REVIEW: Anti-ischemic Activity of Carnosine
S. L. Stvolinsky1* and D. Dobrota2
1Laboratory of Clinical Neurochemistry, Institute of Neurology, Russian Academy of Medical Sciences, Volokolamskoe Shosse 80, Moscow, 123367 Russia; fax: (7-095) 490-2408; E-mail: sls@bio.inevro.msk.ru2Department of Biochemistry, Jessenius Faculty of Medicine, Komensky University of Bratislava, Martin 036 45, Slovakia; E-mail: dusan.dobrota@jfmed.uniba.sk
* To whom correspondence should be addressed.
Received December 13, 1999
This review summarizes the data on anti-ischemic activity of carnosine. The pronounced anti-ischemic effects of carnosine in the brain and heart are due to the combination of antioxidant and membrane-protecting activity, proton buffering capacity, formation of complexes with transition metals, and regulation of macrophage function. In experimental cerebral ischemia, carnosine decreases mortality and is beneficial for neurological conditions of the animals. In cardiac ischemia, carnosine protects cardiomyocytes from damage and improves contractility of the heart. The data indicate that carnosine can be used as an anti-ischemic drug.
KEY WORDS: carnosine, brain, heart, experimental ischemia, anti-ischemic activity
A high rate of oxidative metabolism is characteristic for the brain. Under normal conditions, the adult brain consumes 3-4 ml O2/min per 100 g tissue, and this can represent up to 20% of all inhaled oxygen. When cerebral circulation is altered, oxygen consumption in the ischemic zone is dramatically reduced, thus inducing significant neuronal injury [1].
Glucose is the main energy substrate of the brain. During ischemia, the amount of glucose arriving to the brain is in excess because oxidative metabolism takes place under oxygen deficiency. This imbalance induces the transition of the brain towards anaerobic metabolism, resulting in accumulation of lactate in the final stages of glycolysis. Progressive acidosis damages cerebral tissue [2, 3].
ATP deficiency results from the transition to the less efficient type of energy metabolism and is another factor of pathogenesis of ischemia. ATP deficiency decreases the membrane potential of neurons and glial cells [4]. The presynaptic potential-dependent Ca2+-channels are activated and excitatory amino acids are secreted to the extracellular medium. On the other hand, energy depletion suppresses the reverse transport of excitotoxic amino acids, thus extending the time of activation of glutamate receptors. This decreases intracellular level of K+ and dramatically increases the intracellular levels of Ca2+, Na+, and Cl-. Ca2+ entry inside the cell activates proteolytic enzymes, endonucleases, phospholipase A2, cyclooxygenase, NO-synthase, and other enzymes. Activation of phospholipase A2 and cyclooxygenase induces generation of reactive oxygen species and lipid peroxide radicals, damaging cellular membranes, mitochondria, and DNA. NO-synthase significantly contributes to the production of cytotoxic radicals.
Free radicals induce the formation of inflammatory mediators, which in turn activate microglia and initiate infiltration of leukocytes from the blood stream.
Thus, development of cerebral ischemia involves interrelated metabolic processes, and it is difficult to determine the main pathogenic factor. Hence, there is no universal protocol for therapy of cerebral ischemia; the main approach is administration of drugs improving rheological characteristics of the blood stream [5]. Efficient medication should be neuroprotective and should decrease cerebral metabolism, antagonize glutamate neurotoxicity, neutralize free radicals, inhibit Ca2+ entry, and improve cerebral reperfusion. It is very unlikely that a single compound would combine all these properties. Thus, development of new anti-ischemic substances is of special importance.
Carnosine and related histidine-containing dipeptides are abundant in vertebrate tissues including brain (carnosine and homocarnosine) and heart (acetylated forms of carnosine, homocarnosine, and anserine), and these compounds are involved in various physiological processes [6]. Carnosine has several functions: considering its anti-ischemic activity, antioxidant and membrane-protecting properties, complexing of heavy metal ions, and regulation of macrophage activity are the most important.
V. P. Skulachev has discussed the function of carnosine as a mobile buffer and carrier of H+ released by lactate accumulation during glycolysis in working muscle [7]. The calculations indicate that the contents of carnosine in the muscle are enough for maintenance of intracellular pH within the required limits (see the review of H. Abe in this issue).
Carnosine and related compounds efficiently bind divalent ions of transition metals [8] including iron [9]; this is of special importance considering the role of iron in production of reactive oxygen species and development of peroxidation during ischemia, i.e., when iron deposition mechanisms are altered. Complexes of carnosine with Cu2+ and Zn2+ have superoxide dismutase activity [10, 11], which contributes to the antioxidant effects of the dipeptide.
Membrane-protecting and antioxidant properties of carnosine were described in 1984 [12]. A number of in vitro experimental evidences indicate that carnosine and related histidine-containing dipeptides can scavenge superoxide and quench singlet oxygen and hydroxyl radical [13].
In vitro, it was shown that carnosine and related histidine-containing dipeptides modify the activity of leukocytes during respiratory burst, decreasing the levels of hypochlorite and increasing the half-life of superoxide radical [14]. In this case, carnosine not only traps the radicals but also modifies the activity of the enzyme systems responsible for the production of reactive oxygen species [15].
In vivo, antiradical activity of carnosine was demonstrated in the models of immobilization and electric pain stress, which are associated with activation of synthesis of oxygen radicals and lipid peroxidation (LPO) in the brain [16, 17]. Carnosine enhanced the resistance of animals towards ionizing radiation [18, 19], hypobaric hypoxia [20], and supercooling [21], lowering mortality of the animals and improving their recovery.
Experimental and clinical therapy with carnosine demonstrated that it is an efficient immunomodulator and anti-inflammatory agent [22-24]. The effects could be due to the antioxidant, membrane-stabilizing, and pH-buffering activities of carnosine [6]. The diverse pharmacological activities of carnosine are of interest for protection of the brain against ischemia and hypoxia.
Joint studies performed in the Institute of Neurology (Russian Academy of Medical Sciences) and Department of Physiology of the School of Biology (Lomonosov Moscow State University) investigated the effect of carnosine administered to rats during acute hypobaric hypoxia. It was demonstrated (Table 1) that injection of carnosine before hypoxia enhanced the resistance of the animals to acute oxygen deficiency and accelerated the recovery of respiratory function and mobility after hypoxia, suppressing accumulation of LPO products in the serum [23, 25, 26]. V. B. Koshelev, A. A. Krushinsky (Department of Physiology), T. V. Ryasina, T. S. Korshunova (Institute of Neurology), and S. L. Stvolinsky studied antispasmodic and angioprotector effects of carnosine in a model of audiogenic epilepsy in the rat strain developed by Krushinsky and Molodkina; these rats are genetically predisposed to audiogenic epilepsy associated with development of extended hemorrhage of cerebral vessels. Injection of carnosine before initiation of acoustic stress suppressed development of epilepsy, decreased the frequency of cerebral hemorrhages, and normalized the functions of the pituitary-adrenal system (Table 2). These results prompted studies of anti-ischemic activity of carnosine.
Table 1. Effect of carnosine
(250 mg/kg) on resistance of rats to acute hypobaric hypoxia
(PO2 = 27 mm Hg)
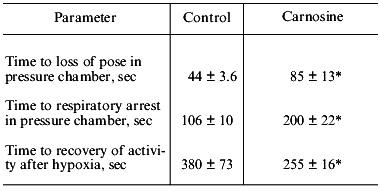
*p < 0.05 versus control.
Table 2. Effect of carnosine (250 mg/kg) on
antispasmodic activity of Krushinsky-Molodkina rats during acoustic
stress
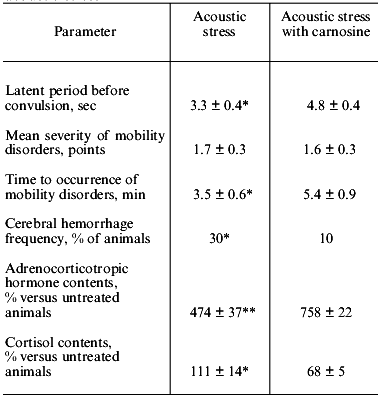
*p < 0.05 versus control.
**p < 0.02 versus control.
The prophylactic effect of carnosine was studied in various models of ischemia.
When ischemia was induced in rats by bilateral occlusion of the carotid arteries, intraperitoneal injection of carnosine decreased mortality of the animals by 2-fold (Table 3), and in all survivors the severity of neurological symptoms was reduced. Effective reduction of mortality by carnosine was also observed in another model of ischemia in rats induced by occlusion of four major cephalic vessels including the spinal and common carotid arteries (Table 3).
Table 3. Effect of carnosine on survival of
rats during experimental cerebral ischemia (number of survivors, % of
animals in the group)
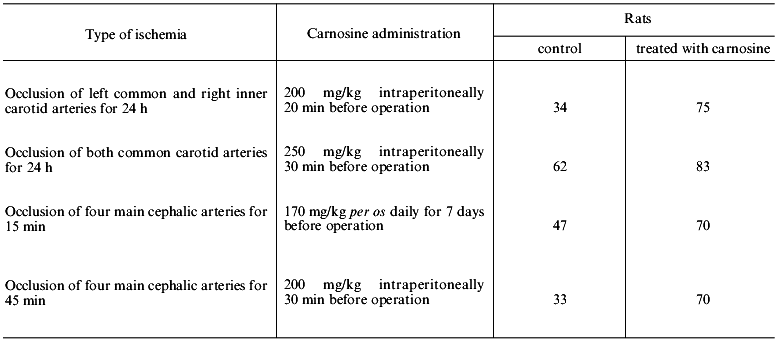
The data of Table 3 suggest that there is a limit of survival (70-80%) of the rats subjected to profound ischemia, and this limit can be provided by full functioning of the endogenous defense system. Apparently, carnosine can compensate for the development of deficiency of this system independently of the reasons inducing its damage.
Occlusion of the common carotid arteries was used to decrease cerebral blood flow by 50% for 15-30 min, and then the blood flow was restored [27]. In rats without neurological symptoms that did not receive carnosine, the cerebral blood flow was completely restores in 24 h, whereas in animals with neurological symptoms the blood flow did not exceed 50-70% of the baseline. Administration of carnosine lowered the decrease in the cerebral blood flow during the early stages of development of ischemia. In all animals that received carnosine before the operation, the cerebral blood flow was restored to 85-120% of the baseline, and the restoration was more complete in rats without neurological symptoms [27]. Electroencephalographic (EEG) studies of electrical activity of the brain indicate that 15 min after the occlusion of the common carotid arteries, the amplitude of the theta rhythm was increased; high electric activity of the brain was detected during the first 30-45 min of ischemia development, and it remained high for 24 h after ischemia. Carnosine administered 25-30 min before occlusion of common carotid arteries stabilized the amplitude of the theta rhythm of the EEG, and the effects were more pronounced in animals highly sensitive to ischemia [27].
It would be of interest to compare the anti-ischemic effects of carnosine to biochemical characteristics of the brain of animals in the case of ischemia.
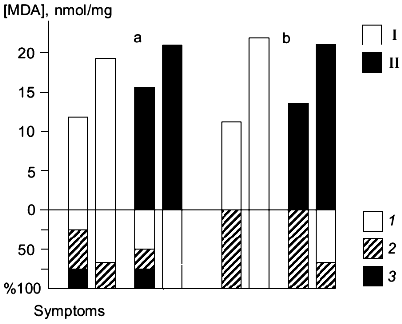
Synaptosomes were isolated from the brains of animals subjected to ischemia for 24 h (the ischemia was induced by occlusion of the common carotid arteries); in these synaptosomes, activation of LPO by the iron/ascorbate system was studied [28]. In rats highly sensitive to ischemia, it was shown that accumulation of peroxidation products during 60 min of LPO activation was lower in the control group versus that in animals treated with carnosine 30 min before the operation (intraperitoneal injection, 250 mg/kg). Animals with high and low susceptibility to LPO induction were detected in the control and carnosine-treated groups. It was suggested that administration of carnosine before ischemia protects the lipid components of the brain membranes from peroxidation developing during ischemia. This effect was more pronounced in animals with high sensitivity to ischemia (Fig. 1a). The neurological symptoms were less expressed in animals whose membrane lipids were protected from peroxidation. For example, 25% of rats with low LPO induction (not treated with carnosine) surviving occlusion of the common carotid arteries did not develop neurological symptoms; 50% of these rats developed light symptoms and 25% developed severe neurological symptoms. In rats pretreated with carnosine, 50% of animals did not develop neurological symptoms; 25% developed light symptoms, and 25% developed severe neurological symptoms. In rats with high induction of LPO surviving the operation, neurological symptoms were less expressed. In animals not treated with carnosine, approximately 30% developed light neurological symptoms, and in carnosine-treated rats, neurological symptoms were not detected (Fig. 1a). In animals with low sensitivity to ischemia, the severity of neurological symptoms was also inversely correlated to induction of LPO in brain synaptosomes (Fig. 1b); rats with low LPO induction did not develop neurological symptoms and rats with high LPO induction developed light symptoms. Carnosine did not influence induction of LPO in brain synaptosomes of rats with low sensitivity to ischemia [28].
The system of endogenous antioxidants was analyzed in the brain of rats after 24 h of ischemia; the data indicate that the pool of homocarnosine was better preserved in animals pretreated with carnosine which had less pronounced neurological symptoms [28]. On the other hand, these experiments demonstrated that the levels of carnosine were not increased after 24 h of ischemia in the brain of rats intraperitoneally injected with carnosine before surgery. Thus, the neuroprotective effect of carnosine is not limited to the direct antioxidant action. It has been shown that after intraperitoneal injection of carnosine in rats, peak levels of carnosine are observed 30-45 min from the injection, and then carnosine rapidly disappears from the brain [16].Fig. 1. Correlation between induction of membrane lipid peroxidation of the brain and neurological symptoms 24 h after occlusion of the common carotid arteries in rats with high (a) and low (b) sensitivity to ischemia; I) ischemia; II) ischemia after pretreatment with carnosine. The upper part of the figure shows the level of LPO products (malonic dialdehyde (MDA), nmol/mg protein) accumulated in 60 min of LPO induction. The lower part of the figure shows the percentage of animals with neurological disorders of various severities: 1) no neurological symptoms; 2) light neurological disorders; 3) severe neurological disorders.
Oxygen radicals produced by polymorphonuclear leukocytes and monocytes-macrophages significantly contribute to the development of cerebral ischemia [29]. We have shown that, under normal conditions, injection of carnosine into rats inhibits respiratory burst of leukocytes, and the suppression of the hypochlorite formation was pronounced to the highest extent [15]. The effect of carnosine was preserved for 24 h either after a single oral administration (1 h before cells were isolated) or after regular (daily for 5 days) administration in drinking water before the leukocyte activity was determined. This suggests that carnosine has an indirect ultimate effect when its concentration in the blood is decreased to levels that are not efficient in direct in vitro experimental measurements of leukocyte burst [15].
In rats with ischemia induced by bilateral occlusion of the common carotid arteries for 24 h, leukocyte respiratory burst was observed and this burst was efficiently suppressed by carnosine injected 30 min before occlusion. The number of polymorphonuclear leukocytes and production of reactive oxygen species by these cells were decreased by 2.5-3-fold [30]. Thus, carnosine can influence the development of ischemic injury of the brain via modification of the activity of the leukocyte system.
Accumulation of lactate and acidosis are among the most important pathogenic factors of hypoxic and ischemia injury of the brain. Similar processes associated with accumulation of H+ and lactate occur when energy metabolism of the muscle is switched to glycolysis [7]. According to NMR spectroscopy, pretreatment with carnosine decreases lactate accumulation in the muscle of rats when glycolysis is activated by physical activity load [31]. Thus, it was suggested that injection of carnosine during ischemia could result in binding of excessive protons, suppressing lactate accumulation in the brain (similar to oxygen deficiency in muscle), thus decreasing the pathogenetic effect of these factors of ischemic injury of the brain. According to 1H-NMR spectroscopy, injection of carnosine into rats subjected to ischemia (occlusion of four cephalic arteries) inhibits lactate accumulation in the brain [32]. In control rats, the levels of lactate were increased in the ischemic injury zone by 4-5-fold during 45 min, whereas in carnosine-pretreated animals, the level of lactate was increased by only 2-fold; the effect was preserved for long periods of time (up to 90 min and more). In the control group, the level of lactate in the brain was still increasing (Fig. 2). These data cannot be considered as direct evidence of the role of carnosine in the anti-ischemic defense system as a proton buffer, but the effect indicates that carnosine is a very promising medication removing one of the important pathogenic factors of ischemic injury, i.e., accumulation of lactate in the ischemic zone; this may be due to improved microcirculation in the ischemic zone [27].
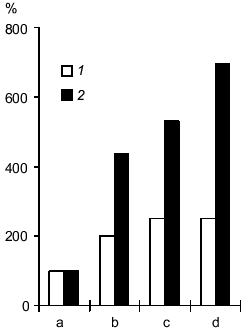
Development of ischemic injury induces alterations in enzymes and receptors of the brain. The effect of carnosine on the activities of Na,K-ATPase, monoamine oxidase B (MAO B), and glutamate receptors was studied in experimental ischemia induced by occlusion of cephalic vessels.Fig. 2. Effect of carnosine on lactate accumulation in the ischemic injury zone of the brain induced by occlusion of four main cephalic arteries (data of H1-NMR spectroscopy): 1) ischemia in rats pretreated with carnosine; 2) ischemia in control rats. The levels of lactate before arterial occlusion (a) and in 35-45 min (b), 90-100 min (c), or 150-170 min after occlusion (d).
Na,K-ATPase is one of the first and the most susceptible targets of oxygen radicals [33]. Inhibition of its activity alters ion transport and induces membrane depolarization; neurons lose their most important function, i.e., electrical conductivity. In vitro, carnosine efficiently protects Na,K-ATPase from the effects of oxygen radicals [34].
Studies of Na,K-ATPase from the brain of Mongolian jerboas subjected to ischemic injury indicate that enzyme activity is reduced by 22% [35]. Similar inactivation of Na,K-ATPase was detected in a model of profound ischemia of rat brain (occlusion of four main cephalic arteries) [30, 36]. In this model, injection of carnosine before the occlusion suppressed inhibition of enzyme activity. In ischemic rats not pretreated with carnosine, Na,K-ATPase activity was approximately 80% of that in the intact animals but in carnosine-treated rats, enzyme activity did not significantly differ from that in the intact animals [30].
Uncontrolled increase in excitotoxic compounds (glutamate, aspartate) in the interneuronal medium is an important pathogenic factor of development of ischemic injury of the brain. These excitotoxic compounds induce sustained activation of the corresponding receptors providing for pleiotropic effects of their activation in the cell, thus altering intracellular homeostasis. Activation of glutamate receptors by kainate (KA) or N-methyl-D-aspartate (NMDA) is associated with Ca-dependent formation of reactive oxygen species inducing massive cell death. Pretreatment of neuronal suspension with carnosine protects the cells from KA- and NMDA-dependent injury. Suppression of glutamate receptor-dependent toxicity was dose-dependent, and IC50 was 0.75 mM; this value is similar to the physiological concentrations of carnosine in the vertebrate brain [37].
In vivo, in a model of ischemia of rat brain induced by prolonged (5 days) occlusion of the common carotid arteries, the activity of the glutamate receptors was studied as well as the effect of carnosine on their function. Ischemic injury of the tissue was characterized by activation of NMDA receptors, and this activation was suppressed by carnosine. After prolonged ischemia of the brain, binding of NMDA by the glutamate receptors was 2-fold higher versus that in the intact animals. In rats pretreated with carnosine before the occlusion of the carotid arteries, binding of NMDA by brain synaptosomes (determined at the end of ischemia) was similar to that in the intact animals.
Excessive production of dopamine in ischemia and dopaminergic hyperactivity during recirculation in the ischemic zone play an important role in neuronal death. Oxidation by monoamine oxidases A and B is the main pathway of catecholamine and other monoamine neurotransmitter catabolism. In the case of ischemia of the brain, activity of monoamine oxidases is decreased by 20-30% [38]. In a rat model of global ischemia of the brain (occlusion of cephalic vessels), MAO B activity is decreased to 75-80% of the levels detected in the intact brain. Pretreatment with carnosine inhibited this suppression of the MAO B activity [39]. These data do not suggest a direct mechanism of MAO B protection by carnosine in the ischemic brain but indicate that carnosine can compensate for the pathogenic effects of catecholamines. Another possible pathway of carnosine-dependent regulation of catecholamine levels in the ischemic brain may involve its effects on tyrosine hydroxylase, which is the key enzyme of synthesis of these neuromediators. In vitro, it was shown that micromolar concentrations of carnosine suppress the activity of tyrosine hydroxylase (isolated from rat brain) by 75% in a model system [40]. Thus, the influence of carnosine on the levels of catecholamines in the brain can involve limitation of the synthesis of these excitotoxic compounds and protecting pathways of their oxidative conversion.
Data on protection of the heart from ischemia by carnosine are of interest considering the reviewed problem. In cardiac muscle, the concentration of the histidine-containing dipeptides is up to 10 mM, and most of these dipeptides are acetylated [41]. During short-term hypoxia of the heart, carnosine promoted preservation of the coronary blood flow, decreased loss of lactate dehydrogenase from cardiomyocytes, and suppressed the development of ischemic contracture [42]. In cardiac reperfusion after lethal blood loss, carnosine administered in nutrient blood-substituting medium protected cardiomyocyte membranes from injury and normalized the functioning of the enzymes localized in these membranes [43]. Carnosine decreased the development of contracture in ischemia-reperfusion. At the same time, release of myoglobin and nucleosides from cardiomyocytes was decreased, suggesting that carnosine has membrane-protecting activity [44]. Acetylated carnosine was more efficient in suppressing ischemic contracture, and its influence on cardiac contractility was more pronounced. Moreover, acetylcarnosine restored myocardial contractility during ischemia [45]. Hence, carnosine has anti-ischemic activity in the heart as well as in the brain.
Thus, carnosine is a very promising candidate for an anti-ischemic medication due to a combination of several effects. The effects of carnosine on the production and levels of reactive oxygen species can play the key role in anti-ischemic activity of this dipeptide. However, significant impact of other activities of carnosine should not be neglected, including its function as a mobile proton buffer and iron ion chelator. Further studies will resolve this problem, but even at present clinical studies of anti-ischemic effects of carnosine can be considered.
The authors are indebted to Professor A. A. Boldyrev for valuable comments during preparation of the manuscript and for helpful discussions.
REFERENCES
1.Sims, N. R., and Zaidan, E. (1995) Int. J.
Biochem. Cell Biol., 27, 531-550.
2.Crumrine, R. C., and LaManna, J. C. (1991) J.
Cerebr. Blood Flow Metab., 11, 272-282.
3.Graham, G. D., Blamire, A. M., Howseman, A. M.,
Rothman, D. L., Fayad, P. B., Brass, L. M., Petroff, O. A. C., Shulman,
R. G., and Prichard, J. W. (1992) Stroke, 23,
333-340.
4.Katsura, K., Kristian, T., and Siesjo, B. K. (1994)
Biochem. Soc. Trans., 22, 991-996.
5.Hungerhuber, E., Zausinger, S., Baethmann, A.,
Reulen, H.-J., and Schmid-Elsaesser, R. (1999) in Maturation
Phenomenon in Cerebral Ischemia. III. Defensive Mechanisms Versus
Apoptosis. Neuronal Recovery and Protection in Cerebral Infarction
(Ito, U., Fieschi, C., Orzi, F., Kuroiwa, T., and Klatzo, I., eds.)
Springer-Verlag, Berlin-Heidelberg, pp. 159-167.
6.Boldyrev, A. A. (1999) Carnosine and Defence of
Brain from Oxidative Stress [in Russian], MGU-Dialog Publisher,
Moscow.
7.Skulachev, V. P. (1992) Biokhimiya,
57, 1311-1316.
8.Brown, C. E., and Antholine, W. E. (1979) J.
Phys. Chem., 83, 3314-3319.
9.Vladimirov, Y. A. (1996) in Proc. Int. Symp.
Natural Antioxidants. Molecular Mechanisms and Health Effects
(Parcker, L., Traber, M. G., and Xin, W.,eds.) AOCS Press,
Champaing, Illinois, pp. 125-144.
10.Gulyaeva, N. V. (1987) Biokhimiya,
52, 1216-1220.
11.Kohen, R., Misgav, R., and Ginsburg, I. (1991)
Free Rad. Res. Commun., 12/13, Pt. 1, 179-185.
12.Dupin, A. M., Boldyrev, A. A., Arkhipenko, Yu.
V., and Kagan, V. E. (1984) Byul. Eksp. Biol. Med., 98,
No. 8, 186-188.
13.Boldyrev, A. A., and Abe, H. (1999) Cell. Mol.
Neurobiol., 19, 163-175.
14.Stvolinsky, S. L., Souza Pontesh, E., Sergienko,
V. I., and Boldyrev, A. A. (1996) Biol. Membr. (Moscow),
13, 299-306.
15.Boldyrev, A., Abe, H., Stvolinsky, S., and
Tyulina, O. (1995) Comp. Biochem. Physiol., 112B,
481-485.
16.Gulyaeva, N. V., Obidin, A. B., Levshina, I. P.,
Filonenko, A. I., Dupin, A. M., and Boldyrev, A. A. (1989) Biol.
Nauki, No. 8, 5-16.
17.Gulyaeva, N. V., Dupin, A. M., Levshina, I. P.,
Obidin, A. B., and Boldyrev, A. A. (1989) Byul. Eksp. Biol.
Med., 107, No. 2, 144-147.
18.Severin, S. E., Boldyrev, A. A., Stvolinsky, S.
L., Bordyukov, M. M., Goncharenko, E. N., Deev, L. I., Malinina, M. E.,
and Kudryashov, Yu. B. (1990) Radiobiologiya, 30,
765-768.
19.Naumova, O. V., Goncharenko, E. N., and Deev, L.
I. (1992) Biokhimiya, 57, 1373-1377.
20.Korobov, V. N., Doliba, N. M., and Telegus, Ya.
V. (1993) Biochemistry (Moscow), 58, 488-490.
21.Goncharenko, E. N., Deev, L. I., Akhalaya, M.
Ya., Antonova, S. V., Baizhumanov, A. A., and Naumova, O. V. (1995)
Vestnik MGU, Ser. 16, Biologiya, No. 2, 37-41.
22.Nagai, K., and Suda, T. (1988) Meth. Findings
Exp. Clin. Pharmacol., 10, 497-507.
23.Stvolinsky, S., Kotlobai, A., and Boldyrev, A.
(1995) Eksp. Klin. Farmakol., 58, No. 2, 66-74.
24.Mzhel'skaya, T. I., and Boldyrev, A. A. (1997)
Zh. Evolyuts. Biokhim. Fiziol., 33, 688-698.
25.Boldyrev, A. A., Stvolinsky, S. L., Tyulina, O.
V., Koshelev, V. B., Hori, N., and Carpenter, D. (1997) Cell. Mol.
Neurobiol., 17, 259-271.
26.Suslina, Z. A., Fedorova, T. N., Stvolinsky, S.
L., Ryasina, T. V., Maksimova, M. Yu., and Boldyrev, A. A. (1998)
Abst. I Far-Eastern Conf. New Medical Technologies in Far East,
Far-Eastern Medical University, Khabarovsk, pp. 83-94.
27.Kuklei, M. L., and Gannushkina, I. V. (1997)
Dokl. RAN, 352, 416-419.
28.Boldyrev, A. A., Kukley, M. L., Stvolinsky, S.
L., and Gannushkina, I. V. (1996) in Proc. Int. Symp. on Natural
Antioxidants. Molecular Mechanisms and Heals Effects (Parcker, L.,
Traber, M. G., and Xin, W., eds.) AOCS Press, Champaign, Illinois, pp.
600-614.
29.Kochanek, P. M., and Hallenbeck, J. M. (1992)
Stroke, 23, 1367-1379.
30.Stvolinsky, S. L., Kukley, M. L., Dobrota, D.,
Matejovichova (Vachova), M., Tkac, I., and Boldyrev, A. A. (1999)
Cel. Mol. Neurobiol., 19, 45-56.
31.Stvolinsky, S. L., Dobrota, D., Mezeshova, V.,
Liptai, T., Pronaiova, N., Zalibera, L., and Boldyrev, A. A. (1992)
Biokhimiya, 57, 1317-1323.
32.Dobrota, D., Tkac, I., Mlynarik, V., Liptaj, T.,
Boldyrev, A. A., and Stvolinsky, S. L. (1997) in Neurochemistry.
Cellular, Molecular and Clinical Aspects (Teelken, A., and Korf,
J., eds.) Plenum Press, N. Y., pp. 177-182.
33.Mintorovitch, J., Yang, G., Shimizu, H.,
Kucharczyk, J., Chan, P., and Weinstein, Ph. (1994) J. Cereb. Blood
Flow Metab., 14, 332-336.
34.Boldyrev, A. A. (1998) in Phosphate Metabolism
in Neuronal Disorders. Proc. of WHO Meet. (Murer, H., Knochel, J.
P., Prilipko, L., and Bolis, C. L., eds.) Word Health Organization,
Geneva, pp. 99-113.
35.Dobrota, D., Matejovichova (Vachova), M.,
Kurella, E. G., and Boldyrev, A. A. (1999) Cel. Mol. Neurobiol.,
19, 141-149.
36.Kurella, E. G., Kukley, M. L., Tyulina, O. V.,
Dobrota, D., Matejovicova, M., Mezesova, V., and Boldyrev, A. A.
(1997) Ann. N. Y. Acad. Sci., 534, 661-665.
37.Boldyrev, A. A., Song, R., Lawrens, D., and
Carpenter, D. O. (1999) Neuroscience, 94, 571-577.
38.Gorkin, V. Z. (1981) Amine Oxidases and Their
Role in Medicine [in Russian], Meditsina, Moscow.
39.Dobrota, D., Stvolinsky, S., Kukley, M., Tkac,
I., Boldyrev, A., and Mezesova, V. (1997) Acta Neurobiol. Exp.,
57 (Suppl.), 53.
40.Mineeva, M. F., and Stvolinsky, S. L. (1996)
Byul. Eksp. Biol. Med., 121, 420-422.
41.O'Dowd, A., O'Dowd, J. J., O'Dowd, J. J. M.,
McFarlan, N., Abe, H., and Miller, D. (1992) J. Chromat.,
577, 347-353.
42.Prokop'eva, V. D., Laptev, B. I., and Afanas'ev,
S. A. (1992) Biokhimiya, 57, 1389-1392.
43.Rusakov, V. V., and Dolgikh, V. T. (1992)
Biokhimiya, 57, 1393-1397.
44.Alabovsky, V. V., Boldyrev, A. A., Vinokurov, A.
A., and Shchavratsky, V. Kh. (1997) Biochemistry (Moscow),
62, 77-87.
45.Alabovsky, V. V., Boldyrev, A. A., Vinokurov, A.
A., Gallant, S., and Chesnokov, D. N. (1999) Byul. Eksp. Biol.
Med., 127, 290-294.