Interaction of Na,K-ATPase Catalytic Subunit with Cellular Proteins and Other Endogenous Regulators
O. D. Lopina
Department of Biochemistry, School of Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-3955; E-mail: od_lopina@mail.ru
Received April 29, 2001; Revision received June 21, 2001
Some mechanisms of regulation of Na,K-ATPase activity in various tissues including the phosphorylation of the catalytic subunit of the enzyme by different protein kinases (PKA, PKC, and tyrosine kinase) and the interaction of the alpha-subunit with different proteins (Na,K-ATPase beta- and gamma-subunits, ankyrin, phosphoinositide-3 kinase, and AP-2 protein) and endogenous digitalis-like factors are considered. Special attention is given to the search for possible protein-partners including melittin-like protein and to the mechanism of enzyme regulation connected with the change of Na,K-ATPase quaternary structure. A recently discovered role of Na,K-ATPase as a receptor providing signal transduction inside the cell not only by changing the concentration of biologically significant cations but also using direct interaction of the enzyme with the protein-partners is discussed.
KEY WORDS: Na,K-ATPase, signal transduction, protein kinases, beta- and gamma-subunits, conserved domains, melittin, digitalis-like factors
Na,K-ATPase (Na-pump) is an enzyme built into the plasma membrane which catalyzes ATP hydrolysis and coupled with it Na+ and K+ transfer through the membrane against the electrochemical gradient in accord with the following equation:
ATP + 3Nain+ + 2Kout+ --> ADP + Pi + 3Naout+ + 2Kin+,
where Nain+ and Kin+ are intracellular ions and Naout+ and Kout+ are extracellular ions. Thus, Na,K-ATPase, which is present in all animal cells, performs the electrogenic exchange of intracellular Na+ for extracellular K+.
Na,K-ATPase is a member of the family of P-type ATPases. ATP hydrolysis by these ATPases includes a step of transfer of the terminal phosphoryl group of ATP onto the carboxyl group of an aspartic acid residue that is located in the active site of the enzyme. After this step the hydrolysis of the acyl-phosphate bond occurs, which results in the release of Pi. Other ion pumps that transport biologically significant cations, in particular, gastric mucosa H,K-ATPase, a close relative of Na,K-ATPase, Ca-ATPases of plasma membrane and sarco(endo)plasmic reticulum (SERCA ATPases), H-ATPases from fungi and plants, and some ATPases of procaryotes (in particular, Kdp-ATPase from E. coli, which provides K+-accumulation) are also members of the P-type ATPase family [1].
Discovered more than 40 years ago, Na,K-ATPase continues to attract the attention of the researchers today. However, whereas soon after the discovery of this enzyme most efforts were directed to the study of the structure and function of the enzyme, now more attention is given to the problem of its regulation. This is due to the following circumstances. First, significant successes have been achieved in the study of Na,K-ATPase structure and function during recent years. Today we know the amino acid sequences of Na,K-ATPase alpha- and beta-subunits, we have data concerning the secondary structure of these polypeptides and their membrane topology. We now have a clear understanding of the mechanism of the enzyme: intermediate steps of ATP hydrolysis and the principal enzyme conformations that are present during different steps of the catalytic cycle have been revealed and the conformations with “occluded” Na+ and K+ are identified. Site-directed mutagenesis and chemical modification techniques have been used to identify the amino acids participating in ATP hydrolysis and cation transport. The data are presented in reviews devoted to Na,K-ATPase (see [1-4]). Also, Na,K-ATPase plays an important role in the functions of various tissues (see later), and this also attracts attention to the regulation of the enzyme.
STRUCTURE OF THE Na,K-ATPase PROTOMER
Na,K-ATPase consists of two polypeptide chains: the catalytic alpha-subunit and the beta-subunit, which is a glycoprotein that does not participate in the catalysis directly. The molecular masses of the alpha- and beta-subunit are 110 and 55 kD, respectively. Both subunits are integral membrane proteins and are bound to each other by noncovalent interactions: in purified preparations of Na,K-ATPase they are present in molar ratio 1 : 1.
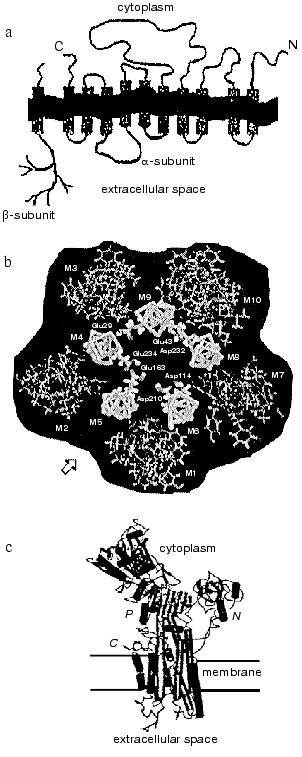
The polypeptide chain of the Na,K-ATPase alpha-subunit is packed into the lipid bilayer forming 10 transmembrane alpha-helical segments; the N- and C-termini of the chain are located in the cytosol (see reviews [2, 3]). Most amino acid residues are also located inside the cell forming a large cytoplasmic domain; significantly less of the alpha-subunit polypeptide chain is exposed outside the cell (figure, panel (a)). The transmembrane segments embedded in the membrane are in a close proximity; for example, there are data showing that even transmembrane segments M1-M2 and M7-M8, which are located in polypeptide chain very far from each other, can interact with each other [5]. Transmembrane segments appear to participate in the formation of the channel through which cations cross the membrane. The proposed arrangement of alpha-subunit transmembrane segments forming the channel is presented in the figure (panel (b)) [6]. High resolution X-ray analysis of Na,K-ATPase has not yet been carried out; however, recent X-ray analysis of sarcoplasmic Ca-ATPase [7], a protein closely related to the Na,K-ATPase alpha-subunit (the homology in amino acid sequence is only about 20%, but hydropathy profiles and apparently three-dimensional structure are very similar) give us an idea of a plausible look for the Na,K-ATPase alpha-subunit and its membrane topology (figure, panel (c)).
The polypeptide chain of the beta-subunit crosses the membrane only once [8]; a small N-terminal segment of the chain is located in the cytoplasm, whereas most of the subunit is located outside the cell (figure, panel (a)) (see reviews [3, 4]). Three disulfide bonds are formed on the extracellular part of the beta-subunit [8]. Beside this, on the extracellular part of the beta-subunit there are three sites of N-glycosylation to which branched carbohydrate chains are attached [9] (the molecular mass of the carbohydrate parts consists of about one third of the molecular mass of the entire beta-subunit). Amino acid residues located on the extracellular part of the protein in close proximity to the C-terminal fragment of the beta-subunit appear to participate in the association of the beta-subunit with the alpha-subunit [3, 10], as does its transmembrane fragment, which interacts with transmembrane fragments 9-10 of the alpha-subunit [5].a) Scheme illustrating the location of the polypeptide chains of Na,K-ATPase alpha- and beta-subunits in the membrane; b) proposed location of transmembrane segments of the alpha-subunit in the plane of the membrane (starting from the arrow from left to right: inner cycle: M5, M4, M9, M8, M6; outward cycle: M2, M3, M10, M7, M1); c) outlook of SR Ca-ATPase molecule (a close relative of Na,K-ATPase alpha-subunit) that is based on data of high resolution (2.6 Å) X-ray analysis.
QUATERNARY STRUCTURE OF Na,K-ATPase
There are numerous experimental data demonstrating that membrane Na,K-ATPase is an oligomer (most likely an asymmetric tetramer [11]) consisting of 2-4 alphabeta-protomers. Both negative [11] and positive [4] cooperative interactions are characteristic for this structure. Data on Na,K-ATPase oligomeric structure have been obtained using various methods including steady-state and pre-stationary state kinetics, titration of binding sites for different ligands (analysis of curves reflecting ligand binding and the binding stoichiometry), cross-linking of Na,K-ATPase subunits, and different physical methods (radioinactivation, rotational mobility measurements, fluorescence energy transfer). All these data are analyzed in recently published reviews [4, 11].
The formation of oligomeric complexes of Na,K-ATPase is due to the interaction between the alpha-subunits of the enzyme. This was determined by cross-linking methods [12, 13] and immunoprecipitation of native and chimeric molecules of the alpha-subunit. A peptide containing about 150 amino acid residues and located within the large cytoplasmic domain (Pro561-Gln709) appears to play a significant role in the process of association of alpha-subunits [14].
Solubilized non-denatured Na,K-ATPase is a mixture of associating-dissociating protomers, the relative amount of which depends on protein and ligands concentrations [15-17]. However, there is evidence showing that the lipid bilayer in which enzyme is embedded [11] and the interaction with protein-partners (for example, ankyrin, see later) control the interactions between Na,K-ATPase protomers.
ROLE OF Na,K-ATPase IN THE FUNCTIONING OF DIFFERENT
TISSUES
The regulation of Na,K-ATPase attracts attention because of the significant and various roles which Na,K-ATPase plays in the functioning of different tissues. Na,K-ATPase creates a gradient of Na+ and K+ concentrations that is used in different way in different tissues. The density of Na,K-ATPase in the cellular plasma membrane differs in different tissues: it varies from several molecules per square micrometer in erythrocytes [18] to several thousands molecules per square micrometer in nephron epithelium [19] or in the cells of excitable tissues. In all animal cells Na,K-ATPase participates in the creation of resting membrane potential and in the maintenance and regulation of cell volume [20]. However, Na,K-ATPase also performs specific functions that are characteristic for particular tissues.
Na,K-ATPase plays a significant role in the maintenance of ion conductivity in excitable tissues (neurons and muscle tissues). Voltage-gated ion channels (first Na- and then K-channels) become open during the propagation of action potential along the membrane. This results in the movement of these cations through the membrane in accordance with the gradient of their concentrations and to reversed membrane depolarization. Immediately after the passing of an action potential the change of Na+ and K+ concentrations is compensated by Na,K-ATPase because its activity during rest is limited by intracellular Na+ concentration (because of low intracellular Na+ concentration, the activity of this pump consists of only a few percent of the maximally possible activity; this means that there is significant reserved activity of this pump in excitable tissues [21]).
A Na-gradient is used in many tissues to provide the activity of different ion-exchange transport systems. The increase of intracellular Na+ concentration results in activation of Na+/Ca2+ exchanger that is embedded in the plasma membrane of different cells including neurons and cardiac and smooth muscle [22]. This may lead to the increase of intracellular Ca2+ concentration that in turn induces an increase of cardiac muscle contraction and vessel constriction [21]. Na+/H+ exchanger providing the exchange of intracellular H+ for extracellular Na+ plays an important role in the regulation of pH inside cardiac muscle [23], kidney epithelium [24] and other cells.
Na,K-ATPase is located in basolateral membrane of collecting duct epithelial cells and accomplishes Na+ transports from the cell to the extracellular medium [25]. It maintains low intracellular Na+ concentration that in turn is necessary for normal function of Na-transport systems located in the apical membrane of these cells including Na+,K+/Cl- cotransporter, Na+/Ca2+ and Na+/H+ exchangers, and Na-channels [24]. Thus the Na,K-ATPase located in basolateral membrane of kidney epithelial cells provides transepithelial Na+ transport from the nephron lumen inside the kidney.
Na+ concentration gradient is used in other tissues not only to provide the function of ionic exchangers mentioned above but also to accomplish transport of some metabolites (sugars, amino acids) into the cell. Inhibition of Na,K-ATPase and dissipation of Na+ concentration gradient results in a decreased rate of amino acid transport into the cell. That in turn may lead to inhibition of cell growth [26].
Thus the Na,K-ATPase which creates Na+ and K+ gradients through the plasma membrane is involved in different tissues in accomplishing different functions. This is a reason why the study of Na,K-ATPase regulation is so important to understand the regulation of the functions of various tissues.
Na,K-ATPase ISOENZYMES
The idea of the existence of different functions of Na,K-ATPase depending on tissue type is supported by the discovery of Na,K-ATPase isoenzymes. Four isoforms of the alpha-subunit (alpha1, alpha2, alpha3, and alpha4) [27-31] and three isoforms of the beta-subunit (beta1, beta2, and beta3) [32-35] that are encoded by different genes are found in vertebrates. These subunits may associate into alphabeta-dimers in different combinations. The resulting isoenzymes possess different kinetic properties [36] and have different patterns of distribution in various tissues and within one tissue during ontogenesis [37]. Synthesis of both corresponding mRNAs and the isoforms encoded by these mRNAs is under the control of various hormones [37, 38].
PHOSPHORYLATION OF Na,K-ATPase BY DIFFERENT PROTEIN
KINASES
Regulation of Na,K-ATPase activity by increase of biosynthesis of new isoforms of Na,K-ATPase subunits is a quite long process which requires several hours and even days. More rapid regulation of Na,K-ATPase activity may be realized using signal from membrane receptors for hormones. In many cases the phosphorylation of target proteins by different protein kinases is involved in this type of signal transduction.
The alpha1-isoform of Na,K-ATPase is the predominant form in kidney [39], where especially fine regulation of Na,K-ATPase activity is required. This isoform, also present in other tissues including heart, skeletal muscle, and neurons, can be phosphorylated by cAMP-dependent (PKA) [40-48], cGMP-dependent (PKG) protein kinases [49], Ca-phospholipid-dependent (PKC) protein kinase [40-43, 45, 50-55], and by tyrosine kinase [56].
PKA phosphorylates the only serine residue of the alpha-subunit, which is located in the C-terminal part of the molecule exposed to the cytosol: it is Ser943 in Na,K-ATPase alpha-subunit from rat kidney (Ser938 if you do not take into account first 5 amino acids that are removed during posttranslational modification) [43-45]. This site is conserved in all Na,K-ATPase isoforms and in gastric H,K-ATPase. The phosphorylation of Na,K-ATPase alpha-subunit in vitro is accomplished by both the catalytic subunit of PKA [41, 42, 45-47] and the complex of PKA (catalytic subunit-regulatory subunit) [42]. However, the consequences of this phosphorylation for enzyme activity are still not known. This is because of a technical problem that appeared during the study of this phenomenon: in vitro phosphorylation of Na,K-ATPase by PKA was achieved only in the presence of detergents [41-47]. We have found that in the presence of Triton X-100 PKA phosphorylated also the alpha-subunit of H,K-ATPase [57], which shares significant homology in amino acid sequence with Na,K-ATPase alpha-subunit (about 60%). Also, the phosphorylation of Na,K-ATPase by PKG in vitro also occurs only in the presence of the detergent [49].
Numerous attempts to find how PKA-mediated phosphorylation of Na,K-ATPase in vitro affects the enzyme activity gave controversial results: inhibition [40], activation [46], and the absence of any effect of phosphorylation on the Na,K-ATPase activity were found [43, 46]. One explanation of these contradictions was the following: the effect of Na,K-ATPase phosphorylation by PKA depends on the type of alpha-isoform that is phosphorylated. This idea was supported by the data of experiments where different isoforms of Na,K-ATPase were expressed in SF-9 cells of insects and endogenous PKA was activated in these cells. In this case, the activity of alpha1beta1 and alpha2beta1 isoenzymes was inhibited, whereas the activity of alpha3beta1-isoenzyme was activated after the phosphorylation [58].
Experiments with cell culture obtained from monkey kidney (line COS) have shown that activation of endogenous PKA by dibutyryl cAMP results in the inclusion of Pi into Ser943 of the alpha1-subunit and in the inhibition of wild type Na,K-ATPase that was expressed in these cells [44]. When a mutant form of Na,K-ATPase alpha-subunit in which Ser943 was replaced by Ala was expressed in COS cells, the activation of endogenous PKA does not lead to the inhibition of Na,K-ATPase activity. Thus, there is powerful evidence that the phosphorylation of Na,K-ATPase alpha1-subunit by PKA results in the suppression of Na,K-ATPase activity.
Successful choice of experimental conditions let us show that in vitro phosphorylation of duck salt gland Na,K-ATPase by the catalytic subunit of PKA in the presence of detergent leads to the inhibition of ATPase activity. A correlation was found between the inclusion of Pi into the alpha-subunit and the loss of enzyme activity under these conditions [47]. In this case we observed mainly phosphorylation of serine residues. Although the amino acid sequences of duck salt gland Na,K-ATPase subunits are still unknown, kinetic and immunological data [59, 60] and subunit N-terminal sequences [61] show that this enzyme is an alpha1beta1-isoenzyme. This conclusion is supported by data on the physiological function of this enzyme: Na,K-ATPase is purified from salt glands after keeping ducks on a diet with high content of NaCl that induces enzyme synthesis [62]. As a result of salt loading, not only kidney but also salt glands of birds, where a large amount of Na,K-ATPase is expressed, begin to participate in the process of salt excretion [63]. Thus, the Na,K-ATPase in salt glands has a function which is homological to the function of Na,K-ATPase in kidney epithelium, where this enzyme is mainly the alpha1beta1-isoenzyme. Finally, it should be noted that our data obtained in vitro are in good agreement with data obtained with live cells: the result of phosphorylation of Na,K-ATPase alpha1beta1-isoenzyme by PKA decreases its activity.
It remains unclear why PKA does not phosphorylate Na,K-ATPase in the medium without the detergent. We suggested that some component affecting the access of PKA to the phosphorylation site is removed during the purification procedure [47]. A recently published paper supports this suggestion [48]. It was shown that Na,K-ATPase from basolateral membrane of parotid glands is phosphorylated by PKA in medium without detergent but only in the presence of the PKA regulatory subunit and cAMP. Under these conditions association of the catalytic and regulatory (RII) PKA subunits is observed and the formed complex binds to membranes. This binding is accomplished by the interaction of the PKA regulatory subunit with the anchor protein AKAP, which has a molecular mass of about 150 kD. This experiment also showed that phosphorylation of Na,K-ATPase by PKA results in the loss of the ATPase activity [48].
Na,K-ATPase is phosphorylated by tyrosine kinase: in this case Pi is bound to Tyr10 of rat Na,K-ATPase alpha-subunit (numbering of amino acid residues is given for the alpha1-subunit that has not undergone posttranslational modification which results in the loss of the first five amino acids) [53]. Phosphorylation of Tyr10 in Na,K-ATPase alpha-subunit in kidney nephrons is induced by insulin and is accompanied by an increase of Na,K-ATPase activity. Phosphorylation does not proceed in the presence of genistin, a tyrosine kinase inhibitor.
PKC also phosphorylates Na,K-ATPase [40-43, 50-55], Pi being bound to two serine residues located in the N-terminal part of the alpha1-subunit: Ser11 and Ser18 (numbering of amino acid residues is given for the rat alpha1-subunit after removal of the first five amino acids) [51, 52]. Phosphorylation of Ser18 in vitro does change the Na,K-ATPase activity [52]. However when the phosphorylation of Ser18 occurred in nephron epithelium cells in response to the addition of dopamine, the activity of Na,K-ATPase was inhibited [53]. This is due to the decrease of the amount of Na,K-ATPase molecules in the membrane as result of endocytosis of phosphorylated molecules. If Ser11 and Ser18 are phosphorylated together (this is observed when PKC-beta is activated by phorbol ester), the activation of Na,K-ATPase that is due to the embedding of a new molecule of enzyme into the membrane is observed in the same cells [54].
Endocytosis of Na,K-ATPase induced by phosphorylation of Ser18 is accompanied by activation of phosphoinositide-3 kinase. This is due to the binding of the kinase to the N-terminal domain of the Na,K-ATPase alpha-subunit containing phosphorylated Ser18. The binding is provided by the interaction of the SH3-domain of the kinase with a region of the alpha-subunit enriched with proline (TPPPTTPE87) [55]. The binding and activation of phosphoinositide-3 kinase facilitates the binding of the alpha-subunit with adaptor AP-2 protein providing the inclusion of Na,K-ATPase into clathrin vesicles. However, Ser18, in contrast to Ser11, is found only in rat alpha1-subunit and is not present in pig and dog alpha1-subunit. This indicates that the described mechanism of Na,K-ATPase regulation is not universal but is species specific.
Thus, the phosphorylation of Na,K-ATPase by PKC may be a signal for endo- and exocytosis: the phosphorylation of Ser18 leads in some species to endocytosis and combined phosphorylation of Ser11 and Ser18--to exocytosis. Endocytosis may result from consecutive binding with Na,K-ATPase at first phosphoinositide-3 kinase and then the adaptor protein.
INTERACTION OF Na,K-ATPase alpha-SUBUNIT WITH OTHER
PROTEINS
The phosphorylation of Na,K-ATPase by PKC demonstrates that Na,K-ATPase is able to perceive signals that usually are transmitted in the cell with participation of cell proteins containing different conserved domains like the SH2- or SH3-domain, the plekstrin domain, and some others. Such interactions of proteins inside cells are now considered one of the important routes of signal transduction [64]. It should be noted that regulation of Na,K-ATPase via interactions with different protein-partners is still not well enough studied. Besides phosphoinositide-3 kinase and AP-2 mentioned above, the list of proteins for which interactions with the catalytic alpha-subunit of Na,K-ATPase has been described includes also the beta- and gamma-subunits of Na,K-ATPase and the cytoskeleton protein ankyrin.
beta-SUBUNIT
This subunit is a part of the Na,K- and H,K-ATPase but is not included in other P-type ATPases, for example, in Ca-ATPase of both plasma membrane and sarcoplasmic reticulum. The sensitivity of Na,K-ATPase to the cations Na+ and K+ is known to change depending on the isoform of the beta-subunit included in the alphabeta complex [64] because the beta-subunit affects the activation of Na,K-ATPase by extracellular K+ [66]. It was mentioned above that the beta-subunit, which does not participate in the process of catalysis directly, is nevertheless tightly bound to the alpha-subunit and is required for the insertion of the catalytic subunit into the membrane and the delivery of both subunits to the plasma membrane [67]. However, it appears that the role of the beta-subunit is not limited to these functions. The composition of sugars present in the carbohydrate moiety of the Na,K-ATPase beta-subunit is tissue specific [68]. The beta-subunit has also been identified in nervous tissue as a factor responsible for cell adhesion [69]. There is also old data about the influence of lectins (in particular, concanavalin A [70]) on the activity of Na,K-ATPase. All this information together suggests that the beta-subunit may interact with galectins (various lectins of animal origin), some of which regulate cell adhesion [71] and provide in this way signal transmission to the catalytic subunit of Na,K-ATPase. However the problem of interaction of galectins with Na,K-ATPase requires further investigation.
gamma-SUBUNIT AND RELATED PROTEINS
It was found that the sensitivity of Na,K-ATPase to monovalent cations in nephrons, where mainly the alpha1beta1-isoenzyme is present, depends on the region of the nephron. Because mainly one isoenzyme is present along the whole nephron this phenomenon might be explained by the presence of a 7.4-kD peptide (the gamma-subunit)that is bound to Na,K-ATPase in some regions of the nephron [72]. This subunit was found in preparations of purified Na,K-ATPase in the late 70s [73]. Because before that time this peptide was not found in preparations of purified Na,K-ATPase obtained from other sources, it was thought that Na,K-ATPase consists of only two subunits.
The gamma-subunit is a member of the membrane protein family that consists of proteins having only one transmembrane domain. Expressed in oocytes, such proteins serve as ion channels [74]. It was found that the addition of gamma-subunit to Na,K-ATPase changes its affinity to monovalent cations: the alpha1beta1gamma complex has lower affinity to Na+ and K+ than the alpha1beta1 complex [72]. In addition, there is apparently a family of proteins that are relatives (isoforms) of this subunit: two of them found in rat kidney have seven different amino acid residues in the N-terminal domain, and one of these two forms can be acylated [75]. Isoforms of the gamma-subunit have different electrophoretic mobility and differently affect the affinity of the alpha1beta1gamma complex for Na+ [72, 75]. These data suggest that the gamma-subunit in kidney is a factor regulating the sensitivity of Na,K-ATPase to transported cations.
A 15-kD protein sharing significant homology with the gamma-subunit and phospholeman was found in purified preparations of Na,K-ATPase from shark rectal glands. This protein is a target for phosphorylation by PKA and PKC. Antibodies against the alpha-subunit precipitate complex of this protein with Na,K-ATPase, and purified 15-kD protein is able to form complex with alpha-subunit [76]. Thus there appears to be a family of low molecular weight proteins that are regulators of the sensitivity of Na,K-ATPase to monovalent cations.
ANKYRIN
Ankyrins compose a family of proteins that are linkers between the integral membrane proteins and the proteins of cytoskeleton (spectrin and fodrin fibrils) Different isoforms of ankyrins are present in different tissues. They differ also depending on the type of membrane to which the cytoskeleton is attached (see review [77]). The more studied ankyrin from red cells is present in several isoforms, two of which having molecular masses of about 210 and 200 kD consist of three domains. One domain binds ankyrin to integral membrane proteins, another to spectrin (fodrin), and the third domain is a regulatory domain.
The membrane-binding domain of ankyrin contains so-called ankyrin repeats--conserved sequences that are present in numerous regulatory proteins. Ankyrin interacts with Na,K-ATPase alpha-subunit. Two sites of the alpha-subunit participate in the ankyrin binding [78]. A loop is formed in one of these sites where peptide Lys(154)-Ile-Met-Glu(157) is located; this loop binds to one of the beta-hairpins formed by ankyrin repeats through ionic interactions. A phenylalanine residue (Phe158) that is located in the beginning of the loop interacts with another beta-hairpin of the same ankyrin repeat through hydrophobic forces [79]. The binding of ankyrin is also provided by a cluster containing four amino acid residues: Ala(455)Leu-Leu-Lys(458) that is located in the third cytoplasmic domain of Na,K-ATPase [78].
It was mentioned above that Na,K-ATPase is incorporated into basolateral membrane of kidney and salivary gland epithelium. In choroid plexus (a single-layer secretory epithelium covering the surface of the brain ventricles) and in retinal pigment epithelium Na,K-ATPase is located in apical membrane. The location of Na,K-ATPase in certain region of cell plasma membrane is apparently due to the interaction of the enzyme with ankyrin and fodrin because Na,K-ATPase is precipitated by antibodies in a complex with these proteins. However, Na,K-ATPase is moved into the corresponding part of the membrane only after the epithelial cells become polarized [80-85].
There is still are no information on whether or not ankyrin binding affects Na,K-ATPase activity. In our experiments the addition of red cell ankyrin to purified Na,K-ATPase from duck salt glands resulted in the formation of an ankyrin-Na,K-ATPase complex: ankyrin was cosedimented with the enzyme during centrifugation and decreased the fluorescence of FITC-labeled Na,K-ATPase in the presence of K+. We did not find any effect of ankyrin on the enzyme activity although ankyrin was bound with Na,K-ATPase in molar ratio 1 : 4, this suggest that ankyrin may induce oligomerization of the enzyme. This effect of ankyrin on integral membrane proteins is quite well studied in experiments with band 3 protein from erythrocytes. Removal of ankyrin from the ankyrin-band 3 protein mixture was shown to result in the gradual transformation of band 3 tetramers into dimers and then into monomers. Dissociation of band 3 tetramers leads to the disappearance of most ankyrin-binding sites on the red cell membrane [86]; in other words, ankyrin interacts mainly with oligomeric complexes of band 3. Ankyrin appears to affect the Na,K-ATPase in a similar way.
MELITTIN AND MELITTIN-LIKE PROTEIN
Melittin, a bee venom peptide consisting of 26 amino acid residues and forming amphipatic alpha-helix with positive charge concentrated on the C-terminal region interacts with numerous proteins and with negatively charged lipid bilayers. In particular, it specifically reacts with calmodulin, apparently imitating the site of protein targets of calmodulin that participate in the binding of calmodulin [87]. Melittin also interacts with other proteins: calsequestrin [88], myosin light chain kinase [89], and phospholipase A2 [90]. This peptide inhibits P-type ATPases: sarcoplasmic reticulum (SR) Ca-ATPase [91], Na,K-ATPase [92], and gastric mucosa H,K-ATPase [93]. It was suggested at first that the inhibition of P-type ATPases by melittin is due to the aggregation of these integral proteins in the membrane that in turn results in the loss of enzyme activity [94]. However, careful analysis that we made using SR Ca-ATPase [95] and duck salt gland Na,K-ATPase (unpublished results) has shown that there is no correlation between the inhibition of these enzymes by melittin and the aggregation of these proteins induced by the peptide. It was shown also that P-type ATPases bind melittin specifically at sites located on the catalytic subunit of the enzyme [96, 97]. This is a peptide with sequence MI/LDPPR [98] which is present in the alpha-subunit of H,K-ATPase (M(603)DPPRAT) and in all isoforms of Na,K-ATPase alpha-subunit (MI(591)DPPRAA) and SERCA ATPases (M(599)LDPPRKE). This suggests that the site of melittin binding is a binding site of some intracellular proteins, and melittin imitates a module of these proteins that participate in binding.
An endogenous activator of phospholipase A2 [99] with molecular mass of about 26 kD was purified earlier using antibodies against melittin. Its C-terminal sequence is identical to the melittin sequence, and the N-terminal domain is very similar to a domain present in regulatory proteins from the family of beta-transducin [100]. A 68-kD protein interacting with antibodies against melittin was also found in gastric mucosa cells [101] and in homogenate from rabbit skeletal muscle (our unpublished data). Activation of HCl secretion in gastric parietal cells is accompanied by the movement of this protein from the cytoplasmic to the membrane fraction [101]. A melittin-like protein was purified from gastric mucosa using immunoaffinity chromatography [102]. This protein is present in gastric mucosa in rather high concentration (about 1% of the total protein content). The purified melittin-like protein stains poorly with Coomassie brilliant blue and is able to interact with Na,K- and SR Ca-ATPase (our unpublished data). It could be one of the cytoskeleton proteins interacting with P-type ATPases.
DIGITALIS LIKE FACTORS
Cardiac glycosides, specific inhibitors of Na,K-ATPase, are steroid O-glycosides containing in 17 position an unsaturated ring: gamma-lactone with 5 carbon atoms (cardenelides) or sigma-lactone with 6 carbon atoms (bufadienolides). A hydrocarbon chain located in 3beta-position usually consists of 1-5 monosaccharides. Digoxin and ouabain, substances of plant origin, are well known members of this group. These compounds were first used for treatment of cardiac failure more than 200 years ago because at low (nontoxic) concentrations they improve the function of cardiac muscle (so-called positive inotropic effect).
According to modern understanding, the mechanism of the effect of cardiac glycosides on cardiac muscle contraction is the following. Partial inhibition of Na,K-ATPase by these compounds leads to some increase of intracellular Na+ concentration that in turn activates Na+/Ca2+ exchanger. The consequence of this is a significant increase of intracellular Ca2+ concentration and increase of cardiac muscle contractility [21]. The site of cardiac glycoside binding is located on the extracellular part of the Na,K-ATPase alpha-subunit [103]. Sensitivity to these inhibitors differs depending on animal species (minimal sensitivity is characteristic for the enzyme from rat) and on the type of alpha-subunit that is present in the protomer of Na,K-ATPase (for enzyme obtained from one animal species I50 for inhibition of Na,K-ATPase containing alpha1-isoform is 500-1000-fold higher than the I50 for enzyme containing alpha2-isoform). This is connected mainly with substitution of certain amino acids in the ouabain-binding site of the Na,K-ATPase alpha-subunit.
The fact that substances of plant origin specifically affect the activity of an important animal cell enzyme leads to the idea that animals there may be endogenous digitalis (ouabain)-like factors that can specifically regulate the activity of this enzyme (see review [104]). This hypothesis is now proven: digitalis-like factors were found in hypothalamus and adrenocortical glands of animals [104]. The digitalis-like factor from hypothalamus was identified as an isomer of ouabain (see [105]). It was found also that the concentration of these factors in blood is increased during the development of some diseases, for example hypertension [106, 107], cataracts [108].
SIGNAL-TRANSDUCING FUNCTION OF Na,K-ATPase
A study of the effect of ouabain on cardiomyocytes and on other cultivated cells by Askari and coauthors recently led them to the astonishing conclusion that Na,K-ATPase is not only an ion pump maintaining Na+ and K+ concentration gradient, but also a receptor for digitalis-like factors participating in signal transduction from the surface inside the cell to mitochondria and nucleus using various signal transduction pathways. It was found at first that partial inhibition of Na,K-ATPase of non-muscle cells by ouabain or by low K+ concentration amplify the expression of protooncogenes c-fos and c-jun [109]. The increase of the level of c-fos and c-jun mRNAs and transcriptional factor AP-1 was observed in response to cell treatment by ouabain. After that an increase of hypertrophic growth occurred [110]. This response was proportional to the increase of intracellular Ca2+ level, was absent in medium without Ca2+, and was enhanced after the treatment of the cells by phorbol esters regulating the activity of PKC.
Further investigation of this effect has shown that partial Na,K-ATPase inhibition by ouabain led to activation of Ras and p42/44 mitogen-activated protein kinase (MAPK) [111]. Induction of alpha-actin synthesis was observed only after activation of Ras-dependent signal transduction pathway but not the p42/44 MAPK-dependent pathway. Repression of Na,K-ATPase alpha3-isoform synthesis required the activation of the Ras-p42/44 cascade and induction of c-fos and atrial natriuretic factor synthesis was mediated partially via the Ras-p42/44 cascade and partially via signal-transduction pathways that are independent of Ras and p42/44 MAPK. All these effects of ouabain were enhanced by inhibitors of PKC and by calmodulin antagonists. Thus Ca2+ and PKC are involved in a signal-transduction pathway connected with the inhibition of Na,K-ATPase, which then branches to independent cascades.
However, this complex mechanism does not represent all effects induced by Na,K-ATPase inhibition. Some of these effects are connected with generation of reactive oxygen species (ROS) inside the cell and are inhibited by antioxidants [112]. Also, in cardiomyocytes and in rat cells of A7r5 line the inhibition of Na,K-ATPase by ouabain induced the activation of Src kinase, its movement into a Triton-insoluble fraction, association with receptor for epidermal growth factor, and phosphorylation of this receptor in a site that differs from the main site of autophosphorylation [113]. This effect of ouabain as well as its effect on the generation of ROS does not depend on the change of intracellular Na+ and Ca2+ concentration. Also, these effects were found to only partially (about 50%) inhibit Na,K-ATPase, and this does not effect the viability of the cells.
Thus, in cells Na,K-ATPase can be an acceptor and transducer of signal using two pathways: 1) accomplishing its main function--creation of Na+ and K+ concentration gradients, and 2) by unknown ways that include activation of several systems of secondary messengers. These new data may explain why Na,K-ATPase itself is subject to regulation via various ways, some of which are now known, but others require further investigation.
This work was supported by the Russian Foundation for Basic Research, grant 01-04-48237.
REFERENCES
1.Moller, J., Juul, D., and le Maire, M. (1996)
Biochim. Biophys. Acta, 1286, 1-51.
2.Fambrough, D., Lemas, V., Takeyasu, K., Renaud, K.
J., and Inman, E. M. (1994) Current Topics in Membranes,
41, 45-69.
3.Lopina, O. D (1999) Biol. Membr. (Moscow),
16, 584-603.
4.Therein, A., and Blostein, R. (2000) Am. J.
Physiol., 279, C541-C566.
5.Sarvazyan, N. A., Modynov, N. N., and Askari, A.
(1995) J. Biol. Chem., 95, 270, 26528-26532.
6.Luzhkov, V. B., and Surkov, N. F. (1998) J. Mol.
Model., 4, 61-72.
7.Toyoshima, C., Nakasako, M., Nomura, H., and Ogawa,
H. (2000) Nature, 405, 647-655.
8.Kirley, T. (1989) J. Biol. Chem.,
264, 7185-7192.
9.Schalzing, G., and Gloor, S. (1994) Cell
Physiol. Biochem., 4, 96-114.
10.Beggah, A. T., Beguin, P., Jaunin, P., Peitsch,
M. C., and Horisberger, J. D. (1993) Biochemistry, 32,
14117-14124.
11.Askari, A. (2000) in Na,K-ATPase and Related
ATPases (Taniguch, K., and Kaya, S., eds.) Elsevier Science,
Amsterdam, pp. 17-26.
12.Askari, A. (1987) J. Bioenerg. Biomembr.,
19, 359-374.
13.Periyasama, S. M., Huang, W.-Y., and Askari, A.
(1983) J. Biol. Chem., 258, 9878-9885.
14.Koster, J. C., Blanko, G., and Mercer, R. W.
(1995) J. Biol. Chem., 270, 14332-14339.
15.Hayashi, Y., Mimura, K., Matsui, H., and Takagi,
T. (1989) Biochim. Biophys. Acta, 983, 217-229.
16.Mimura, K., Matsui, H., Takagi, T., and Hayashi,
Y. (1993) Biochim. Biophys. Acta, 1145, 63-74.
17.Hayashi, Y., Kameyama, K., Kobayashi, T.,
Hagiwara, E., Shinji, N., and Takagi, T. (1997) Ann. N. Y. Acad.
Sci., 834, 19-29.
18.Erdmann, E., and Hasse, W. (1975) J. Physiol.
(London), 251, 671-682.
19.Jorgensen, P. L. (1982) Biochim. Biophys.
Acta, 694, 27-68.
20.Kabakov, A. Yu. (1994) J. Theor. Biol.,
169, 51-64.
21.Akera, T., and Ng, Y. C. (1991) Life.
Sci., 18, 135-142.
22.Moore, E. D., Etter, E. F., Philipson, K. D.,
Carrington, W. A., Fogarty, K. E., Lifshitz, L. M., and Fay, F. S.
(1993) Nature, 365, 657-660.
23.Avkiran, M., and Snabaitis, A. K. (1999) J.
Thromb. Thrombolysis, 8, 25-32.
24.Greger, R. (2000) Am. J. Med. Sci.,
319, 51-62.
25.Nelson, W. J., and Veshnock, P. (1987)
Nature, 328, 533-537.
26.Shank, B. B., and Smith, N. E. (1976) J. Cell.
Physiol., 87, 377-387.
27.Schneider, J. W., Mercer, R. W., Caplan, M.,
Emanuel, J. R., Sweadner, K. J., Benz, E. J., Jr., and Levenson, R.
(1985) Proc. Natl. Acad. Sci. USA, 82,
6357-6361.
28.Shull, G. E., Greeb, J., and Lingrel (1986)
Biochemistry, 25, 8125-8132.
29.Sverdlov, E. D., Monastyrskaya, G. S., Broude, N.
E., Ushkaryov, Yu. A., Allikmets, R. L., Melkov, A. M., Smirnov, Yu.
V., Malyshev, I. V., Dulobova, I. E., and Petrukhin, K. E. (1987)
FEBS Lett., 217, 275-278.
30.Shamraj, O. I., and Lingrel, J. B. (1994)
Proc. Natl. Acad. Sci. USA, 91, 12952-12956.
31.Blanko, G., Melton, R., Sanchez, G., and Mercer,
R. W. (1999) Biochemistry, 38, 13661-13669.
32.Martin-Vasallo, P., Dackowski, W., Emanuel, J.
R., and Levenson, R. (1989) J. Biol. Chem., 264,
4613-4618.
33.Gloor, S., Antonicek, Sweadner, K. J., Pagliusi,
S., Frank, R., Moos, M., and Schachner, M. (1990) J. Cell.
Biol., 110, 165-174.
34.Malik, N., Canfield, V. A., Backers, M. C., Gros,
P., and Levenson, R. (1996) J. Biol. Chem., 271,
22754-22758.
35.Besirli, C. G., Gong, T. W., and Lomax, M. I.
(1997) Biochim. Biophys. Acta, 1350, 21-26.
36.Blanko, G., Koster, J. C., Sanchez, G., and
Mercer, R. W. (1995) Biochemistry, 34, 319-325.
37.Mobasheri, A., Avila, J., Cozar-Castellano, I.,
Brownleder, M. D., Trevan, M., Francis, M. J., Lamb, J. F., and
Martin-Vasallo, P. (2000) Biosci. Rep., 20, 51-91.
38.Sweadner, K. J. (1989) Biochim. Biophys.
Acta, 988, 185-220.
39.Barlet-Bas, C., Arystarkhova, E., Cheval, Marsy,
S., Sweadner, K. J., Modyanov, N., and Doucet, A. (1993) J. Biol.
Chem., 268, 11512-11515.
40.Bertorello, A. M., Aperia, A., Walaas, S. I.,
Nairn, A. C., and Greengard, P. (1991) Proc. Natl. Acad. Sci.
USA, 88, 11359-11362.
41.Chibalin, A. V., Vasilets, L. A., Hennekes, H.,
Pralong, D., and Geering, K. (1992) J. Biol. Chem., 267,
22378-22384.
42.Chibalin, A. V., Lopina, O. D., Petukhov, S. P.,
and Vasilets, L. A. (1993) J. Bioenerg. Biomembr.,
25, 61-66.
43.Feschenko, M. S., and Sweadner, K. J. (1994)
J. Biol. Chem., 269, 30436-30444.
44.Fisone, G., Cheng, S., Nairn, A. C., Czernik, A.
J., Hemmings, H. C., Hoog, J.-O., Bertorello, A. M., Kaizer, R.,
Bergman, T., Jornvall, H., Aperia, A., and Greengard, P. (1994) J.
Biol. Chem., 269, 9368-9373.
45.Beguin, P., Beggah, A. T., Chibalin, A. V.,
Burgener-Kairuz, P., Jasser, F., Mathews, P. M., Rossier, B. C.,
Cottecchia, S., and Geering, K. (1994) J. Biol. Chem.,
269, 24437-24445.
46.Cornelius, F., and Logvinenko, N. (1996) FEBS
Lett., 380, 277-280.
47.Murtazina, D. A., Petukhov, S. P., Rubtsov, A.
M., Storey, K. B., and Lopina, O. D. (2001) Biochemistry
(Moscow), 66, 865-874.
48.Kurihara, K., Nakanishi, N., and Ueha, T. (2000)
Am. J. Physiol., 279, C1516-C1527.
49.Fotis, H., Tatjanenko, L. V., and Vasilets, L. A.
(1999) Eur. J. Biochem., 260, 904-910.
50.Lowdens, J. M., Hokin-Neaverson, M., and Bertics,
P. J. (1990) Biochim. Biophys. Acta, 1052, 143-151.
51.Feschenko, M. S., and Sweadner, K. J. (1995)
J. Biol. Chem., 270, 14072-14077.
52.Feschenko, M. S., and Sweadner, K. J. (1997)
J. Biol. Chem., 272, 17726-17733.
53.Chibalin, A. V., Ogimoto, G., Pedemonte, C. H.,
Pressley, T., Katz, A., Feraile, Berggren, P.-O., and Bertorello, A. M.
(1999) J. Biol. Chem., 274, 1920-1927.
54.Efendiev, R., Bertorello, A. M., Pressley, T.,
Rousselot, M., Feraile, E., and Pedemonte, C. H. (2000)
Biochemistry, 39, 9884-9892.
55.Yudowski, G. A., Efendiev, R., Pedemonte, C. H.,
Katz, A., Berggren, P.-J., and Bertorello, A. (2000) Proc. Natl.
Acad. Sci. USA, 97, 6556-6561.
56.Feraille, E., Carranza, M. L., Gonin, S., Beguin,
P., Pedemonte, C., Rousselot, M., Caverzasio, J., Geering, K., Martin,
P. V., and Favre, H. (1999) Mol. Biol. Cell, 10,
2847-2859.
57.Murtazina, D. A., Petukhov, S. P., Storey, K. B.,
Rubtsov, A. M., and Lopina, O. D. (1999) Biosci. Rep.,
19, 109-114.
58.Blanko, G., Sanchez, G., and Mercer, R. (1998)
Arch. Biochem. Biophys., 359, 139-150.
59.Lopina, O. D., Sarvazyan, N. A., Askari, A., and
Boldyrev, A. (1995) Arch. Biochem. Biophys., 321,
429-433.
60.Martin, D. W., and Sachs, J. R. (1999)
Biochemistry, 38, 7485-7497.
61.Boldyrev, A. A., Lopina, O. D., Kenny, M., and
Johnson, P. (1995) Biochem. Biophys. Res. Commun., 216,
1048-1053.
62.Hopkins, B. E., Wagner, H., and Smith, T. (1976)
J. Biol. Chem., 251, 4365-4371.
63.Lingham, R. B., Stewart, D. J., and Sen, A. K.
(1980) Biochim. Biophys. Acta, 601, 229-234.
64.Pawson, T. (1995) Nature, 373,
573-580.
65.Blanko, G., and Mercer, R. W. (1995)
Biochemistry, 34, 319-325.
66.Jasser, F., Canessa, C. M., Horisberger, J. D.,
and Rossier, B. (1992) J. Biol. Chem., 267,
16895-16903.
67.Lutsenko, S., and Kaplan, J. H. (1992) Ann. N.
Y. Acad. Sci. USA, 671, 147-155.
68.Vladimirova, N. M., Murav'eva, T. I.,
Ovchinnikova, T. V., Potapenko, N. A., and Khodova, O. M. (1998)
Biol. Membr. (Moscow), 15, 349-352.
69.Gloor, S., Stonicek, H., Sweadner, K. J.,
Pagliusi, S., Frank, R., Moos, M., and Schachner, M. (1990) J. Cell.
Biol., 110, 165-174.
70.Swann, A. C. (1975) Biochim. Biophys.
Acta, 401, 299-306.
71.Ochieng, J., Warfield, P., Green-Jarvis, B., and
Fentie, I. (1999) J. Cell. Biochem., 75, 505-514.
72.Arystarkhova, E., Wetzel, R., Asinovski, N. K.,
and Sweadner, K. J. (1999) J. Biol. Chem., 274,
33183-33185.
73.Forbush, B., Kaplan, J. H., and Hoffman, J. F.
(1978) Biochemistry, 17, 3667-3676.
74.Minor, N. T., Sha, Q., Nichols, C. G., and
Mercer, R. W. (1998) Proc. Natl. Acad. Sci. USA, 95,
6521-6525.
75.Kuster, B., Shainskaya, A., Pu, H. X.,
Goldshleger, R., Blostein, R., Mann, M., and Karlish, S. J. (2000)
J. Biol. Chem., 275, 18441-18446.
76.Mahmmoud, Y. A., Vorum, H., and Cornelius, F.
(2000) J. Biol. Chem., 275, 35969-35977.
77.Rubtsov, A. M., and Lopina, O. D. (2000) FEBS
Lett., 482, 1-5.
78.Devarajan, P., Scaramuzzino, and Morrow, J. S.
(1994) Proc. Natl. Acad. Sci. USA, 91, 2965-2969.
79.Gorina, S., and Pavlevich, N. P. (1996)
Science, 274, 1001-1005.
80.Marrs, J. A., Napolitano, E. W., Murphy-Erdos,
C., Mays, R. W., Reichardt, and Nelson, W. J. (1993) J. Cell.
Biol., 123, 149-164.
81.Alper, S. I., Stuart-Tilley, A., Simmons, C. F.,
Brown, D., and Drenckhalhn, D. (1994) J. Clin. Invest.,
93, 1430-1438.
82.Gundersen, D., Orlovski, J., and
Rodriguez-Boulan, E. (1991) J. Cell. Biol., 112,
863-872.
83.Gundersen, D., Powell, S. K., and
Rodriguez-Boulan, E. (1993) J. Cell. Biol., 121,
335-343.
84.Nelson, W. J., and Hammerton, R. W. J. (1989)
J. Cell. Biol., 108, 893-902.
85.Dubreuil, R. R., Maddux, P. B., Grushko, T. A.,
and MacVicar, G. R. (1997) Mol. Biol. Cell, 8,
1933-1942.
86.Van Dort, H. M., Moriyama, R., and Low, P. S.
(1998) J. Biol. Chem., 273, 14819-14826.
87.Kataoka, M., Head, J. F., Seaton, B. A., and
Engelman, D. M. (1989) Proc. Natl. Acad. Sci. USA, 86,
6944-6948.
88.He, Z., Dunker, A. K., Wesson, C. R., and
Trumble, W. R. (1993) J. Biol. Chem., 268,
24635-24641.
89.Malencik, D. A., and Anderson, S. R. (1988)
Biochemistry, 27, 1941-1949.
90.Fletcher, J. F., and Jang, M. S. (1993)
Toxicon, 31, 669-695.
91.Voss, J., Birmachu, W., Hussey, D. M., and
Thomas, D. D. (1991) Biochemistry, 30, 7498-7506.
92.Murtazina, D. A., Mast, N. V., Rubtsov, A. M.,
and Lopina, O. D. (1992) Biokhimiya, 67, 54-61.
93.Cuppoletti, J., Blumenthal, K. E., and
Malinowska, D. H. (1989) Arch. Biochem. Biophys., 283,
249-257.
94.Birmachu, W., and Thomas, D. D. (1990)
Biochemistry, 29, 3904-3914.
95.Shorina, E. A., Mast, N. V., Rubtsov, A. M., and
Lopina, O. D. (1997) Biochemistry, 36, 13455-13460.
96.Cuppoletti, J., and Malinowska, D. H. (1992)
Mol. Cell. Biochem., 114, 57-63.
97.Cuppoletti, J., and Abbot, A. (1990) Arch.
Biochem. Biophys., 278, 405-415.
98.Cuppoletti, J., Blumenthal, K. E., and
Malinowska, D. H. (1996) Arch. Biochem. Biophys., 278,
263-270.
99.Clarke, M., Conway, T. M., Shorr, R. G., and
Crooke, S. T. (1987) J. Biol. Chem., 262, 4402-4406.
100.Peitsch, M. C., Borner, and Tschopp, J. (1993)
TIBS, 18, 292-293.
101.Cuppoletti, J., Huang, P., Kaetzel, M. A., and
Malinowska, D. H. (1993) Am. J. Physiol., 264,
G637-G644.
102.Cuppoletti, J., Rubtsov, A. M., Behbehani, G.,
and Lopina, O. D. (1995) Biophys. J., 68, A235.
103.Lingrel, J. B., van Huysse, J., O'Brien, W.,
Jewell-Motz, E., Askew, R., and Schultheis, P. (1994) Kidney
Int., 45, S32-S39.
104.Doris, P. (1994) PSEBM, 205,
202-212.
105.Buckalew, V. M., Jr., and Gonick, H. C. (1998)
Clin. Exp. Hypertens., 20, 481-488.
106.Rogaeva, E. A., Perova, N. V., Alexandrov, A.
A., Oganov, R. G., Lopina, O. D., and Boldyrev, A. A. (1987) Vopr.
Med. Khim., 4, 34-39.
107.Haddy, F. J. (1984) J. Cardiovasc.
Pharmacol., 6, S439-S456.
108.Tao, Q.-F., Hollenberg, N. K., and Graves, S.
W. (1999) Hypertension, 34, 1168-1174.
109.Nakagawa, Y., Rivera, V., and Larner, A. C.
(1992) J. Biol. Chem., 267, 8785-8788.
110.Peng, M., Huang, L., Xie, Z., Huang, W.-H., and
Askari, A. (1996) J. Biol. Chem., 271, 10372-10378.
111.Kometiani, P., Li, J., Gnudi, L., Kahn, B.,
Askari, A., and Xie, Z. (1998) J. Biol. Chem., 273,
15249-15256.
112.Xie, Z., Kometiani, P., Liu, J., Li, J.,
Shapiro, J. I., and Askari, A. (1999) J. Biol. Chem.,
274, 19323-19328.
113.Haas, M., Askari, A., and Xie, Z. (2000) J.
Biol. Chem., 275, 27832-27837.