REVIEW: A Role for Carnosine in Cellular Maintenance
R. Holliday* and G. A. McFarland
CSIRO Division of Molecular Science, North Ryde, Sydney, N. S. W. 1670, Australia; fax: 61-2-9490-5010; E-mail: gail.mcfarland@molsci.csiro.au or RandL.Holliday@bigpond.com* To whom correspondence should be addressed.
Received November 22, 1999
The dipeptide L-carnosine has beneficial effects on cultured human fibroblasts. Physiological concentrations in standard media prolong their in vitro lifespan and strongly reduce the normal features of senescence. Late passage cells in normal medium are rejuvenated when transferred to medium containing carnosine, and become senescent when carnosine is removed. In the absence of pyruvate, carnosine is cytotoxic to neoplastic and transformed human and rodent cells. None of these effects are seen with its optical isomer, D-carnosine.
KEY WORDS: carnosine, ageing, human fibroblasts, transformation
For the first time, the effects of physiological concentrations of L-carnosine (beta-alanyl-L-histidine) on normal human diploid cells grown in cell culture have been documented. We have used the system first developed by Hayflick and Moorhead [1, 2], which demonstrates the long-term growth of human fibroblasts followed by their eventual senescence. We have shown that continuous growth of these cells in carnosine not only increases their lifespan in population doublings and period of growth, but also can reverse the normal features of senescent cells [3, 4]. In other experiments, we have shown that carnosine is cytotoxic to neoplastic cells in culture under conditions in which normal cells survive and proliferate [5, 6].
THE SENESCENCE OF HUMAN DIPLOID CELLS
Hayflick [2] clearly demonstrated that human fibroblasts grow at a constant rate, with a similar yield of cells per flask, over many population doublings (PDs). Typically, this is in the range 50-70 PDs, but it is well established that early passage ampoules of the same strain have variable growth potential [7]. At the end of exponential growth, the yield of cells per flask declines, as does the time required for the population to double its size. The period of senescence has then been reached. Hayflick [2] proposed that this is a manifestation of cellular ageing in vitro, and that these cells provide excellent material for experimental investigations of human ageing at the cellular level. This has been accepted by innumerable laboratories, which over a period of more than 3 decades have published a very large number of experimental studies on cellular senescence of fibroblasts, and a lesser number using other types of cells, which also undergo senescence in vitro (reviewed in [8]).
In view of these extensive studies with fibroblasts, it is surprising that there is no consensus about the general features of their in vitro senescence. Two schools of thought have emerged, and to understand the effects of carnosine on the senescence of human diploid cells, it is necessary to describe the two different ways senescence has been described in the literature. In their original studies, Hayflick and Moorhead [1, 2] demonstrate that if senescent cells are split when they become confluent, the yield per flask declines to a point where the cells no longer reach confluence. These cells are abnormal. They are variable in shape and size; they contain excessive granular material and vacuoles; some cells fail to attach, or become detached, to form debris in the medium; others remain attached, but will never divide again. At an earlier stage of senescence, when the cells are able to reach confluence, they have lost their ability to line up in the parallel arrays which is very characteristic of young fibroblast populations, and they also have a grainy or granular appearance. In studies over 30 years, mainly with the diploid strain MRC-5, all these features of cell senescence have invariably been seen. However, there is another school of thought that emphasizes the fact that the primary defect in senescent human cells is a block in cell division. It has frequently been stated that senescent human cells, when confluent, can survive for a very long period provided the medium is changed weekly. These cells do not divide (or only very occasionally [9]), but are active in RNA and protein synthesis (reviewed in [10]). It is evident that the two terminal states seen by different investigators are largely due to the way the cells are handled. If they are continually split, until they no longer become confluent, then the abnormal cells described above are seen. If they are held confluent and not split, they will survive for a long period of time.
GROWTH OF HUMAN DIPLOID CELLS IN MEDIUM CONTAINING
CARNOSINE
Human muscle contains 20 mM carnosine, and this was the concentration we initially used in experiments with fetal lung strain MRC-5, and human foreskin strain HFF-1 (kindly provided by Dr. R. Reddel, Children's Medical Research Institute, Westmead Hospital, Sydney). Both grew at the normal rate in MEM (Minimal Essential Medium, containing 0.1% glucose, and no pyruvate) or DMEM (Dulbecco's modification of Minimal Essential Medium, containing 0.45% glucose and 1 mM sodium pyruvate). However, at higher concentrations of carnosine (30 mM) we found that the cells grew better in DMEM, and that this difference is largely, or entirely, due to the additional glucose and pyruvate. We also found that only HFF-1 would grow in DMEM containing as much as 50 mM carnosine (approximately 1.25% w/v). This slow growth continued for a very long period, as is shown in Table 1. The final appearance of these cells is remarkably normal, as shown in the figure. There are no granules or vacuoles, no debris in the medium, and little variability in size or shape, which is in sharp contrast to cells grown to their final passage in unsupplemented DMEM. In another experiment, HFF-1 cells were grown in MEM with and without 20 mM carnosine [3]. In this case the control and treated cells grew at the same rate for 44 PDs, but the cells grown in carnosine continued growing for a total of 400 days (last split at 56.8 PDs). In contrast, the untreated cells ceased growth at 310 days (last split of two cultures at 47.4 and 49.4 PDs).
Table 1. Effects of carnosine in increasing
the period of growth prior to termination of cell division in strains
HFF-1 and MRC-5 (all experiments were with DMEM medium)
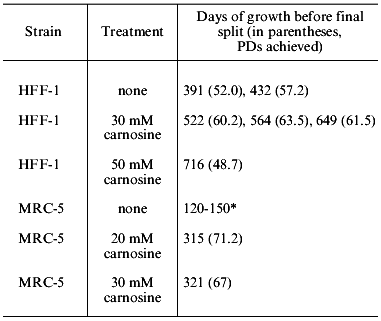
*The characteristic range of lifespans in days for MRC-5 cells split
initially at 1:8, and then at 1:4 and 1:2 in late passages. The single
control in the experiment shown reached 59.8 PDs.
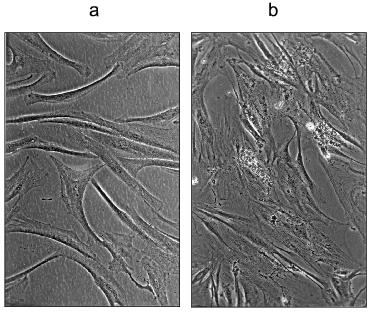
We also transferred MRC-5 cells growing in normal DMEM medium at PD 55.1 to DMEM containing 30 mM carnosine. Remarkably, these cells continued growing slowly for a total of 413 days (the time of last split), whereas two control populations ceased growth at 126 and 139 days (Fig. 5 and Table 1 in [3]). For reasons unknown, other experiments in which late passage cells were transferred from normal medium to carnosine-containing medium did not give the same striking effect. This suggests that there may be a critical period during late passage growth, before which carnosine has a strong effect, and after which it does not. Further experiments are necessary to investigate this.a) HFF-1 fibroblasts grown to their last passage in DMEM containing 50 mM carnosine (these cells failed to become confluent); b) the same cells after transfer, at approximately the same density, to DMEM without carnosine (these cells did not become confluent).
INCREASED LIFESPAN OF CELLS GROWN IN MEDIUM WITH CARNOSINE
We have consistently observed that the addition of 20 and 30 mM carnosine to normal medium increases the lifespan of MRC-5 and HFF-1 in PDs. In these experiments, carnosine is present from early passage, until growth fully ceases. MRC-5 cells in DMEM containing 20 or 30 mM carnosine were subcultured at the same intervals as the controls until approximately PD level 45. Thereafter the control populations slowed down and reached 56.7 and 60.6 PDs. The cells in 20 mM carnosine achieved 69.7 and 70.7 PDs and those in 30 mM achieved 64.7 and 64.3 PDs. In experiments with HFF-1 cells grown in DMEM medium, 30 mM carnosine increased the lifespan by an average of 7.1 PDs. With 20 mM carnosine in MEM medium the lifespan increased by 8.4 PDs. In this experiment, one culture was grown continually in 20 mM D-carnosine. It was not significantly different from the control. It should be noted that an increase of 7 PDs represents an increase in cell mass of approximately 128 times, and an increase of 10 PDs is an increase of approximately 1000 times. In another experiment with MRC-5 cells, the control populations reached PD levels of 79.2 and 76.2 [3]. This is highly unusual and suggests these populations may have had rare clones of long-lived cells [7], which in this case obscured the usual life-extension effect of carnosine.
During the passaging of human cells, the number of cells at confluence is counted after trypsinization (using an electronic Coulter counter), and a proportion of these cells (usually 1/2, 1/4, or 1/8 corresponding to 1, 2, and 3 passages) is seeded in fresh medium into new flasks. When these cells become confluent they are again counted, and from the growth increment the exact increase in PDs is calculated. This assumes 100% of the seeded cells attach to their substrate. It was possible that carnosine was having an effect on attachment, and thereby producing a change in the calculated PDs, in comparison to controls. We therefore carried out experiments in which the proportion of cells that attached at every passage was measured in control and carnosine treated cells. Carnosine did not in fact affect attachment of the cells, but again their lifespan was increased. In two experiments with MRC-5 cells in MEM and DMEM the lifespan was increased by 11.0 and 11.4 PDs, respectively, when the medium contained 20 mM carnosine [4]. All our results on lifespan extension are summarized in Table 2.
Table 2. Increases in the lifespan in
population doublings (PDs) induced by continuous growth in medium
containing L- and D-carnosine (see [3, 4])

EFFECT OF CARNOSINE ON THE PLATING EFFICIENCY OF MRC-5
CELLS
When MRC-5 fibroblasts are plated at fairly high density, as in a 1:2, 1:4 or 1:8 split, they begin to divide after about 18 h. However, at lower density, the attached cells move over the surface, but may not divide until a small cluster of cells is formed. Plating 103 early passage cells in a standard 9-cm dish typically yields about 20 colonies, representing a “plating efficiency” of only 2%. (If a feeder layer of non-dividing irradiated or mitomycin C-treated cells is also added, the plating efficiency is very much higher). When we plated young MRC-5 cells in DMEM containing 20 mM carnosine, the plating efficiency was very significantly elevated, to about 10% [4]. When 103 late passage cells are seeded in a 9-cm dish, no colonies are expected because the cells have almost reached the end of their lifespan. However, cells at PD level 56, when plated in DMEM containing 20 mM carnosine, produced a number of colonies visible after staining with Giemsa, and none in a control plate. These colonies represent about 15 additional PDs. Clearly, in this and other experiments the late passage cells are rejuvenated by carnosine.
REVERSIBILITY OF THE SENESCENT PHENOTYPE BY CARNOSINE
We have consistently observed that late passage cells grown in the presence of carnosine have a more normal appearance than late passage control cells. The carnosine treated cells line up in parallel arrays, to form the characteristic whorls of young normal fibroblasts. They lack the typical granular appearance of untreated senescent cells, and there is little debris in the medium. We have also examined in many experiments the effect of transferring these late passage cells from carnosine medium to unsupplemented medium and the reverse switch, in some cases with several successive switches over a long period. The results are fully documented by photographs in references [3, 4], and can be summarized as follows. 1) Late passage cells with normal morphology in medium containing carnosine revert in 7-10 days to a senescent phenotype when carnosine is removed. In some cases, the senescent phenotype becomes more evident after splitting the cells. 2) Late passage cells in normal medium with a senescent phenotype are rejuvenated to a normal phenotype when a high concentration of carnosine is added. Again, this effect may be more clearly seen after subculturing the cells in the presence of carnosine. 3) Sequential transfer of late passage cells to and from unsupplemented and carnosine-supplemented medium is always accompanied by a switch in cell morphology from senescent to normal, and the reverse. 4) These effects are most clearly seen in late passage HFF-1 cells, using DMEM medium with and without 30 or 50 mM carnosine. The figure shows subconfluent HFF-1 cells in 50 mM carnosine at their last passage and the effect of switching such cells to unsupplemented medium.
CARNOSINE KILLS TRANSFORMED OR NEOPLASTIC CELLS
In early experiments, we had one human foreskin culture that proved to be contaminated with transformed 3T3 cells. These quickly overtook the growth of the human cells. However, we had added 30 or 50 mM carnosine to some flasks, and we noticed that in these the human cells grew with no sign of the 3T3 contamination cells. Later, we followed up the observation in a series of experiments with human or rodent transformed and neoplastic cells [5]. We found that carnosine was cytotoxic to these cells in MEM medium, but not in DMEM medium. This difference was traced to the 1 mM pyruvate in DMEM, although the lower glucose was also in part responsible for the cytotoxic effect of carnosine in MEM. We also found that dialyzed fetal calf serum, in which low molecular weight compounds have been removed, enhanced the cytotoxic effect of carnosine. For seven transformed or neoplastic human cell lines, and two rodent lines, we defined the carnosine-containing media that killed the cells. (In most cases, cytotoxicity was observed as the rounding up and detachment of the cells, but we also demonstrated the decline in survival of HeLa and CHO cells, by plating carnosine-treated cells in normal medium). We also tested all these media, and others containing carnosine, on normal human cells. In some the cells grew well, but in others the medium was cytostatic to the cells. They remained attached to the substrate and had a normal morphology. When the carnosine was removed by changing the medium, the fibroblasts resumed growth. It is very well known that HeLa contamination quickly results in the loss of any contact-inhibited cell population, since the HeLa cells quickly produce overgrowth. We devised a procedure whereby HeLa cells contaminating an MRC-5 culture are eliminated. When 103 HeLa cells were added to a population of MRC-5 cells, after 5 days these HeLa cells formed individual colonies visible on a background of confluent MRC-5 cells. At this time the carnosine treatment was begun, using MEM with 10% dialyzed serum and 20 mM carnosine. The cells were split 3 times in this medium, with intervening changes of medium. After 27 days, no HeLa cells were visible, and the cells were transferred to normal MEM. They grew to their normal late passage senescence, with no reappearance of HeLa cells [5]. The same experiment was also successfully carried out with other experimental regimes.
We were also interested in comparing mouse embryonic stem cells (ES cells) with mouse embryonic teratocarcinoma cells (EC cells). These are both immortal pluripotent cell lines, but ES cells have a normal pluripotent phenotype. As in the case of diploid somatic cells and transformed cells, we found that carnosine prevents the growth of the EC cells in the absence of pyruvate, whereas the ES cells grow well under the same conditions. Thus, the neoplastic phenotype is again associated with carnosine sensitivity [6].
As well as pyruvate, we also checked the effect of other components of the tricarboxylic cycle in nullifying the cytotoxic effects of carnosine. We found that 1 mM oxaloacetate or 1 mM alpha-ketoglutarate had an effect comparable to pyruvate, but citrate, isocitrate, succinate, fumarate, and malate had no effect. We also demonstrated that D-carnosine in MEM without pyruvate is entirely nontoxic to HeLa cells. We tested homocarnosine, anserine, beta-alanine, and L-histidine on HeLa cells. Only anserine has cytotoxic activity like carnosine (i.e., dependent on the absence of pyruvate), and 20 mM histidine is toxic to HeLa cells irrespective of the presence of pyruvate [5].
DISCUSSION
For such a small molecule, it is remarkable that carnosine has multiple properties. It is an antioxidant, a chelator of toxic metal ions, a buffer, and an inhibitor of non-enzymic glycosylation of proteins. These properties are reviewed and discussed elsewhere in this volume. Its high concentration in skeletal muscle suggests that it may have a role in neutralizing the accumulation of lactic acid, which occurs when muscles obtain energy from glycolysis rather than respiration. It is, however, striking that human muscle contains 20 mM carnosine, whereas mouse muscle contains only 1 mM ([11], J. Michaelis, personal communication). This suggests that carnosine may have a more fundamental role in cellular maintenance, since it is well established that the efficiency of maintenance in mammalian species is correlated with lifespan [8, 12]. The proposed maintenance function could include antioxidant activity, chelation of toxic metals, and the inhibition of non-enzymic glycosylation of proteins and the accumulation of AGEs (advanced glycation end products). These high-molecular-weight aggregates are seen in several tissues in senescent animals and obviously accumulate more rapidly in short-lived animals than in long-lived ones.
The effect of carnosine in reversing the normal symptoms of cell senescence, and also increasing cells' growth potential, may well be a reflection of maintenance functions. It is widely believed that the final cessation of cell division after the prolonged growth of diploid somatic cells is due to the induction of a block in cell division (reviewed in [13, 14]). The protein p53 has a central role as “guardian of the genome”. This means that when DNA damage occurs, cell division is blocked through the p53-mediated inhibition of DNA synthesis. One type of DNA damage that has received a great deal of attention is the sequential loss of telomeric DNA. This loss eventually triggers a response involving a block in cellular division. However, in this case, there is no recovery from the damage, so the cell never divides again. It is also possible that other types of change in DNA have the same effect, for instance, the accumulation of defects in DNA structure, or the loss of DNA methylation. Carnosine could operate in one of two ways. First, it could reduce the degree or incidence of the events that eventually trigger the cell cycle block. For example, it could reduce the length of the telomeric DNA fragments lost, or the rate of DNA methylation decline. Second, it could reduce the physiological effects of changes in DNA, so that the point at which a final cell division block occurs is significantly delayed. Such an effect would manifest itself in the rejuvenation of the cellular phenotype, which is seen when senescent cells are grown in or transferred to a medium containing carnosine. Similarly, the removal of carnosine would result in the reversion of a fairly normal cellular phenotype to a visibly abnormal senescent cell. Obviously, a number of experimental approaches to test or distinguish these possibilities would now be possible.
The differential effect of carnosine on normal cells and their transformed derivatives is extremely striking. It is possible that carnosine has anti-cancer activity in vivo. This would certainly fit in with the well-known fact that human cells are much less readily transformed than rodent cells [12], and the twenty-fold higher concentration of carnosine in some human cells, such as those in skeletal muscle, than in the same mouse cells.
It was discovered by Warburg many decades ago that cancer cells have much higher glycolysis activity than normal cells [15]. He thought cancer cells were defective in respiration, which is not correct, but it has been extremely well established that the glycolysis/respiration ratio in the generation of ATP is far higher in cancer cells than normal somatic cells [16]. In other words, transformed or neoplastic cells have uncoupled the normal regulation of glycolysis and respiration. With regard to carnosine, it is well known that it reacts by the Amadori reaction with aldehyde and keto groups [17]. It is particularly reactive with the phosphorylated triose sugar glycolysis intermediates. This immediately suggests that carnosine inhibits glycolysis activity and therefore the generation of ATP in cancer cells. The effect is totally reversed by the addition of pyruvate (and at least two other Krebs cycle intermediates). These would of course stimulate respiration to generate ATP, and allow the tumor cells to grow normally. It is also significant that another chemical that reacts with sugars, known as tenilsetam, also inhibits HeLa cell growth in the absence of pyruvate, but not in its presence [5]. In addition, it is highly significant that we have shown that carnosine has differential effects on ES (embryonic stem) cells and EC (teratocarcinoma) cells. Both these types of cells are immortal cell lines, and both are pluripotent in their capacity to differentiate into many types of cell and tissue. Yet only the EC cells are inhibited by carnosine in the absence of pyruvate [6].
Much recent research on the molecular biology of cell transformation and immortalization completely ignores the fundamental differences in the physiology and biochemistry of cancer cells and normal cells. Apart from glycolysis and respiration, it has long been known that there can also be profound differences in the activities of many important metabolic enzymes in cancer and normal cells (reviewed in [18]; for a recent example, see [19]). There is much documentation of mutations in oncogenes and tumor suppressor genes that predispose or cause normal cells to become malignant. It should not be forgotten that the switch from a normal ES cell to an abnormal EC cell does not depend on gene mutations. Insertion of a mouse embryo into an abnormal environment (such as that beneath an adult kidney capsule) is sufficient to generate teratocarcinomas. This is a heritable epigenetic event, which clearly has fundamental effects on the regulation of cellular metabolism. Carnosine may be a very useful tool to probe further not only the physiological differences between normal and tumor cells, but also the importance of cellular maintenance in delaying ageing or protecting cells against oncogenic transformation.
REFERENCES
1.Hayflick, L., and Moorhead, P. S. (1961) Exp.
Cell Res., 25, 585-621.
2.Hayflick, L. (1965) Exp. Cell Res.,
37, 614-636.
3.McFarland, G. A., and Holliday, R. (1994) Exp.
Cell Res., 212, 167-175.
4.McFarland, G. A., and Holliday, R. (1999) Exp.
Geront., 34, 35-45.
5.Holliday, R., and McFarland, G. A. (1996) Brit.
J. Cancer, 73, 966-971.
6.McFarland, G. A., and Holliday, R. (1999) In
vitro Cell Devel. Biol. - Animal, 35, 15-16.
7.Holliday, R., Huschtscha, L. I., Tarrant, G. M.,
and Kirkwood, T. B. L. (1977) Science, 198, 366-372.
8.Holliday, R. (1995) Understanding Ageing,
Cambridge University Press, Cambridge.
9.Matsumura, T., Zerrudo, Z., and Hayflick, L. (1979)
J. Gerontol., 34, 328-334.
10.Smith, J. R., and Lincoln, D. W. (1984) Int.
Res. Cytol., 89, 151-177.
11.Mannion, A. F., Jakeman, P. M., Dunnett, M.,
Harris, R. C., and Willian, P. L. (1992) Eur. J. Appl. Physiol.,
64, 47-50.
12.Holliday, R. (1996) in Genetic Instability in
Cancer (Lindahl, T., ed.) Cold Spring Harbor Laboratory Press, N.
Y., pp. 103-115.
13.Reddel, R. (1998) BioEssays, 20,
977-984.
14.Wynford-Thomas, D. (1999) J. Pathol.,
187, 100-111.
15.Warburg, O. (1956) Science, 123,
309-314.
16.Aisenberg, A. C. (1961) The Glycolysis and
Respiration of Tumors, Academic Press, N. Y.
17.Hipkiss, A. R. (2000) Biochemistry
(Moscow), 65, 771-778.
18.Weber, G. (1983) Cancer Res., 43,
3466-3492.
19.Holliday, R., and Ho, T. (1998) Proc. Natl.
Acad. Sci. USA, 95, 8727-8732.