic from necrotic neurons. An example of such discrimination is presented in Fig. 5. When using these two labels independently, annexin-V positive cells are located in the 4th quadrant, PI positive in the 1st quadrant, and cells labeled with neither of these compounds in the 3rd quadrant. When using PI and annexin-V simultaneously, the same discrimination is carried out, but in addition some PI positive cells move from the annexin-V negative quadrant 1 to the annexin-V positive quadrant 2, thus demonstrating affinity to both labels simultaneously. This means that the cells are under heavy necrosis and their membranes are penetrable for both low-molecular-weight PI and high-molecular-weight annexin-V. Another words, cells in the 1st quadrant correspond to the beginning step of necrosis (light necrosis) and those in 2nd quadrant to heavy necrosis.
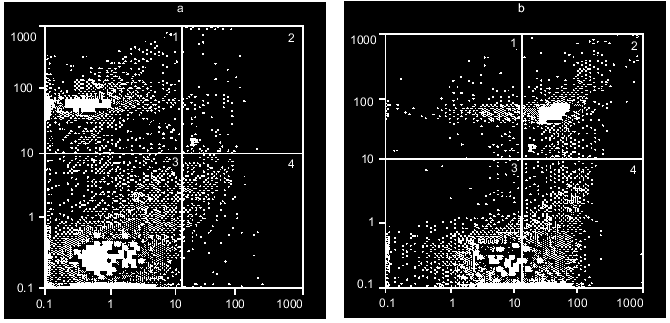
Figure 6 shows an example of the excitotoxic effect of 2 mM NMDA on neurons; it is seen that 3-h exposure results in both apoptotic and necrotic cell death. Moreover, light necrosis is replaced by heavy necrosis. Lower concentrations of this and other glutamate agonists induce apoptosis with no increase in the population of necrotic cells. The earliest stages of apoptosis appear 1-3 h after beginning of the incubation [24].Fig. 5. Discrimination of neurons using double staining with PI and FITC-labeled annexin-V. Distribution of cells in the absence of dyes added (a), with PI (b), with FITC-labeled annexin-V (c) or both dyes added simultaneously (d).
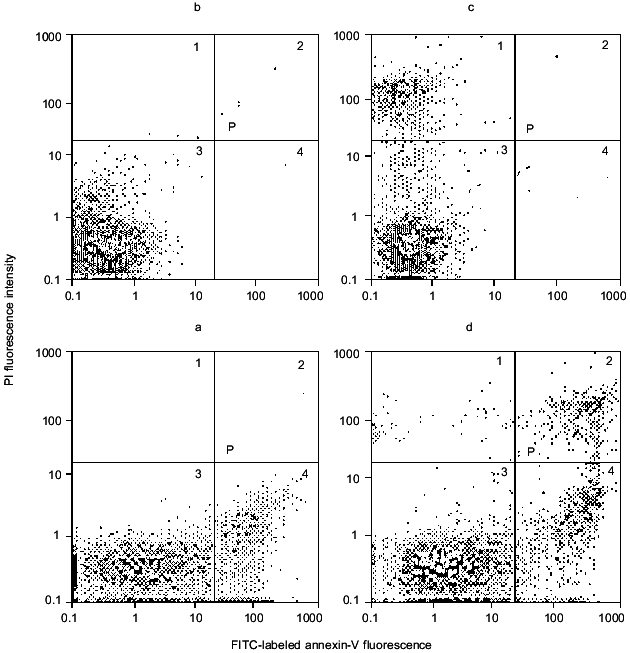
Fig. 6. Distribution of cerebellar granule cells between populations of intact, apoptotic, and necrotic cells before (a) and after (b) 3 h incubation with 2 mM NMDA.
EFFECT OF BIOLOGICALLY ACTIVE COMPOUNDS ON THE VIABILITY OF NEURONS
Many biologically active compounds are known to increase the resistance of cells against oxidative stress. As a rule, they are antioxidants, like carnosine and related compounds [11]. Actually, carnosine can protect neurons against ROS induced damage [6], thus an attempt to use carnosine to prevent apoptosis seems reasonable.
We have characterized the effect of carnosine and its several natural derivatives on the viability of rat cerebellum neurons exposed to the excitotoxic compounds NMDA and KA. Neurons were incubated for 1 h with 10 mM carnosine, anserine, N-acetylcarnosine, or homocarnosine, then the suspension was diluted 10 times and exposed to the above-mentioned glutamate agonists inducing excitotoxic damage of cells. After different time of exposure the measurement of apoptotic, necrotic, and intact neurons was made as described above and compared with the same values in the control sample. Additionally, a separate cell sample pre-loaded with DCF-DA was used to measure intracellular ROS level. All of the tested compounds were found to decrease DCF fluorescence to the same level in relation with their well-known antioxidant properties [11]. These data suggest that all they will protect neurons from apoptosis with similar efficiency. However, the degree of protection found was far from this expectation (Table 2).
Table 2. Quantification of apoptosis and
necrosis of rat cerebellum neurons exposed to the excitotoxic ligands
NMDA and KA (% distribution between different subpopulations of neurons
is presented)
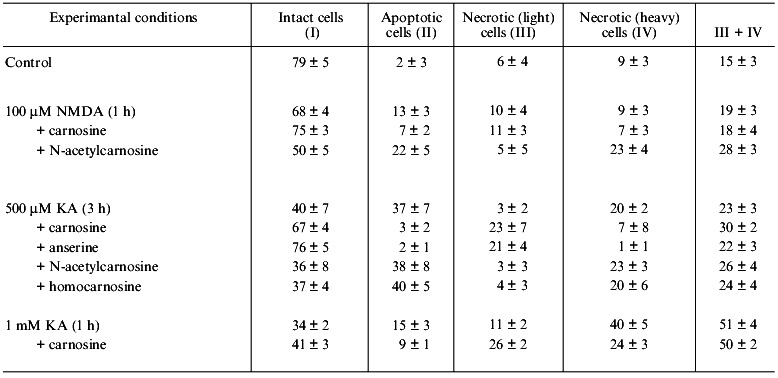
Table 2 shows that exposure of neurons to a low concentration of excitotoxic ligand results in their activation, and increase in concentration of the stimulus also induces necrosis. In these experiments carnosine and anserine both prevented apoptosis of neurons and the transformation of light into heavy necrosis. However, neither homocarnosine nor N-acetylcarnosine prevented the apoptosis of neurons, the latter actually stimulating the experimental neuronal death.
These observations indicate additional effects of carnosine and anserine which are not directly related to their ROS quenching ability. Independent data describing non-antioxidant action of carnosine on the cell cycle [25] as well as its ability to induce expression of specific proteins when added to cultured fibroblasts [26] and to prevent protein glycation [27] demonstrate that carnosine is able to modify cell metabolism in a broader way than antioxidants usually do. It is likely that some of these properties of carnosine (and its N1-methylated derivative anserine) may block the reactions of programmed cell death to prevent apoptosis. Other carnosine derivatives do not possess such regulatory abilities as shown earlier when protection of cellular proteins from glycation was tested; neither N-acetylcarnosine nor homocarnosine were able to prevent the glycation of muscle actin [28] or liver histones [22].
Moreover, the data of Table 2 demonstrate that several carnosine-related compounds can increase the probability of apoptotic transformation of neurons induced by excitotoxic compounds. The molecular mechanisms of this effect are obscure. However, in vertebrate tissues including brain, carnosine is under continuous turnover, resulting in the formation of several derivatives, some of which can prevent (anserine) and some stimulate (N-acetylcarnosine) neuronal apoptosis induced by excitotoxic compounds. This fact may reflect the existence of additional mechanisms both regulating cell viability and stimulating apoptotic death under unfavorable conditions. Such mechanism can additionally regulate flexible cell response to environmental stimuli, which is especially important for mature brain neurons, which die preferably via apoptosis (rather than necrosis) after being exposed to serious ischemic injury as from stroke.
Thus, apoptosis is one of the most intriguing and mechanistically complex natural processes of cell death. Methodology for studying its earliest stages is limited by lack of proper approaches. For this reason, the method described here seems to be useful for studies of the initiation of apoptotic processes.
It is my great pleasure to cordially thank David Carpenter (School of Public Health, State University of New York at Albany, NY, USA), in whose laboratory this study was started with financial support from the Fulbright Foundation.
REFERENCES
1.Skulachev, V. P. (1999) Biochemistry
(Moscow), 64, 1418-1426.
2.Peskin, A. V. (1997) Biochemistry (Moscow),
62, 1341-1347.
3.Boldyrev, A. A. (2000) Byul. Eksp. Biol.
Med., in press.
4.Smithies, J. (1999) Eur. J. Pharmacol.,
370, 1-7.
5.Boldyrev, A., Carpenter, D., Huentelman, M.,
Peters, C., and Johnson, P. (1999) Biochem. Biophys. Res.
Commun., 256, 320-324.
6.Boldyrev, A., Johnson, P., Wei, Y., Tan, Y., and
Carpenter, D. (1999) Neurosci. Lett., 263, 169-172.
7.Hanbauer, I., Cox, G. W., and Wink, D. A. (1994)
Ann. N.Y. Acad. Sci., 738, 173-180.
8.Sen, Ch., and Packer, L. (1996) FASEB J.,
10, 709-720.
9.Halliwell, B. (1997) Nutr. Rev., 55,
S44-S51.
10.Hancock, J., and Neill, S. (1999)
Biochemist, No. 8, 33-37.
11.Boldyrev, A. A. (1999) Carnosine and
Protection of Tissues Against Oxidative Stress [in Russian], Moscow
University Publishing House-Dialog, Moscow.
12.Boldyrev, A. A. (1995) Biochemistry
(Moscow), 60, 1173-1178.
13.Boldyrev, A., Yuneva, M., Song, R., Lawrence, D.,
and Carpenter, D. (1999) Abst. 29th Meet. on Neurochemistry,
Vol. 2, Miami, USA, p. 1193.
14.Oyama, Y., Carpenter, D. O., Chikahisa, L., and
Okazaki, E. (1996) Brain Res., 728, 121-124.
15.Sureda, F. X., Camins, A., Trullas, R., Camarasa,
J., and Escubedo, E. (1996) Brain Res., 723, 110-114.
16.Boldyrev, A., Johnson, P., and Carpenter, D.
(2000) Trends Biochem. Physiol., in press.
17.Maftah, A., Huet, O., Gallet, P.-F., and
Ratinaud, M.-H. (1993) Biol. Cell, 78, 85-93.
18.Gunarsekar, P. G., Kanthasamy, A. G., Borowitz,
J. L., and Isom, G. E. (1995) J. Neurosci. Meth., 61,
15-21.
19.Chikahisa, L., Oyama, Y., Okazaki, E., and Noda,
K. (1996) Jap. J. Pharmacol., 71, 299-305.
20.Boldyrev, A., Djatlov, V., Song, R., Lawrence,
D., and Carpenter, D. (2000) Cell Molec. Neurobiol., in
press.
21.Fabisjak, J., Kagan, V. E., Ritov, V. B.,
Johnson, D., and Lazo, J. (1997) Am. J. Physiol., 272,
C675-C684.
22.Boldyrev, A. A., Carpenter, D. O., Kovalenko, Z.,
Kuleva, N., Lawrence, D., and Song, R. (1998) in Maturation
Phenomenon in Cerebral Ischemia. Defensive Mechanisms versus Apoptosis,
Neuronal Recovery and Protection in Cerebral Infarction (Ito, U.,
Fieschi, C., Orzi, E., Kuroiwa, T., and Klatzo, J., eds.) Springer,
Pozzili, Italy, pp. 325-326.
23.Boldyrev, A., Song, R., Lawrence, D., and
Carpenter, D. (1999) Neuroscience, 94, 571-577.
24.Boldyrev, A., Lawrence, D., and Carpenter, D.
(1999) in Peptide Science - Present and Future, Kluwer Academic
Publishing, Dordrecht-Boston-London, pp. 413-415.
25.Gille, J. J., Pasman, P., van Berkel, C. G., and
Joenje, H. (1991) Mutagenesis, 6, 313-318.
26.Ikeda, D., Wada, S., Yoneda, C., Azbe, H., and
Ochi, H.(1999) Cell Struct. Funct., 24, 79-87.
27.Hipkiss, A. (2000) Biochemistry (Moscow),
65, 771-778.
28.Kuleva, N. V., and Kovalenko, Z. S. (1997)
Biochemistry (Moscow), 62, 1119-1123.