H2O2 Sensors of Lungs and Blood Vessels and Their Role in the Antioxidant Defense of the Body
V. P. Skulachev
Department of Bioenergetics, Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-0338; E-mail: skulach@genebee.msu.su
Received April 2, 2001; Revision received August 2, 2001
This paper considers the composition and function of sensory systems monitoring H2O2 level by the lung neuroepithelial cells and carotid bodies. These systems are localized in the plasma membrane of the corresponding cells and are composed of O2*--generating NADPH-oxidase and an H2O2-activated K+ channel. This complex structure of the H2O2 sensors is probably due to their function in antioxidant defense. By means of these sensors, an increase in the H2O2 level in lung or blood results in a decrease in lung ventilation and constriction of blood vessels. This action lowers the O2 flux to the tissues and, hence, intracellular [O2]. The [O2] decrease, in turn, inhibits intracellular generation of reactive oxygen species. The possible roles of such systems under normal conditions (e.g., the effect of O2*- in air) and in some pathologies (e.g., pneumonia) is discussed.
KEY WORDS: reactive oxygen species, H2O2 sensor, neuroepithelial cells, lung, carotid bodies
I am very much obliged to Sergei Eugenievich Severin. I had a chance to take a course of his bright lectures on the biochemistry and gases of blood, he was the advisor of my bachelor, master, and Ph.D. studies and then--just a wise adviser and friend who was always ready to help. It was Sergei Eugenievich who attracted my attention to the issue of the biological functions of oxygen in 1955. This problem became one of the main topics of my biochemical studies.
It is obvious that oxygen simultaneously performs several functions essential for aerobic life. It plays the role of terminal electron acceptor for the respiratory chain which is the major energy-providing mechanism for respiring cells. Moreover, O2 is a substrate of oxygenases as well as of oxidases alternative to cytochrome oxidase of the respiratory chain. Some of these oxidases produce reactive oxygen species (ROS), namely O2*- and H2O2, instead of the inert H2O that is formed by cytochrome oxidase. ROS can be used by the organism as a tool to attack pathogens or as a signal. In the majority of cases, this is the signal for self-elimination of organelles, cells, organs, or even the entire organism. ROS are also formed nonenzymatically by “parasitic” chemical reactions of one-electron reduction of O2 by the respiratory chain electron carriers and some other natural reductants [1-3].
It is noteworthy that H2O2 can be reduced by Fe2+ and Cu+ to extremely dangerous hydroxyl radical (OH*), which is able to oxidize almost all cellular compounds including DNA. The high toxicity of ROS is due especially to OH*.
Higher organisms possess a multilevel system of anti-ROS defense. The first line of this system is to decrease the intracellular [O2] to a level still saturating cytochrome oxidase but insufficient for non-enzymatic ROS formation. One of the great achievements of evolution of aerobic life was the invention of cytochrome oxidase, an enzyme able to reduce O2 at a high rate at O2 levels even 100-fold lower than that in water under normal atmospheric pressure. As to ROS, their nonenzymatic formation parallels the decrease in [O2] according to the mass action law. This is why a decrease in intracellular [O2] over wide limits does not affect the cytochrome oxidase reaction but strongly inhibits nonenzymatic one-electron reduction of oxygen [1-4].
The strategy of higher organisms is that the rate of O2 delivery to a tissue, being high in the state of active work, decreases dramatically during rest. This effect is achieved first of all by means of a decrease in lung ventilation and constriction of blood vessels at the work-to-rest transition.
It should be emphasized that reactive oxygen species, rather than O2 per se, are dangerous. Therefore, it would be desirable for organisms to have a sensor monitoring the level of OH*, the most aggressive ROS. However, this is hardly possible since OH*, in fact, is too aggressive. On the way to the active site of the sensor, it would be discharged, spoiling thereby any cellular component including the hypothetical OH* sensor itself. On the other hand, it would be much easier to monitor the level of H2O2, a direct precursor of OH* in the chain of ROS interconversion reactions.
There are some indications that mammals possess at least two H2O2 sensors. One is located in cells of the lung neuroepithelial bodies, being responsible for constriction of the lung airways when the H2O2 level rises [5-7]. The other performs the same function in the blood vessels, being found in cells of the carotid body [8-10].
The two H2O2 sensors have very similar mechanisms as shown in the figure. They are composed of two independent protein systems, one H2O2-forming and another responding to H2O2. H2O2 is formed by an NADPH-oxidase which is of the same type as that found in the plasma membrane of phagocytes, where this enzyme forms O2*- to suppress pathogens (for review, see [11]). The enzyme oxidizes intracellular NADPH, transporting electrons through the membrane to its outer surface (FAD and special two-heme cytochrome b are involved). Here one-electron reduction of O2 to O2*- occurs. Two O2*- molecules dismutate to form O2 and H2O2. The latter interacts with the outer part of a K+ channel protein located in the plasma membrane. As a result, the channel is stabilized in its open conformation. If the O2 concentration drops, the rate of the NADPH-oxidase reaction decreases, [H2O2] decreases, the K+ channel closes, the membrane potential on plasma membrane decreases, and the cell is excited. The excitation gives rise to release of intracellular serotonin to the extracellular medium. Serotonin operates as a mediator of opening of the lung airways (it is significant that the neuroepithelial cells are located in places of branching of these airways). This situation is typical for periods of active work when mitochondrial cytochrome oxidase consumes large amounts of oxygen:
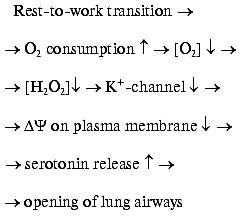
The work-to-rest transition results in a decrease of the O2 consumption in cells, a rise in the blood O2 concentration, and consequent lowering of O2 diffusion from the lungs to the blood. As a result, [O2] outside the neuroepithelial cells rises, NADPH oxidase is activated, [H2O2] increases, K+ channel opens, and serotonin is not released. The final event will be a decrease in the O2 supply to the body due to a constriction of airways [8].H2O2 sensor in the plasma membrane of the lung neuroepithelial and carotid cells [11]
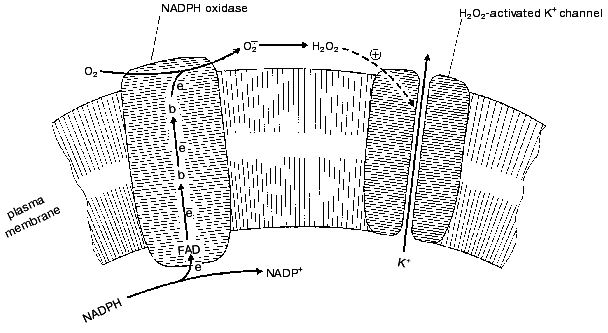
Similar events occur in the carotid cells. The only difference is that they release catecholamines instead of serotonin, causing dilatation of the blood vessels.
It is generally assumed that the lung neuroepithelial cells as well as carotid cells are O2 sensors [8]. From this point of view, however, it is difficult to understand why the O2 sensors of animals are organized in such a complex manner. It is known that the O2 sensors are already inherent in bacteria, where they are much simpler than in animals and are competent in [O2] monitoring by means of a direct O2 binding [Footnote: One of them was quite recently described by Alam and coworkers in Halobacterium salinarium and Bacillus subtilis. This is a single protein composed of two domains. The first (175 amino acid residues) is homologous to the animal myoglobin, whereas the second (amino acids 222-489) is very similar to the bacterial methyl-accepting proteins taking part in chemotaxis. They assume that the O2 binding by the first domain results in a conformational change transmitted to the second domain participating in transduction of the signal to the bacterial flagellum [12]. There are some reasons to suggest that heme-containing O2 sensors that bind O2 without its subsequent reduction operate also in animals, but their role consists in regulation of some events at the level of the cell rather than the organism (for review, see [13-15])].
These relationships might be explained by suggesting that the major function of the sensors in neuroepithelial and carotid cells is that of the antioxidant defense of the organism. The very fact that it is [H2O2] rather than [O2] that is monitored by these sensors allows the organism to effectively perform such a function. The described organization of the sensors causes constriction of the airways and the blood vessels due to an increase in [H2O2] independently of the reasons causing this increase. The reasons may be not only elevation of [O2] because of a decrease in O2 consumption in the tissues, but also activation of H2O2 production or inhibition of the H2O2 decomposition. This means that any damage to the antioxidant system of the body will actuate such an effective defense mechanism as a decrease in the O2 supply to tissues and cells. Such a response would be impossible if in the above-mentioned sensory cells a simple bacterial-type O2 sensor would be employed.
Quite recently, Weintraub and coworkers [16] reported that H2O2 activates an O2*--generating NAD(P)H oxidase in a non-phagocytic cell type of vascular origin (smooth muscle cells and fibroblasts). This means that production of H2O2 by, say, carotid cells can initiate a feed-forward mechanism amplifying the H2O2 signal. It is quite obvious that such a cascade may strongly reinforce the ability of ROS to down-regulate the O2 delivery to the tissues.
The concept described above can explain a number of physiological and pathological phenomena. For example, bronchospasms in the case of pneumonia may be a consequence of an increased O2*- production by the phagocyte NADPH oxidase in the inflamed regions [11], an event erroneously interpreted by the organism as a signal of oxygen danger. The same situation may take place as a result of a viral infection in lungs due to activation of xanthine oxidase. As reported by Maeda and coworkers [17-19], the influenza virus causes strong (by 2-3 orders of magnitude) activation in lungs of xanthine oxidase, an enzyme forming O2*- and H2O2 from O2 (concerning the possible significance of this effect for suppression of the viral infection, see [20]).
It seems possible that O2*- in the air (so-called negative aeroions) may regulate the work of the lungs. An increase in [O2*-] in the consumed air may be interpreted as a signal to decrease lung ventilation. Respectively, a decrease in [O2*-] will lead to hyperventilation, tissue [O2] increase, stimulation of ROS production, and, as a consequence, acceleration of ageing [2].
In this context, it should be noted that one of the most aggressive types of cancer, small cell lung carcinoma, represents, in fact, a result of malignant transformation of the lung neuroepithelial cells. These tumor cells still produce the same neuromediators [21, 22] and contain both NADPH oxidase and the H2O2-stimulated K+ channels [8]. Even more, malignant transformation was found to be oxygen-dependent [23].
It seems possible that the favorable effects of O2*--generating devices (see, e.g., [24, 25]) is also mediated by some H2O2 sensor(s). As shown by Goldstein and coworker [24], mice and rats die when kept under O2*--free conditions for 16 and 23 days, respectively. Most probably, the death is a consequence of deregulation of some functions of vital importance occurring due to the absence of signals from O2*-- and/or H2O2-sensors normally reporting about the level of these reactive oxygen species in airways. These signals might be produced by either lung neuroepithelial bodies (see above) or the so-called vomeronasal organ [26, 27]. There are indications that in large cities the air [O2*-] is strongly reduced due to antioxidant actions of products of decomposition of rubber and some other polymers [28, 29]. The air O2*- deficiency could be compensated by artificial O2*--generators. However, here we should be very careful since hyperproduction of O2*- can lead to catastrophic consequences due to constriction of the lung airways. This is why the use of O2*- generators should be considered only after detailed investigation of their effects on lung function. In any case, it is very probable that air O2*- monitoring should be useful to improve conditions of existence of humans in the modern world.
REFERENCES
1.Skulachev, V. P. (1995) Mol. Biol. (Moscow),
29, 709-715.
2.Skulachev, V. P. (2000) Oxygen and Programmed
Death Phenomenon. First Severin Lecture [in Russian], Russian
Biochemical Society, Moscow.
3.Skulachev, V. P. (1999) Mol. Asp. Med.,
20, 139-184.
4.Skulachev, V. P. (1994) Biochemistry
(Moscow), 59, 1433-1434.
5.Skulachev, V. P. (1996) Quart. Rev.
Biophys., 29, 169-202.
6.Lopez-Barneo, J. (1994) Trends Neurosci.,
17, 133-134.
7.Fu, X. W., Wang, D., Nurse, C. A., Dinauer, M. C.,
and Cutz, E. (2000) Proc. Natl. Acad. Sci. USA,
97, 4374-4379.
8.Wang, D., Youngson, C., Wong, V., Yeger, H.,
Dinauer, M. C., de Miera, E. V.-S., Rudy, B., and Cutz, E. (1996)
Proc. Natl. Acad. Sci. USA, 93, 13182-13187.
9.Youngson, C., Nurse, C., Yeger, H., and Cutz, E.
(1993) Nature, 365, 153-155.
10.Wyatt, C. N., Wright, C., Bee, D., and Peers, C.
(1995) Proc. Natl. Acad. Sci. USA, 92, 295-299.
11.Jones, O. T. G., and Hancock, J. T. (2000) in
Free Radicals and Inflammation (Winyard, P. G., Blake, D. R.,
and Evans, C. H., eds.) Birkhauser Verlag, Basel, pp. 19-44.
12.Hou, S., Larsen, R. W., Boudko, D., Rilley, C.
W., Karatan, E., Zimmer, M., Ordal, G. W., and Alam, M. (2000)
Nature, 403, 540-543.
13.Semenza, G. (1999) Cell, 98,
281-284.
14.Duchen, M. R. (1999) J. Physiol.,
516, 1-17.
15.Wenger, R. H. (2000) J. Exp. Biol.,
203, 1253-1263.
16.Li, W.-G., Miller, F. J., Jr., Zhang, H. J.,
Spitz, D. R., Oberley, L. W., and Weintraub, N. L. (2001) J. Biol.
Chem., 276, 29251-29256.
17.Akaike, T., Suga, M., and Maeda, H. (1998)
Proc. Soc. Exp. Biol. Med., 217, 64-73.
18.Akaike, T., Ando, M., Oda, T., Doi, T., Ijiri,
S., Araki, S., and Maeda, H. (1990) J. Clin. Invest., 85,
739-745.
19.Maeda, H., and Akaike, T. (1998) Biochemistry
(Moscow), 63, 854-865.
20.Skulachev, V. P. (1998) Biochemistry
(Moscow), 63, 1438-1440.
21.Gazdar, A. F., Helman, L. J., Israel, M. A.,
Russel, E. K., Linnoila, R. I., Mulshine, J. L., Schuller, H. M., and
Park, J. G. (1988) Cancer Res., 48, 4078-4082.
22.Moody, T. W., Pert, C. B., Gazdar, A. F., Carney,
D. N., and Minna, J. D. (1981) Science, 214,
1246-1248.
23.Schuller, H. M. (1991) Exp. Lung Res.,
17, 837-852.
24.Goldstein, N. I., and Arshavskaya, T. V. (1997)
Z. Naturforsch., 52c, 396-404.
25.Norman, G. E. (2001) Biokhimiya,
66, 123-126.
26.Meredith, M. (1991)J. Steroid Biochem. Mol.
Biol., 39, 601-614.
27.Berliner, D. L., Monti-Bloch, L., Jennings-White,
C., and Diaz-Sanchez, V. (1996) J. Steroid Biochem. Mol. Biol.,
58, 259-265.
28.Belyakov, V. A., Burlakova, E. B., Vasil'ev, R.
F., Arkhipova, G. V., Fedotova, I. B., and Chernyavskaya, L. I. (1994)
Dokl. RAN, 336, 124-126.
29.Belyakov, V. A., Vasil'ev, R. F., Trofimov, A.
V., and Fedorova, G. F. (1997) in Free Radicals in Biology and
Environment (Minisci, F., ed.) Kluwer Academic Publishers,
Dordrecht, pp. 233-250.