Functional Flexibility of the Transketolase Molecule
G. A. Kochetov
Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-3181; E-mail: kochetov@genebee.msu.su
Received April 27, 2001; Revision received May 24, 2001
Transketolase is the simplest representative of the thiamine diphosphate-dependent enzymes. It was the first of these enzymes for which X-ray analysis was performed. Based on the data of X-ray studies and using the mutagenesis technique, the nature of functional groups of the enzyme involved in the interaction with substrates and cofactors and in the coenzyme activation was defined. Thus, considerable achievements have been made in studying the structure of transketolase. However, there is relatively little information on the conformational flexibility of the enzyme molecule while it is functioning, i.e., during its interaction with cofactors and substrates and in the course of intermediate product formation. This review summarizes mainly the results obtained in the author's group, as well as those rare data on this subject that could be found in literature.
KEY WORDS: transketolase, conformational flexibility of enzymes, thiamine diphosphate, active site, active site nonequivalence, circular dichroism, thiamine diphosphate-dependent enzymes
Abbreviations: TK) transketolase; ThDP) thiamine diphosphate.
1. INTRODUCTION
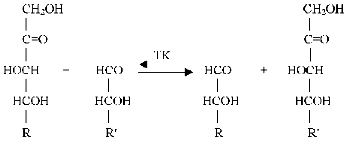
Transketolase catalyses transfer of a two-carbon fragment, a glycolaldehyde residue, from a ketose (donor substrate) to an aldose (acceptor substrate) (see Fig. 1).
Xylulose 5-phosphate, fructose 6-phosphate, sedoheptulose 7-phosphate, erythrulose, etc. serve as the donor substrates. Ribose 5-phosphate, erythrose 4-phosphate, glyceraldehyde 3-phosphate, glycolaldehyde, etc. are used as the acceptor substrates. Common features for the donor substrates are the following: a keto group adjacent to the splitting C-C-bond, a hydroxyl group on the C1 atom, and trans-orientation of the hydroxyl groups at C3 and C4 asymmetrical carbon atoms. Hydroxypyruvate and dioxyacetone, which contain no asymmetrical carbon atoms, are the exceptions. The transketolase reaction is reversible; however, the process becomes irreversible in the case hydroxypyruvate, which is subjected to decarboxylation.Fig. 1. Scheme of transketolase reaction.
Transketolase is a member of the group of transferases. It is a homodimer with molecular weight of 148.4 kD [1-3], and it has two active sites of equal catalytic activity [4, 5]. Its cofactors are thiamine diphosphate (Fig. 2) and bivalent metal ions. Native holoenzyme isolated from baker's yeast (the enzyme that we are considering further) contains only calcium (2 g-atoms per mol protein) [6]. Bivalent cations such as magnesium, manganese, cobalt, and some others [7, 8] can also serve as cofactors. Changing one cation for another does not alter significantly catalytic activity of the enzyme, but it does affect the character of interaction between the coenzyme and the apoenzyme (this will be described in detail later).
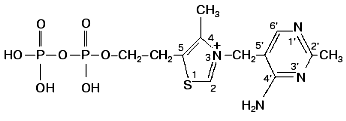
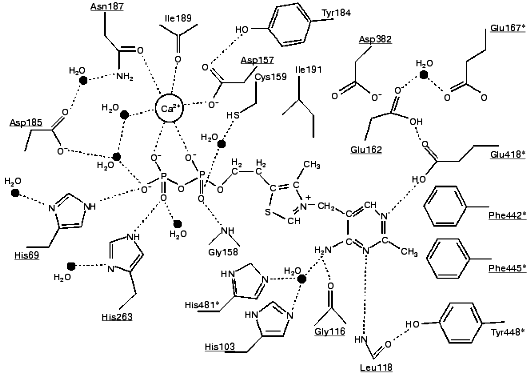
According to X-ray studies, each of the subunits of the dimeric transketolase molecule consists of three domains: N-, or PP-domain (residues 3-322); the middle, or Pyr-domain (residues 323-538), and the C-domain (residues 539-680) [9, 10]. The first two domains are involved in coenzyme binding, while the function of the C-domain remains unclear. The dimer structure of the enzyme is maintained mainly by the interactions between the PP- and Pyr-domains. The coenzyme is located in a deep cleft between the subunits. Only the C2 atom of the thiazole ring involved in the binding of the donor substrate is exposed to the solvent. The diphosphate group of the coenzyme binds to the apoenzyme both directly (forming hydrogen bonds with histidine residues 69 and 263, and with glycine residue 158) and by implication of the calcium ion (Fig. 3).Fig. 2. Thiamine diphosphate structure.
The thiazole ring of thiamine diphosphate within holoTK is bound to both subunits of the enzyme mainly by hydrophobic interactions. The amino pyrimidine ring is located in a hydrophobic pocket formed particularly by aromatic amino acid residues--tryptophans 442 and 445 and Tyr448 (Fig. 3). One of the functions of the hydrophilic Asp382 residue located in proximity to the thiazole ring is supposed to be compensation of positive charge of the thiazole ring [12].
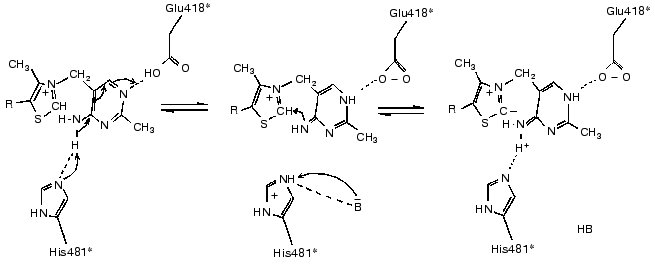
The basis of thiamine catalysis (or, to be exact, the mechanism of coenzyme activation), which is common for all thiamine diphosphate-dependent enzymes, includes two closely connected processes [11, 13]: a deprotonation of C2 of the ThDP thiazole ring (which is involved in the subsequent binding of the substrate) and functioning of the amino pyrimidine part of the coenzyme supporting the deprotonation of C2 of the thiazole ring (Fig. 4). The necessary condition for the mobility of the amino group proton in the pyrimidine ring is protonation of its N1´ atom [14], which is provided by the participation of glutamine acid residue 418 [15]. As a result, the pyrimidine ring acquires the ability to function as a proton donor as well as a proton acceptor, binding a proton by one nitrogen atom, and giving it up from another nitrogen atom, thus functioning as a proton transfer system (Fig. 4).Fig. 3. Scheme of the interaction of ThDP with the coenzyme-binding site of apoTK [11]. Conservative residues are underlined; residues of the second subunit are shown by asterisks; presumed hydrogen bonds are indicated by dash lines.
The present review summarizes the results of investigations related (directly or indirectly) to conformational flexibility of the transketolase molecule. Actually, detailed studies on this subject are beginning just now. Thus, it seems useful to summarize and to sort out the scattered experimental material accumulated to date.Fig. 4. Mechanism of deprotonation of C2 in ThDP thiazole ring [9].
2. OPTICAL CHARACTERISTICS OF TRANSKETOLASE
The interaction of ThDP with apoTK and the formation of catalytically active holoenzyme results in the appearance of a new band in the near UV absorption and CD spectra that is absent in the spectra of the original components [4, 16-19] (Fig. 5). Although the band was discovered a long time ago, its nature has not been determined yet. Initially it was attributed to the formation of a charge transfer complex between the coenzyme and a tryptophan residue of apoTK [17, 18]. The available experimental facts fit this hypothesis fairly well.
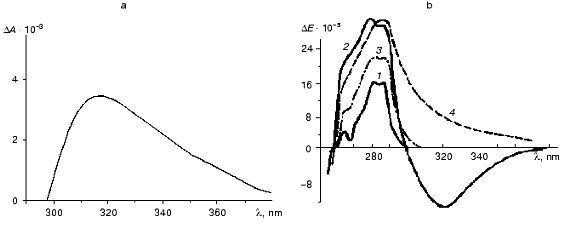
The emergence of the broad induced band in the absorption spectrum of holoTK with a diffuse maximum and a long “tail” extending to the visible region of the spectrum is one piece of evidence in favor of charge transfer complex formation [21]. Similar spectra were observed in a model system in the case of interaction of ThDP with tryptophan and its analogs, but not with other amino acids [17, 22, 23].Fig. 5. Differential absorption spectrum of holoTK against the sum of the spectra of apoTK and ThDP (a) [17] and CD spectra of transketolase with the coenzyme and the substrates (b) [20]: 1) apoTK; 2) holoTK; 3, 4) holoTK + fructose 6-phosphate or hydroxypyruvate, respectively.
Using perturbation UV spectrophotometry, four tryptophan residues exposed to the solvent were revealed within apoTK. The addition of ThDP results in twofold decrease in their quantity (also reducing the quantity of tyrosine residues accessible to the solvent by twofold) [24]. Thus, coenzyme binding to apoTK leads to a conformational change in the enzyme molecule and results in a decrease in the number of surface tryptophan residues by two (as noted before, TK has two active sites).
Reagents for tryptophan modify three residues of this amino acid in apoTK and only one in holoTK, i.e., the coenzyme prevents the modification of two tryptophan residues. The modification of two tryptophan residues in apoTK results in inactivation of the enzyme. The activity does not change if ThDP is added to apoTK before the treatment by the modifier [25].
The idea concerning the participation of a tryptophan residue in the formation of the charge transfer complex within holoTK was shaken when X-ray analysis revealed no tryptophan residues in the active site of the enzyme [9]. Then this function was attributed to phenylalanine 445, which was located in the active site of holoTK in the proximity of the ThDP pyrimidine ring. However, a mutant form of TK containing isoleucine instead of the phenylalanine appeared to retain catalytic activity, and its CD spectrum was identical to that of the wild type holoenzyme [11].
The possibility was also considered that the charge transfer complex is formed due to an interaction between chromophoric groups of apoTK approaching each other in the course of conformational changes of the apoenzyme induced by the coenzyme binding [26].
In any case, this optical effect appeared to be valuable in terms of methodology. Specific alteration of the spectrum caused by ThDP binding allowed determination of active site number in the TK molecule [4]; these data were confirmed by X-ray studies [9]. This approach was also successfully used for investigation of a rather complex process of the interaction between the coenzyme and the apoenzyme (see below). The addition of ketose (the donor substrate) to holoTK results in disappearance (or inversion--in the case of the irreversibly splitting substrate, hydroxypyruvate) of the band with a maximum at 315 nm (curves 3 and 4 at Fig. 5, respectively). These changes are caused by the formation of an intermediate product of the transketolase reaction, dihydroxyethyl-ThDP [27, 28]. The subsequent addition of aldose results in the recovery of the original holoenzyme spectrum (not shown). This allows a separate investigation of two consecutive steps of the transketolase reaction: binding and splitting of the donor substrate, and subsequent transfer of the glycolaldehyde residue formed in the course of the first step to the acceptor substrate [29].
Thus, irrespective of the nature of the induced band in the TK molecule, its emergence characterizes the formation of the active site of the enzyme; and the changes in its intensity after the addition of substrates are due to the enzymatic reaction. The cyclic changes in the induced optical activity observed in the course of substrate binding and transformation indicate changes in the microenvironment of the coenzyme, i.e., in the relative orientation of the coenzyme, substrates, and functional groups of the enzyme within the active site during the catalytic cycle.
3. INTERACTION OF THIAMINE DIPHOSPHATE WITH
APOTRANSKETOLASE
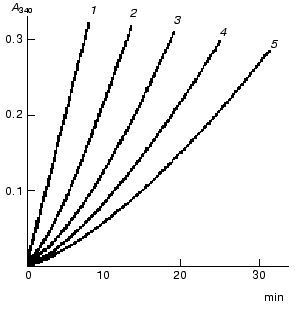
With a saturating concentration of the coenzyme, the rate of the transketolase reaction is constant. If the concentration of the coenzyme is low, a lag phase precedes the steady state. The length of the lag phase increases on decreasing the coenzyme concentration (Fig. 6). The lag period is attributed to a low rate of the interaction between ThDP and the apoenzyme. After the incubation of apoTK with the coenzyme, the length of the lag phase decreases; however, it does not disappear even at high coenzyme concentrations [30, 31].
Subsequent experiments showed that the interaction between apoTK and the coenzyme involved at least two steps (scheme):Fig. 6. Rate of the transketolase reaction at various concentrations of ThDP: 6.5 (1), 1.3 (2), 0.51 (3), 0.26 (4), and 0.13 (5) µM [29]. The activity was determined by the rate of NAD reduction using glyceraldehyde 3-phosphate dehydrogenase as a coupling enzyme.
In the first step of the reaction, easily dissociating and catalytically inactive complex TK.ThDP is formed, which is then slowly transformed into the stable and catalytically active holoenzyme [30].
Originally, the second slow step was accounted for as a slow association of inactive subunits into the catalytically active holodimer, which was supposed to be induced by the coenzyme. In other words, the coenzyme interacts with the separate subunits first, which then slowly associate yielding a dimer. In fact, in the absence of ThDP and at low concentrations of TK, which is normally used for the activity assay, the enzyme exists as a monomer. However, an increase in TK concentration leads to a decrease in the lag phase, but not to its disappearance [31]. Even at the concentrations of TK, when all of the enzyme exists in dimeric form, a pronounced lag phase is observed.
Thus, even if the dimerization process contributes to the existence of the lag phase, it cannot be the only cause of the low rate of interaction between the apoenzyme and the coenzyme yielding catalytically active holoenzyme. Later it was shown that the slow step in the formation of catalytically active holoTK was due to a conformational rearrangement of the enzyme molecule [32]. It is quite likely that the dimerization of TK subunits in the case of low enzyme concentration may also result in the conformational rearrangement. The fact of the fast exchange of the coenzyme of holoTK and free ThDP of the solution is directly related to the problem of conformational flexibility [33]. As mentioned above, the interaction of ThDP with apoTK proceeds in at least in two steps. The first one, fast and easily reversible, yields the catalytically inactive form of TK.ThDP (scheme). Then a slow conformational rearrangement of the TK molecule occurs yielding catalytically active holoenzyme, TK*-ThDP. This step is quasi-irreversible, and it needs tens of minutes for complete removal of ThDP from holoTK [31]. However, the exchange between thiamine diphosphate bound to holoenzyme and free ThDP proceeds within 20 sec [33]. In the first case of the coenzyme removal, the holoenzyme undergoes the slow step of conformational changes (TK*-ThDP --> TK.ThDP, see scheme), while in the second case it does not need to: free ThDP interacts directly with the apoenzyme in the conformation (TK*), which is prepared by the previous binding of the coenzyme and which differs from that of the original apoenzyme (TK in the scheme).
4. NONEQUIVALENCE OF ACTIVE SITES TOWARDS COENZYME BINDING
The values of the apparent (total) dissociation constants determined in the presence of calcium differ approximately by an order of magnitude [4, 5, 31, 34]. The active sites exhibit negative cooperativity towards ThDP [31, 34, 35]. If magnesium is used as a cofactor instead of calcium, similar behavior is observed although the difference is less pronounced.
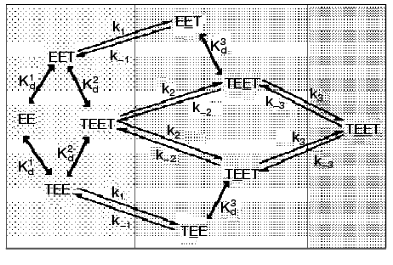
To clarify the cause of active site nonequivalence towards ThDP binding, a kinetic model of interaction between the coenzyme and apoTK was developed [32]. Figure 7 presented a scheme of this process allowing for the functioning of two active sites and also the fact that the coenzyme binds to each active site in two steps: initial fast binding, and then slow multipoint binding accompanied by conformational changes of the enzyme molecule [30, 36, 37]. According to X-ray studies, the original structures of both active sites are equivalent [9, 10], so ThDP can bind (the initial binding) to each of them with equal probability. Using the kinetic model considering the cooperative interaction of two originally identical active sites of apoTK, a two-step mechanism of ThDP binding to each of two active sites of the enzyme was analyzed. Kinetic parameters of the individual steps of this process were determined [32]. It was shown that the nonequivalence of the active sites towards coenzyme binding was due to an increase (approximately by an order of magnitude) of the reverse conformational transition rate constant for the second active site (k-3) compared to that of the first one (k-1). Thus, the nonequivalence of active sites within the holodimer results from conformational instability of one of them. In other words, there is no cooperative interaction between the active sites until ThDP binding is complete in both of them. After that, a destabilization of the secondary complex occurs in one of the sites. The destabilization is reversible and can take place in each of the two active sites with equal probability [32].
Structures of apo- and holoTK differ in the conformation of two loops in each of the subunits (residues 187-198 and 383-394), which is chaotic in apoenzyme and ordered in holoenzyme. Both of the loops are characterized by high flexibility, and they directly contact the coenzyme within holoTK [10, 38]. It is possible that the interdependent antiphase motion of these loops determines the interchangeable destabilization of the secondary complexes between the active sites of TK and the coenzyme [36].Fig. 7. Scheme of two-step process of coenzyme interaction with two active sites of a homodimer molecule of TK [36]. Designations: E) active site of apoTK; T) ThDP; ET) the primary complex; ET) the secondary complex (active holoenzyme); Kd1 and Kd2) dissociation constants of the primary complex of apoTK containing one or two molecules of ThDP; Kd3 = Kd2k1k-2/k-1k2) dissociation constant of the primary complex containing the second catalytically active site; ki and k-i) kinetic constants of direct and reverse conformational transition.
5. SUBSTRATE-INDUCED CHANGE IN HOLOTRANSKETOLASE
CONFORMATION
Conformational change of TK induced by donor substrate was originally discovered by perturbation spectrophotometry. As mentioned above, the coenzyme binding by transketolase resulted in a decrease in the quantity of aromatic residues exposed to the solution; the larger part of them appeared to be buried inside of the protein molecule, i.e., a more compact conformation of the molecule was formed. Subsequent addition of a ketose returned the number of aromatic acid residues exposed to the solvent virtually to the original level characteristic of apoTK [24].
The idea of a loose conformation of apoTK and more compact conformation of holoTK is in good agreement with a number of well-known facts. For example, the holoenzyme is more stable towards some denaturing actions such as low and high pH values [39], trypsin [25], and high temperatures. Presumably, the more compact structure of the holoenzyme maintains its stability in a non-operating state, providing conditions for the interaction with substrates. The looser conformation of the enzyme, which is formed in the course of substrate binding, is characterized by a more flexible surface that provides higher probability of contact between active site functional groups and various ligands. In such a state the enzyme structure is more sensitive to various influences, including regulatory effects.
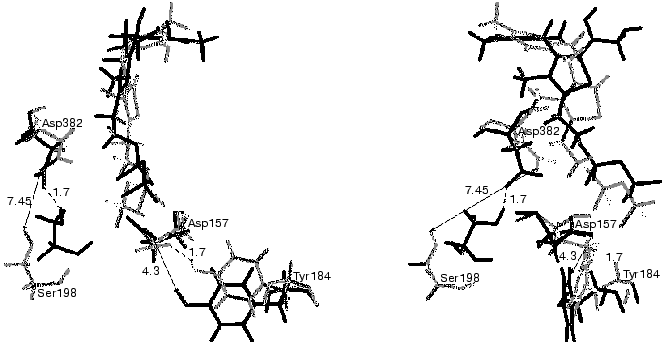
The complexes of holoTK with ketose or with the intermediate of the transketolase reaction have not been crystallized so far. The problem is that the substrate splits during crystallization of the holoTK-ketose complex, yielding the intermediate product of the transketolase reaction--dihydroxyethyl-ThDP bound to the enzyme. The latter is also unstable [40], and it brakes down during the crystallization yielding glycolaldehyde [41]. In this situation, computer simulation of molecular dynamics appeared to be useful. Figure 8 demonstrates superimposed fragments of the active site structures of holoTK (shown in gray) and a model holoTK-glycolaldehyde complex (shown in black) optimized for water surrounding using SYBYL software. As seen from the figure, the formation of the complex holoTK-glycolaldehyde results in an increase in the distance between the side chains of the functional residues Tyr184 [43, 44] and Asp157 [45] from 1.7 to 4.3 Å. The distance between Ser198 and the functional [10, 23] residue Asp382 decreases from 7.45 to 1.7 Å.
Thus, the hypothesis concerning conformational changes of the enzyme molecule induced by substrate binding/cleavage was confirmed, and the nature of these changes was determined.Fig. 8. Superimposed fragments of active site structures of holoTK (gray) and holoTK containing the bound intermediate (black). The front view in relation to the plane of the ThDP thiazole ring is on the right; the side view, on the left [42].
6. NONEQUIVALENCE OF TRANSKETOLASE ACTIVE SITES TOWARDS DONOR
SUBSTRATE
Nonequivalence of active sites towards substrates was revealed during investigation of inactivation of the enzyme by tetranitromethane. As seen from Fig. 9, holoTK as well as semi-holoTK (i.e., TK containing the coenzyme in one of the active sites only) are inactivated at the same rate, which is significantly lower, than the rate of apoenzyme inactivation (compare the curves 1 and 2 to curve 3). In the presence of ketose (donor substrate), the rate of holoTK inactivation significantly increases and becomes equal to the rate of apoTK inactivation (curves 4 and 3, respectively). However, the reaction stops when the extent of inactivation reaches approximately half of the original activity. The rate of semi-holoTK inactivation also increases in the presence of ketose to the same extent that the rate of holoenzyme inactivation in the presence of ketose. However, in the case of semi-holoTK, no inflection point at the level corresponding to 50% of the original activity (which is characteristic of holoTK inactivation) is revealed in the inactivation curve; the dependence is linear to the end of inactivation (curves 4 and 5).
Thus, ketose increases the inactivation rate of one active site and completely prevents the inactivation of the other active site of TK. This fact explains the two-phase character of holoTK inactivation. Since the initial phase of inactivation curve of holoTK coincides with the inactivation curve of semi-holoTK, we conclude that the active site containing the substrate exhibits increased rate of inactivation. Since the active sites of holoTK have the same catalytic activity, i.e., holoTK activity twofold exceeds the activity of semi-holo TK [4, 46], there is a strong probability that the substrate is contained in both the first active site and the second one (which maintains catalytic activity in the presence of substrate), but, presumably, in the different states--split and unsplit. In this case, the active sites function simultaneously, but in antiphase, i.e., when a ketose molecule is splitting in the first active site (the first step of transketolase reaction), another ketose molecule is being formed in the second one (as a result, the second step of transketolase reaction occurs--condensation of active glycolaldehyde with acceptor substrate) [44]. There are some data in literature supporting this assumption. It was shown that after the incubation of holoTK in the presence of fructose 6-phosphate labeled at the C1 atom, the radioactive label bound to the enzyme is distributed equally among fructose 6-phosphate and glycolaldehyde--the product of fructose 6-phosphate cleavage [47].Fig. 9. Effect of ligands on the kinetics of TK inactivation by tetranitromethane [44]. I) ApoTK (3), apoTK in the presence of fructose 6-phosphate (6) and semi-holoTK in the presence of fructose 6-phosphate (5); II) holoTK (1) and semi-holoTK (2); III) holoTK in the presence of fructose 6-phosphate (4); IV) control sample in the absence of the inhibitor (7).
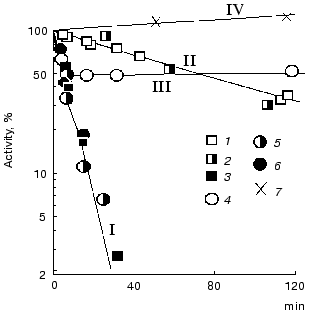
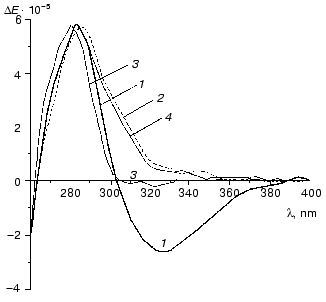
Figure 10 demonstrates CD spectra of holoTK in the presence of irreversibly and reversibly splitting substrates (hydroxypyruvate (curve 2) and xylulose 5-phosphate (curve 3), respectively). As noted above, the alteration of CD spectrum of holoTK in the region of 300-360 nm caused by the addition of ketose was due to its cleavage and formation of dihydroxyethylThDP-glycolaldehyde residue bound to ThDP. As seen from the figure, the amplitude of the spectral changes of holoTK observed in the presence of hydroxypyruvate (irreversibly splitting substrate) approximately twofold exceeds the amplitude of the spectral changes in the presence of xylulose 5-phosphate, a reversibly splitting substrate (Fig. 10, curves 2 and 3). In other words, the second reaction yields twofold less dioxyethylthiamine diphosphate (cleaved substrate) than the first one. This fits the idea that the splitting of xylulose 5-phosphate occurs only in one of two active sites. Hydroxypyruvate, the irreversibly splitting substrate, is decarboxylated in both active sites simultaneously.
The reaction of xylulose 5-phosphate splitting becomes irreversible in the presence of NADH and coupling enzymes, triosephosphate isomerase and glycerolphosphate dehydrogenase, providing the consumption of the first product of xylulose 5-phosphate splitting catalyzed by TK--glyceraldehyde 3-phosphate. In this case, the spectrum of xylulose 5-phosphate + TK becomes identical to the spectrum of hydroxypyruvate + TK (Fig. 10, spectra 2 and 4). Thus, in the latter case, the splitting of xylulose 5-phosphate, as well as hydroxypyruvate, occurs in both active sites yielding the same amount of dioxyethyl-ThDP.Fig. 10. CD spectra of transketolase [28]: 1) holoTK; 2) holoTK + hydroxypyruvate; 3) holoTK + xylulose 5-phosphate; 4) the same as 3, but in the presence of triosephosphate isomerase, glycerolphosphate dehydrogenase, and NADH.
It is noteworthy that, according to X-ray analysis [9, 10], subunits and active sites of TK have similar structure. Their optical characteristics and catalytic activity also do not differ. Consequently, the nonequivalence of active sites described above can only be detected at the functional level.
7. CONCLUSION
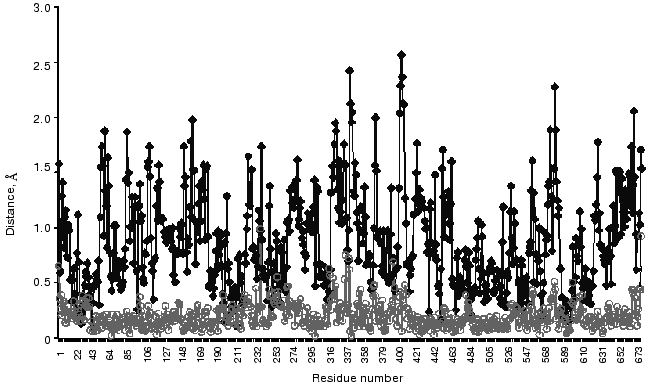
Finishing this review on conformational flexibility of the TK molecule, it is necessary to recall that, due to such flexibility, a crystal structure of an enzyme not always reflects adequately its conformation in solution [48, 49]. So, I would like to present the results obtained by molecular dynamics simulation of structural alterations in a crystal holoTK molecule for an aqueous environment [42]. Figure 11 demonstrates significant differences in the structure of the subunits in solution, while they are negligible in crystal (curves . and omicron). Two of four amino acid residues whose positions are particularly changeable are functionally essential. These residues are Asp157 (the peak 156-158 in Fig. 11) [11] and Tyr370 [50, 51].
The works performed in the author's group were supported in part by the Russian Foundation for Basic Research (grants 96-04-50730 and 99-04-49121) and by the Program of the Science Ministry of Russia “International Projects”.Fig. 11. Distances between C-alpha atoms of two superimposed subunits of crystal holoTK (omicron) and holoTK in an aqueous environment (.) [42].
I am very grateful to I. A. Bykova for great help in preparing the manuscript.
REFERENCES
1.Cavalieri, S. W., Neet, K. E., and Sable, H. Z.
(1975) Arch. Biochem. Biophys., 171, 527-532.
2.Belyaeva, R. C., Chernyak, V. Y., Magretova, N. N.,
and Kochetov, G. A. (1978) Biokhimiya, 43, 545-554.
3.Sundström, M., Lindqvist, Y., Schneider, G.,
Hellman, U., and Ronne, H. (1993) J. Biol. Chem., 268,
24346-24352.
4.Kochetov, G. A., Meshalkina, L. E., and Usmanov, R.
A. (1976) Biochem. Biophys. Res. Commun., 69,
839-843.
5.Meshalkina, L. E., and Kochetov, G. A. (1979)
Biochim. Biophys. Acta, 571, 218-223.
6.Kochetov, G. A., and Philippov, P. P. (1970)
Biochem. Biophys. Res. Commun., 38, 930-933.
7.Kochetov, G. A., and Philippov, P. P. (1970)
Dokl. Akad. Nauk SSSR, 191, 234-236.
8.Heinrich, P. C., Steffen, H., Janser, P., and Wiss,
O. (1972) Eur. J. Biochem., 30, 533-541.
9.Lindqvist, Y., Schneider, G., Ermler, U., and
Sundström, M. (1992) EMBO J., 11, 2373-2379.
10.Nikkola, M., Lindqvist, Y., and Schneider, G.
(1994) J. Biol. Chem., 238, 387-404.
11.Schneider, G., and Lindqvist, Y. (1998)
Biochim. Biophys. Acta, 1385, 387-398.
12.Meshalkina, L., Nilsson, U., Wikner, Ch.,
Kostikova, T., and Schneider, G. (1997) Eur. J. Biochem.,
244, 646-652.
13.Schellenberger, A. (1998) Biochim. Biophys.
Acta, 1385, 177-186.
14.Jordan, F., Chen, J., Nishikawa, S., and Wu, B.
S. (1982) Ann. N. Y. Acad. Sci., 378, 14-29.
15.Wikner, Ch., Meshalkina, L. E., Nilsson, U.,
Nikkola, M., Lindqvist, Y., Sundström, M., and Schneider, G.
(1994) J. Biol. Chem., 269, 32144-32150.
16.Kochetov, G. A., Usmanov, R. A., and Merzlov, V.
P. (1970) FEBS Lett., 9, 265-266.
17.Kochetov, G. A., and Usmanov, R. A. (1970)
Biochem. Biophys. Res. Commun., 41, 1134-1140.
18.Heinrich, C. P., Noack, K., and Wiss, O. (1971)
Biochem. Biophys. Res. Commun., 44, 275-279.
19.Kochetov, G. A., and Usmanov, R. A. (1972)
Dokl. Akad. Nauk SSSR, 202, 471-474.
20.Pustynnikov, M. G., Neif, X., Usmanov, R. A.,
Schellenberger, A., and Kochetov, G. A. (1986) Biokhimiya,
51, 1003-1016.
21.Shifrin, S. (1964) Biochim. Biophys. Acta,
81, 205-210.
22.Biaglow, J. E., Mieyal, J. J., Suchy, J., and
Sable, H. Z. (1969) J. Biol. Chem., 244, 4054-4061.
23.Mieyal, J. J., Suchy, J., Biaglow, J. E., and
Sable, H. Z. (1969) J. Biol. Chem., 244, 4063-4071.
24.Usmanov, R. A., and Kochetov, G. A. (1978)
Biokhimiya, 43, 1796-1804.
25.Heinrich, C. P., Noack, K., and Wiss, O. (1972)
Biochem. Biophys. Res. Commun., 49, 1427-1432.
26.Meshalkina, L. E., Wikner, Ch., Nilsson, U.,
Lindqvist, Y., and Schneider, G. (1996) in Biochemistry and
Physiology of Thiamin Diphosphate Enzymes (Bisswanger, H., and
Schellenberger, A., eds.) A. and C. Intermann, Wissenschaftlicher
Verlag, Prien, pp. 532-542.
27.Usmanov, R. A., and Kochetov, G. A. (1983)
Biokhimiya, 48, 550-558.
28.Solov'eva, O. N., Bykova, I. A., Meshalkina, L.
E., Kovina, M. V., and Kochetov, G. A. (2001) Biochemistry
(Moscow), 66, in press.
29.Usmanov, R. A., and Kochetov, G. A. (1982)
Biochem. Int., 5, 727-734.
30.Kochetov, G. A., and Izotova, A. E. (1973)
Biokhimiya, 38, 552-559.
31.Egan, R. M., and Sable, H. Z. (1981) J. Biol.
Chem., 256, 4877-4883.
32.Kovina, M. V., Selivanov, V. A., Kochevova, N.
V., and Kochetov, G. A. (1997) FEBS Lett., 418,
11-14.
33.Sidorova, N. N., Usmanov, R. A., Kuimov, A. N.,
and Kochetov, G. A. (1996) Biochemistry (Moscow),
61, 880-886 (Russ.).
34.Kochetov, G. A., Tikhomirova, N. K., and
Philippov, P. P. (1975) Biochem. Biophys. Res. Commun.,
63, 924-930.
35.Philippov, P. P., Tikhomirova, N. K., and
Kochetov, G. A. (1978) in Physical and Chemical Methods of Molecular
Biology [in Russian], MGU Publishers, Moscow, pp. 57-64.
36.Kovina, M. V., Selivanov, V. A., Kochevova, N.
V., and Kochetov, G. A. (1998) Biochemistry (Moscow),
63, 988-995.
37.Kochetov, G. A., Philippov, P. P., Razjivin, A.
P., and Tikhomirova, N. K. (1975) FEBS Lett., 53,
211-212.
38.Sundström, M., Lindqvist, Y., and Schneider,
G. (1992) FEBS Lett., 313, 229-231.
39.Kochetov, G. A., and Usmanov, R. A. (1970)
Biokhimiya, 35, 611-621.
40.Kochetov, G. A., Usmanov, R. A., and Mevkh, A. T.
(1973) Biochem. Biophys. Res. Commun., 54, 1619-1626.
41.Nilsson, U., Meshalkina, L. E., Lindqvist, Y.,
and Schneider, G. (1997) J. Biol. Chem., 272,
1864-1869.
42.Kovina, M. V., Tikhonova, O. N., Bykova, I. A.,
Ivanov, A. S., and Kochetov, G. A. (2000) Biochem. Biophys. Res.
Commun., 275, 968-972.
43.Kovina, M. V., Viryasov, M., Baratova, L., and
Kochetov, G. (1996) FEBS Lett., 392, 293-294.
44.Kovina, M. V., and Kochetov, G. A. (1998) FEBS
Lett., 440, 81-84.
45.Muller, Y. A., Lindqvist, Y., Furrey, W., Schulz,
G. E., Jordan, F., and Schneider, G. (1993) Structure, 1,
95-103.
46.Kuimov, A. N., Meshalkina, L. E., and Kochetov,
G. A. (1986) Biokhimiya, 51, 1908-1918.
47.Datta, A. G., and Racker, E. (1961) J. Biol.
Chem., 236, 624-628.
48.Karpen, M. E., and Brooks, Ch. L. (1996) in
Protein Structure Prediction (Sternberg, M. J. E., ed.) Oxford
University Press, pp. 229-261.
49.Brandt, N. N., Chikishev, A. Yu., Sotnikov, A.
I., Savochkina, Yu. A., Agapov, I. I., Tonevitskii, A. G., and
Kirpichnikov, M. P. (2001) Dokl. Akad. Nauk, 376,
687-689.
50.Kovina, M. V., Kuimov, A. N., and Kochetov, G. A.
(1993) Biochemistry (Moscow), 58, 1330-1340 (Russ.).
51.Kovina, M. V., Kuimov, A. N., and Kochetov, G. A.
(1993) Biochemistry (Moscow), 58, 1341-1350
(Russ.).