Affinity Labeling of RNA-Polymerase II in the Transcriptionally Active Complex by a Phosphorylating Analog of the Initiation Substrate
L. K. Savinkova1*, A. A. Sokolenko1, V. A. Rau1, T. V. Arshinova1, Yu. V. Gerasimova2, N. V. Kudryashova2, and T. S. Godovikova2
1Institute of Cytology and Genetics, Siberian Branch of the Russian Academy of Sciences, Novosibirsk, 630090 Russia; E-mail: savsok@niboch.nsc.ru2Novosibirsk Institute of Bioorganic Chemistry, Siberian Branch of the Russian Academy of Sciences, Novosibirsk, 630090 Russia
* To whom correspondence should be addressed.
Received December 14, 1999; Revision received May 30, 2000
Affinity modification of RNA-polymerase II by a phosphorylating analog of the initiation substrate carrying a zwitterionic 5´-terminal phosphate group with a 4-N,N-dimethylaminopyridine residue (DMAP-pA) was studied during specific transcription initiation controlled by the late adenoviral promotor. Super-selective affinity labeling and standard conditions of affinity modification resulted in labeling a polypeptide with molecular weight corresponding to that of the third subunit of the enzyme, RPB3 (45 kD). The initiation substrate (ATP) protects RNA-polymerase II from modification. The third subunit may be involved in the formation of the substrate-binding site of the enzyme.
KEY WORDS: RNA-polymerase II, promotor, basal transcription factors, transcription initiation, DMAP-derivative of initiation substrate
RNA-polymerase II is composed of 12 subunits [1]. Unlike procaryotic RNA-polymerase, whose subunit topography is known in detail, the structural and functional roles of the eucaryotic RNA-polymerase subunits are incompletely understood. The large enzyme subunits RPB1 and RPB2 are the most characterized. These subunits are structural and functional homologs of the beta- and beta´-subunits of bacterial RNA-polymerase. They participate in binding of the template and substrates and in formation of the enzyme active site [2, 3]. The competent labeling method has been widely used in studies of active he site structure [4-8]. RNA-polymerases II isolated from yeast and wheat seedlings were labeled with photoactivated derivatives of uridine monophosphate and ribonucleotide primer; the labeling data indicate that the largest enzyme subunit RPB1 participates in the formation of the active site [4]. Riva et al. [5, 6] labeled RNA-polymerase from the sprout yeast S. cerevisiae with aldehyde derivatives of ribonucleotides with variable spacer length; it was demonstrated that the second enzyme subunit RPB2 is also involved in the formation of the nucleotide-binding site. In addition, it was shown that the second subunit of RNA-polymerase II isolated from human placenta participates in substrate binding [7]. Wlassoff et al. [8] mapped the catalytic region of the enzyme from proliferating yeast S. pombe with UTP analogs (having an arylazide group attached to C-5 atom of the base) incorporated into the 3´-end of RNA. In the latter study, two subunits, RPB1 and RPB2, were labeled, and labeling efficiency varied depending on the length of the spacer between the photoactivated group and the uracil base.
Substrate derivatives with various chemical groups were used to study the active site of RNA-polymerase II. Reagents with arylazide groups were activated by irradiation at the appropriate wavelength. An additional reagent was required for detection of the products of protein interaction with other reagents. For example, when carbonyl-containing reagents were used for irreversible attachment of an affinity reagent to the protein, the generated Schiff bases were reduced with NaBH4. These studies were performed with highly purified enzyme preparations [5, 6] or with partially purified cell extracts [7]. Denatured DNA from thymus [5, 6], poly[d(A-T)] [7], single-strand synthetic DNA [4], or specifically constructed template containing a deca cytidine sequence at 3´-end of one of the strands [8] were used as the template.
In the present work, a phosphorylating analog of the initiation substrate was used to study the active site of RNA-polymerase II; this analog enables chemical cross-linking during transcription and does not require additional activators. RNA-polymerase II and basal transcription factors were isolated for S. cerevisiae yeast. Supercoiled DNA containing the AdML promotor (-39/+20) was used as the template.
MATERIALS AND METHODS
RNA-polymerase II and basal transcription factors were isolated from the yeast Saccharomyces cerevisiae as described in [9]. Total protein concentration in the preparations of RNA-polymerase II and basal transcription factors was ~40 µg/ml. Analysis of protein composition by native electrophoresis through 4-20% polyacrylamide gel and detection with AgNO3 [10] indicated that the preparation was essentially free from contaminating proteins.
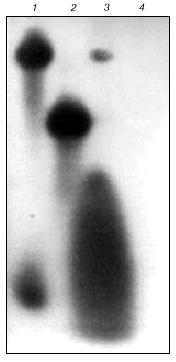
The reaction mixture for the synthesis of RNA in the system of RNA-polymerase II and basal transcription factors (final volume 30 µl) contained 80 mM HEPES-KOH (pH 7.8), 15 mM Mg(CH3COO)2, 3 mM beta-mercaptoethanol, 10% glycerol, 0.2 mM each of ATP, GTP, and UTP, 1 µl 0.1 µM [alpha-32P]CTP (3000 Ci/mmol), ~0.4 µg of the enzyme and basal transcription factors, 1 µg of DNA template, and 5 units of placental RNAse inhibitor. First, RNA-polymerase II was preincubated with DNA template to induce the formation of the promotor complex (including basal transcription factors TBP, TFIIB, and TFIIF) on the TATA box of the AdML promotor; this minimal complex of the basal transcription factors is enough to initiate specific transcription of supercoiled (and melted) DNA [11, 12]. The purification method used in the present work [9] yields RNA-polymerase II and minimal complex of the transcription factors as well as other basal transcription factors (including TFIIE and TFIIH) that should be present in this preparation. Then, the substrates were added to the reaction mixture and one of them (usually ATP) was 32P-labeled. Samples were incubated at 25°C for 30 min. To terminate the transcription, 100 µl of the stopping cocktail were added containing 20 mM Tris-HCl (pH 7.9), 0.5% SDS, and 10 mM EDTA. Transcripts were isolated by phenol-chloroform extraction [13] and analyzed by electrophoresis through 20% SDS-polyacrylamide gel in the presence of urea [13]. Typical results of analysis are shown in Fig. 1 (lane 3). Blockade of RNA synthesis in the presence of 1 µg/ml alpha-amanitin indicates that transcription was performed only by RNA-polymerase II (Fig. 1, lane 4).
Supercoiled DNA of the AdML promotor (-39/+20) incorporated into the pUC119 plasmid (a generous gift from Professor J. Kadonaga [14]) was used as the transcription template. RNA-polymerase II initiates RNA synthesis from this promotor and the 5´-end of RNA contains the pppApCpU sequence. Plasmid DNA was amplified in E. coli strain HB101. Plasmid DNA was isolated and purified by the standard molecular cloning protocols [15].Fig. 1. Autoradiogram of electrophoretically analyzed transcripts (20% polyacrylamide gel, 7 M urea): 1) heterologous 32P-labeled 20-base oligoribonucleotide (upper band) and [alpha-32P]CTP; 2) heterologous 32P-labeled 9-base oligoribonucleotide; 3) transcript; 4) transcription in the presence of alpha-amanitin.
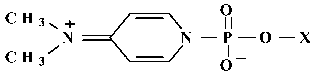
The phosphorylating derivative of AMP (4-N,N-dimethylaminopyridine derivative; (DMAP)pA) was synthesized from the sodium salt of AMP (optical density 10 at 260 nm). AMP was dissolved in 5 µl of water. To this solution, 45 µl of dimethylsulfoxide, 14 µl of a solution of 2,2´-dipyridylsulfide (3.3 mg; 50 µmol) in dimethylformamide, 25 µl of the solution of N,N-dimethylaminopyridine (3.7 mg; 300 µmol) in dimethylformamide, and 14 µl of the solution of triphenylphosphin (3.9 mg; 50 µmol) in dimethylformamide were sequentially added; the mixture was incubated at 37°C for 10 min. Ether (1 ml) was added to the mixture and the pellet was collected by centrifugation, washed with ether (3 times; 1 ml each time), dissolved in 100 µl of water, and immediately used as affinity modifier of RNA-polymerase II. The structure of the synthesized DMAP-derivative of the initiation substrate is shown in Fig. 2. Other AMP analogs (for example, formylphenyl derivatives of AMP) were previously used to study the active site of RNA-polymerase II [7]. We have shown that AMP (instead of ATP) can be used as the initiation substrate in the described transcription system containing RNA-polymerase II from S. cerevisiae (data not shown).
To affinity label RNA-polymerase II, the reaction medium containing the enzyme and basal transcription factors (0.4 µg) was preincubated with DNA template (1 µg) for 10 min at room temperature to assemble the pre-initiating complex on the TATA box of the AdML promotor. Then, initiation substrate analog DMAP-pA was added to 0.2 mM final concentration (the compound was either unlabeled or labeled with 32P) and the reaction mixture was incubated at 25°C for 5 min. Then, unlabeled CTP or [32P]CTP was added, and the incubation was continued for 20 min at the same temperature. The reaction was terminated by addition of the stopping cocktail. Then, 2.5% SDS, 5% beta-mercaptoethanol, 10% glycerol, and 0.02% bromophenol blue were added to the reaction mixture and samples were heated at 100°C for 5 min. The reaction products were analyzed by SDS-polyacrylamide gel electrophoresis in the Laemmli system [16]. Dried gels were exposed to X-ray film at -70°C.Fig. 2. Structure of phosphorylating analog of the initiation substrate.
The following reagents were used: Tris-HCl and beta-mercaptoethanol from Serva (Germany), glycerol and human albumin from Fluka (Switzerland), 2,2´-dipyridylsulfide and triphenylphosphin from Merck (Germany), [alpha-32P]CTP from Biosan Co. (Novosibirsk, Russia), and DMAP from Bergkamen (Germany).
RESULTS
RNA-polymerase II was affinity labeled during specific transcription in the system containing RNA-polymerase II, basal transcription factors, and supercoiled DNA template with AdML promotor. A phosphorylating analog of the initiation substrate with a zwitterionic 5´-terminal phosphate group containing 4-N,N-dimethylaminopyridine (DMAP-pA) residue was used as the affinity reagent. The enzyme was affinity modified by two methods including the classical variant with 32P-labeled DMAP-pA and competent labeling [17, 18]. Competent labeling is based on the fact [19] that in certain cases, an enzyme covalently bound to an affinity reagent can still interact with the second substrate. In the case of RNA-polymerases, this method was designated super-selective labeling [20-23].
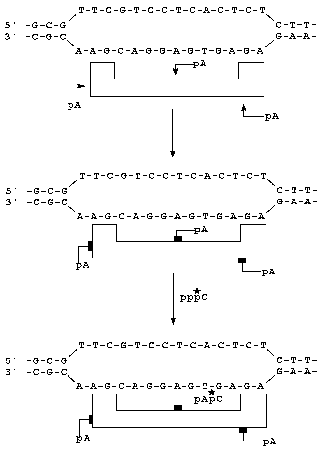
A scheme of super-selective labeling is shown in Fig. 3. An initiation substrate analog was complementary to the corresponding DNA base which is the point of transcription initiation. Radioactive substrate [alpha-32P]CTP used on the second labeling stage was complementary to the subsequent DNA base. Thus, RNA-polymerase was labeled under Watson-Crick complementary conditions.
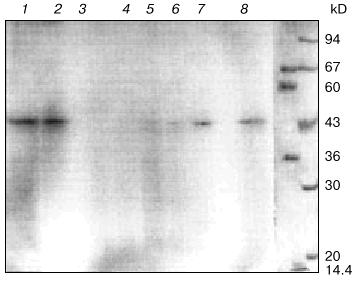
Addition of 32P-labeled DMAP-pA results in covalent attachment of the substrate analog to an amino acid residue of the enzyme contacting the phosphate group. Electrophoresis of the covalent complexes of the product RNA with protein indicates that a 45-kD protein was labeled (Fig. 4, lane 8). The molecular weight of the third subunit of RNA-polymerase II (RPB3) corresponds to the molecular weight of this protein [1, 24]. When the unlabeled initiation substrate analog was used and the second substrate was radioactive ([alpha-32P]CTP; super-selective labeling; Fig. 3), a 45-kD protein was covalently labeled as well (Fig. 4, lane 2). A similar pattern of RNA-polymerase II labeling was detected when the labeled initiation substrate analog (DMAP-[32P]A) was added to the reaction medium and then unlabeled CTP was added (Fig. 4, lane 1). Thus, in all protocols the same protein was labeled.Fig. 3. Scheme of super-selective labeling of RNA-polymerase II in the transcriptionally active complex.
To confirm whether the 45-kD subunit is labeled at the nucleoside triphosphate-binding site of the enzyme, protection with unlabeled ATP from active site modification by DMAP-[32P]A was tested during transcription. RNA-polymerase II and basal transcription factors were preincubated with DNA and 1-, 10-, 20-, and 50-fold molar excess unlabeled ATP was added with respect to the DMAP-[32P]A concentration with subsequent incubation for 5 min. Then, standard concentrations of DMAP-[32P]A and CTP were added and the incubation was continued. The data of Fig. 4 (lanes 4-7) indicate that preincubation with unlabeled ATP at the same concentration as the modified analog insignificantly influences the intensity of the protein labeling (Fig. 4, lane 7). Increasing ATP concentrations decrease the intensity of the labeled band, and when 50-fold molar excess of ATP was used, labeling of the 45-kD protein was undetectable (Fig. 4, lane 4). Labeling was also undetectable when RNA-polymerase II was preincubated with alpha-amanitin (Fig. 4, lane 3) indicating that during transcription, a subunit of transcription-catalyzing RNA-polymerase II was affinity labeled.Fig. 4. Electrophoresis through 10% SDS-polyacrylamide gel in the presence of 7 M urea of the products of affinity labeling of RNA-polymerase II in the transcription system: 1) DMAP-[32P]A + CTP; 2) DMAP-pA + [32P]CTP; 3) alpha-amanitin (1 µg/ml) + DMAP-[32P]A + CTP; 4-7) preincubation of enzyme with 50-, 20-, 10-, and 1-fold excess of ATP and subsequent incubation with DMAP-[32P]A + CTP; 8) DMAP-[32P]A. The two right lanes with legend on the right show the molecular weights of protein markers stained with Coomassie G-250. The molecular weight of the labeled protein (45 kD) was also verified with colored marker proteins (Sigma, USA) and a 45-kD marker protein staining yellow (data not shown).
DISCUSSION
In the present work, the subunit contacts of RNA-polymerase II with an initiation substrate analog were studied during specific transcription initiation. Covalent attachment of the DMAP-derivative of the initiation substrate affinity modifies a protein with molecular weight corresponding to that of the third RNA-polymerase II subunit, RPB3. The labeling can be considered highly specific because only a single protein is modified. Moreover, the protein is not labeled in the absence of the reagent and when the corresponding authentic substrate is substituted for the reagent (data not shown). When actinomycin D (250 µg/ml) was added to the system, the modification was also completely inhibited (data not shown). Natural ligand (ATP) protects the enzyme from modification (Fig. 4, lane 4). This suggests that the modification can be considered as affinity labeling. However, protection by ATP cannot guarantee that the active site is labeled. Apart from the catalytic site, regulatory sites can be modified. Unlike other methods, which do not allow direct conclusions about the structure of the affinity labeled sites, catalytic competence data directly indicate that the modification is specific for the enzyme active site. Since this criterion is fulfilled in our system, this is one of the strongest pieces of evidence supporting affinity modification of the enzyme.
Our data differ from the results of other authors who demonstrated that two large subunits, RPB1 and RPB2, are labeled when the enzyme interacts with initiation substrate analogs [2-8]. This disagreement could arise in several ways; first, use of the DMAP-derivative of the phosphate group without a spacer for covalent labeling provides for modification of amino acid residues directly contacting the alpha-phosphate group of the initiation substrate; second, reactive groups used had different structure; third, RNA-polymerase II was affinity labeled during promotor-controlled specific transcription; fourth, preparations of RNA-polymerase II and basal factors used for specific transcription were of relatively high purity. In other studies, either highly purified enzyme without transcription factors or enzyme in the presence of cellular or nuclear extract proteins were used [4-8]. In all cases, the transcribed DNA region was not promotor-controlled because denatured DNA [5, 6], specifically constructed DNA template with single-strand sequence at a 3´-end of one of the chains [8], or single- or double-stranded synthetic DNAs [4, 7] were used. In our study, the affinity labeling was started by the formation of the promotor complex on the TATA-containing AdML promotor, and this complex catalyzes specific transcription in the presence of the initiation substrate. During the initial stage of transcription initiation, the enzyme is complexed with the basal transcription factors, which can contribute to changes in enzyme conformation and a new conformation can significantly differ from that of the enzyme surrounded by a number of cellular or nuclear proteins. Even a slight change in conformation of the large multi-subunit complex of RNA-polymerase II can influence the accessibility of a particular adjacent amino acid residue (or residues) for covalent modification by the initiation substrate analog.
Differences in modification can be due to the adjacent positions of the regions responsible for the contacts between the first, second, and third enzyme subunits and to the overlapping of the functional regions of these subunits. It was shown that photoactivated UTP analogs covalently modified two large enzyme subunits under certain conditions [8]. Using partial proteolysis of the labeled subunits, the authors have determined the regions of attachment of the 3´-terminal UTP analog at the peptide level. One of these regions of RPB2 (amino acid residues 901-991) is involved in the formation of the active site and also participates in contacts with the third enzyme subunit, i.e., these functional regions overlap. Hence, under our conditions, the amino acid residue contacting the modified phosphate of the initiation substrate is located in the third subunit, and it can be adjacent to the region of contact or can partially overlap with the second subunit. Ishihama et al. [25-27] studied the inter-subunit contacts of the enzyme and demonstrated the presence of the contact regions of RPB3 with large subunits of RNA-polymerase II, RPB1 and RPB2; this suggests that functional regions of the enzyme located on different subunits can overlap.
Two large subunits of RNA-polymerase II, RPB1 and RPB2, are the most completely characterized. Until recently, participation in the formation of the protein-protein contacts during the enzyme assembly was assumed to be the main function of the third subunit (and fifth subunit as well) [25-29]. Temperature-sensitive mutations of various regions of RPB3 alter assembly and stability of the enzyme molecule, thus decreasing the viability of the yeast cell [30]. Recent work by Woychik et al. [31] described a mutant of the third subunit of yeast RNA-polymerase II; this mutation alters enzyme properties so that RNA-polymerase does not catalyze activated transcription, but the level of basal transcription is not influenced. Hence, RPB3 (via protein-protein contacts) can participate in signal reception regulating transcription catalyzed by RNA-polymerase II.
The data of the present work on participation of the third subunit in the formation of the substrate-binding site of the enzyme contribute to the understanding of the structure and function of RNA-polymerase II and mechanisms of enzyme catalysis.
We will continue this work to identify the amino acid residues of the third subunit of RNA-polymerase II participating in the formation of the active site of the enzyme.
The authors are indebted to Academician D. G. Knorre for his constant interest in this work, to Professor J. Kadonaga for a generous gift of plasmid pUC119 containing AdML promotor, to V. V. Bogachev for synthesis of nucleoside-5´-triphosphates, and to D. Semenov for synthesis of radiolabeled nucleotides.
This work was supported by the Russian Foundation for Basic Research (grant No. 97-04-49354).
REFERENCES
1.Young, R. A. (1991) Annu. Rev. Biochem.,
60, 689-715.
2.Kolodziej, P. A., and Young, R. A. (1991) Mol.
Cell. Biol., 11, 4669-4678.
3.Allison, L. A., and Moyle, M. (1985) Cell,
42, 599-610.
4.Cheng, N., Mougey, E. B., Kelly, S., and Dennis, D.
(1993) Biochemistry, 32, 2248-2253.
5.Treich, I., Carles, C., Sentenac, A., and Riva, M.
(1992) Nucleic Acids Res., 20, 4721-4725.
6.Riva, M., Carles, C., Sentenac, A., Grachev, M.,
Mustaev, A., and Zaychikov, E. (1990) J. Biol. Chem.,
265, 16498-16503.
7.Maksimova, T. G., Mustaev, A. A., Zaychikov, E. F.,
Polukhin, A. V., and Rait, V. K. (1990) Bioorg. Khim.,
16, 1145-1148.
8.Wlassoff, W. A., Kimura, M., and Ishihama, A.
(1999) J. Biol. Chem., 274, 5104-5113.
9.Sayre, M. H., Tschochner, H., and Kornberg, R. D.
(1992) J. Biol. Chem., 267, 23376-23382.
10.Wedrychowski, A., Olinski, R., and Hnilica, L. S.
(1986) Anal. Biochem., 159, 323-328.
11.Buratowski, S. (1994) Cell, 77,
1-3.
12.Parvin, J., and Sharp, P. A. (1993) Cell,
73, 533-540.
13.Manly, J. (1987) in Transcription and
Translation Methods (Hames, B., and Higgins, S., eds.) [Russian
translation], Mir, Moscow, pp. 92-98.
14.George, C. P., Lira-DeVito, L. M., Wampler, S.
L., and Kadonaga, J. T. (1995) Mol. Cell. Biol., 15,
1049-1059.
15.Maniatis, T., Frich, E., and Sambruck, J. (1984)
in Methods of Genetic Engineering. Molecular Cloning [Russian
translation], Mir, Moscow, pp. 175-185.
16.Laemmli, U. K. (1970) Nature, 227,
680-685.
17.Groman, E. V., Schults, R. M., and Engel, L. L.
(1975) J. Biol. Chem., 250, 5450-5454.
18.Groman, E. V., Schults, R. M., and Engel, L. L.
(1977) Meth. Enzymol., 46, 54-58.
19.Budker, V. G., Girshovich, A. S., and
Skobel'tsina, L. M. (1972) Dokl. AN SSSR, 207,
215-217.
20.Smirnov, Yu. V., Lipkin, V. M., Ovchinnikov, Yu.
A., Grachev, M. A., and Mustaev, A. A. (1981) Bioorg. Khim.,
7, 1113-1116.
21.Grachev, M. A., and Mustaev, A. A. (1982) FEBS
Lett., 137, 89-94.
22.Grachev, M. A., Hartmann, G. R., Maximova, T. G.,
Mustaev, A. A., Schaffner, A. R., Sieber, H., and Zaychikov, E. F.
(1986) FEBS Lett., 200, 287-290.
23.Grachev, M. A., Kolocheva, T. I., Lukhtanov, E.
A., and Mustaev, A. A. (1987) Eur. J. Biochem., 163,
113-121.
24.Conaway, R. C., and Conaway, J. W. (1996) in
Transcription Factors. Essential Data (Rickwood, D., and Hames,
B. D., eds.) Chichester-New York-Brisbane-Toronto-Singapore, pp.
5-11.
25.Kimura, M., Ishiguro, A., and Ishihama, A. (1997)
J. Biol. Chem., 272, 25851-25855.
26.Miyao, T., Honda, A., Qu, Z., and Ishihama, A.
(1998) Mol. Gen. Genet., 259, 123-129.
27.Kimura, M., Ishiguro, A., and Ishihama, A. (1997)
J. Biol. Chem., 272, 25851-25855.
28.Miyao, T., Yasui, K., Sakurai, H., Yamagishi, M.,
and Ishihama, A. (1996) Genes Cells, 1, 843-854.
29.Svetlov, V., Nolan, K., and Burgess, R. R. (1998)
J. Biol. Chem., 273, 10827-10830.
30.Mitobe, J., Mitsuzawa, H., Yasui, K., and
Ishihama, A. (1999) Mol. Gen. Genet., 262, 73-84.
31.Tan, Q., Linask, K. L., Ebright, R. H., and
Woychik, N. A. (2000) Genes Dev., 14, 339-348.