Structure and Functions of Classical Cadherins
D. B. Ivanov, M. P. Philippova, and V. A. Tkachuk*
Laboratory of Molecular Endocrinology, Institute of Experimental Cardiology, Russian Cardiology Research and Production Association, Ministry of Public Health of the Russian Federation, 3-ya Cherepkovskaya ul. 15a, Moscow, 121552 Russia; fax: (095) 414-6713* To whom correspondence should be addressed.
Received May 17, 2001; Revision received July 10, 2001
Cadherins are a family of membrane receptors that mediate calcium-dependent homophilic cell-cell adhesion. Cadherins play a key role in the regulation of organ and tissue development during embryogenesis. In adult organisms, these proteins are responsible for formation of stable cell-cell junctions and maintenance of normal tissue structure. Disruption in expression or function of cadherins may cause uncontrolled cell migration and proliferation during tumor development. This review focuses on the structure and physiological functions of classical cadherins.
KEY WORDS: cadherins, cell-cell adhesion, morphogenesis, signaling, oncogenesis
Cadherins are a superfamily of adhesion molecules that mediate Ca2+-dependent cell-cell adhesion in all solid tissues of the organism [1-9]. This superfamily involves: 1) classical cadherins that are the major component of cell-cell adhesive junctions; 2) desmosomal cadherins (desmocolins and desmogleins); 3) protocadherins; 4) some other cadherin-related molecules (e.g., the fat protein of Drosophila). Cadherin-mediated cell-cell junctions are formed as a result of interaction between extracellular domains of identical cadherins, which are located on the membranes of the neighboring cells. The stability of these adhesive junctions is ensured by binding of the intracellular cadherin domain with the actin cytoskeleton. Such highly specific homophilic cell-cell adhesion plays a key role in tissue and organ development during embryogenesis and in maintenance of normal tissue structure in the adult organism. In this review, special attention is focused on the structure and functions of classical cadherins.
STRUCTURE OF CLASSICAL CADHERINS
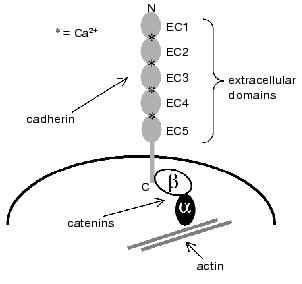
The majority of members of the cadherin superfamily are transmembrane glycoproteins that pass the membrane only once. The N- and C-termini of the cadherin protein chain are located outside and inside the cell, respectively (Fig. 1). The extracellular portion of the cadherin molecule consists of a varying number of so-called cadherin domains that are highly homologous to each other. Each domain is comprised of approximately 110 amino acid residues [10]. Classical cadherins contain five cadherin domains that are commonly designated as EC1-EC5 (beginning with the N-terminus of the molecule). The conformation of the cadherin molecule is stable only in the presence of Ca2+, whose binding with the extracellular portion of the polypeptide chain is prerequisite for cadherin-mediated cell-cell adhesion. Calcium-binding sites consisting of short highly conserved amino acid sequences are located between neighboring extracellular repeats [11]. The cytoplasmic domain of classical cadherins is associated with the cytoplasmic proteins catenins, which, in turn, serve as intermediate linkers between the cadherins and actin filaments [10-12]. It is this cadherin-catenin complex that is required for providing normal cell-cell adhesion. In principle, extracellular cadherin domains per se are capable of homophilic recognition and binding. It was shown that cells that express mutant cadherins lacking the cytoplasmic domains can bind with substrate covered with purified cadherin ectodomains. However, in this case adhesion is much weaker than in the case of cells bearing full-size cadherins [11, 13, 14]. These data indicate that the formation of stable cell-cell junctions depends on the presence in the cadherin molecule of functionally active cytoplasmic domain and association of the latter with the cytoskeleton.
As mentioned above, cadherins mediate homophilic adhesion: during co-culturing of different types of cells, those cells first aggregate that bear identical cadherins on their surfaces [15]. Similar dependence between cell sorting in the developing tissues and expression of different cadherins in them is observed during embryogenesis [16]. The extracellular domains (primarily, the N-terminal domain EC1) play a key role in homophilic recognition between two cadherin molecules. It was shown that cells expressing chimerical E-cadherin, in which the EC1 domain was substituted with EC1 domain of P-cadherin, did not recognize the cells bearing native E-cadherin and aggregated with the P-cadherin-expressing cells [17]. The site responsible for homophilic recognition contains 40 amino acid residues located in the C-terminal region of EC1. Blashchuk et al. [18] assumed that sequence His-Ala-Val located in the C-terminal region of domain EC1 plays a key role in the interaction between cadherins because synthetic peptides containing this sequence effectively blocked mouse embryo blastomere assembling (a process that is mediated by cadherins). However, later it was shown that homophilic recognition also requires the presence of other regions located in the N-terminal domain. In addition, it was discovered that the sequence His-Ala-Val is contained only in the molecule of classical cadherins of type I that involves E- (epithelial), N- (neural), P- (placental), VE- (vascular endothelial), and R- (retinal) cadherins. The corresponding regions of type II classical cadherins that involve recently discovered cadherins designated by numbers 5-12 contain other amino acid residues [9, 10]. Type I and II cadherins also differ from each other in some amino acid residues.Fig. 1. Structure of classical cadherins and their interaction with cytoplasmic proteins [7].
It should be noted that some cadherins can also mediate weak heterophilic interactions. In particular, E- and N-cadherin can bind with the integrin alphaEbeta7 [19] and receptor for fibroblast growth factor [20, 21], respectively.
The role of the four other cadherin repeats (EC2-4) in the cell-cell interaction remains obscure. Possibly, only EC1 domain directly participate in homophilic binding, whereas the remaining domains act as spacers providing the required distance between the junction and cell surface. Nevertheless, they are required for cadherin-dependent adhesion: in the absence of other extracellular domains, the N-terminal domain alone cannot maintain functional binding or adhesive activity [7].
Numerous data that has accumulated to date show that the extracellular cadherin fragments exist in the form of stable parallel lateral dimers. Lateral dimers were revealed by X-ray analysis of N-cadherin EC1 domains [22] and E-cadherin fragments including EC1 and EC2 domains [23]. The existence of dimers was also shown for the whole extracellular fragment of C-cadherin [14]. In the same experiments, it was also shown that the ability of C-cadherin monomers to aggregate significantly decreases compared to the dimers. To date, the mechanism of dimer formation is poorly understood. Apparently, N-cadherin dimers are stabilized by the hydrophobic interactions between the monomers [22], whereas in the case of EC1-EC2 fragment of E-cadherin dimeric structure is maintained by Ca2+ [23].
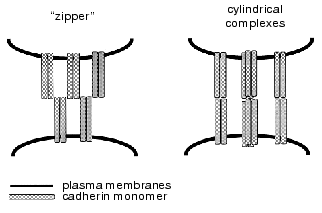
It is still unclear why dimers exert higher activity than monomers during cell-cell interaction. Two main views on this phenomenon exist. The first hypothesis proposes that dimers are bivalent, which increases their avidity. The second hypothesis implies that dimer formation is associated with the occurrence of a unique site ensuring homophilic binding, which is absent from the monomers. The mechanism of interaction of cadherin dimers located on the membranes of different cells has been also the subject of much controversy. Based on the results of X-ray analysis of NCD1, Shapiro et al. [22] proposed the existence of a zipper-like self-assembling structure. This “molecular zipper” model (Fig. 2) logically explains the mechanism whereby numerous weak bonds can ensure highly efficient binding in the cell layer. However, some authors believe that cadherin “zipper” is an in vitro artifact and suggest an alternative hypothesis that was formulated based on the results of electron microscopic analysis of adhesive zone preparations obtained by the freeze-fracture method [24]. Separate protein cylinders extending from one cell surface to another and binding with the similar structures on the neighboring cell are seen on the images. According to the second model, cadherin molecules (dimers or oligomers) act as discrete units and do not form zipper-like ordered structures on the cell surface [10].
Fig. 2. Two models of cadherin molecular organization in adhesive junctions. The “molecular zipper” model based on the results of X-ray analysis of N-cadherin EC1 domain is shown on the left. The model of “cylindrical oligomers” based on the results of electron microscopy of zonula adherens preparations obtained by the freeze-fracture method is shown on the right [7].
INTERACTION OF CADHERINS WITH CYTOPLASMIC PROTEINS
The conclusion that cadherin complexes interact with the cytoskeleton was first made based on the data that cadherins cannot be extracted with non-ionic detergents that effectively solubilized other membrane proteins [13, 25, 26]. It was shown later that the major cytoplasmic proteins associated with the cytoplasmic domain of cadherins and participating in cell adhesion are alpha- and beta-catenins, which mediate the interaction between the cadherins and actin cytoskeleton [11, 13, 25, 27-30]. The catenin-binding site was mapped on E-cadherin. It is located at the distance of 56 amino acid residues from the C-terminus of the molecule [25, 31]. Biochemical analysis with the use of purified catenins and recombinant cytoplasmic domain of cadherins [32, 33] and expression of beta-catenin deletion mutants [34-36] showed that beta-catenin directly binds to the cytoplasmic cadherin fragment and serves as a linker for alpha-catenin attachment.
The crucial role of the cytoplasmic domain of cadherin (and the catenin-binding site, in particular) is corroborated by numerous experiments. It was shown that deletion of the cytoplasmic domain or the catenin-binding site suppresses stable cadherin-mediated adhesion of cultured cells [11, 13]. Alternatively, overexpression of the catenin-binding site in the cultured cells [37], Xenopus laevis embryos [28], or in the intestinal cells of transgenic mice [38] also entails disruption of cell-cell junctions. Such unusual, at first glance, result (at least, in the case of Xenopus laevis) can be, apparently, explained by competition of the expressed catenin-binding site with the endogenous cadherin for catenin binding.
The evidence for participation of alpha-catenin in cell adhesion was obtained on lung carcinoma cell culture that does not contain alpha-catenin and aggregates with each other very weakly despite the presence of cadherins on the cell surface. However, transfection with alpha-catenin cDNA restores cadherin-mediated adhesion in these cells [27, 29]. Rim et al. [12] showed that alpha-catenin directly binds to actin filaments both in vitro and in vivo in the cultured cells. The actin-binding protein alpha-actinin contained in adhesive junctions apparently also interacts with alpha-catenin [39].
Participation of beta-catenin in cell adhesion was confirmed in experiments on Drosophila embryos using mutation analysis of protein armadillo, a homolog of beta-catenin [30]. beta-Catenin is attached to the cytoplasmic domain of cadherin via its central region containing so-called armadillo repeats [34, 36]. These repeats (40 amino acid resides each) were first described in protein armadillo in Drosophila [40, 41]. alpha-Catenin binds to the N-terminus of beta-catenin [32, 34-36]. The role of a linker between cadherin and alpha-catenin is apparently the only function of beta-catenin in cell adhesion. It was shown that a chimerical molecule where the cytoplasmic domain of E-cadherin is substituted with alpha-catenin ensures cell adhesion in the absence of beta-catenin as successfully as the whole protein complex [42].
Plakoglobin (gamma-catenin) sometimes substitutes beta-catenin in the cadherin-catenin complex [34]. However, its physiological role is not completely understood. Plakoglobin is the major component of the desmosomes [43], where it is associated with the desmosomal cadherins [44, 45]. The high extent of homology of plakoglobin to beta-catenin and armadillo [26, 46] implies that these proteins may have similar functions. However, mouse embryo cells lacking beta-catenin due to genetic recombination aggregate very weakly and readily dissociate despite the presence of plakoglobin in them [47]. This is indicative of inability of plakoglobin to completely substitute for beta-catenin in cell adhesion. Deletion of the plakoglobin gene, which was also caused by homologous recombination, entails lethal changes in the heart structure and early death of the embryos, presumably due to disruptions in desmosomal junction formation [48]. Other cytoplasmic proteins directly associated with cadherin are tyrosine phosphatases [49, 50] and the substrate for src-kinase p120cas [51-53].
Interestingly, the level of cadherin expression in the cell may affect catenin expression. Transfection of L-cells with E-, N-, or P-cadherin cDNA results in a significant increase in the catenin content without changing the catenin mRNA content. Hence, the presence of cadherins regulates catenin expression at the post-translation level [54].
It was also reported that cadherin cytoplasmic domain may mediate adhesion independently of catenins. Chimerical cadherin molecules in which cadherin cytoplasmic domain was substituted for the analogous domain of desmoglein-3 (one of desmosomal cadherins) that cannot bind catenins, mediates cadherin-dependent adhesion in the cultured cells [55]. Thus, association with catenins is not the only way of participation of the intracellular cadherin domain in cell-cell adhesion.
CELL-CELL JUNCTIONS CONTAINING CADHERINS
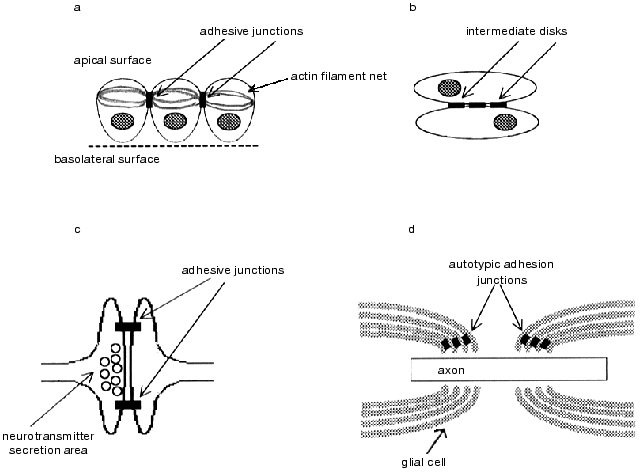
Immunohistochemical analysis of tissues and cultured cells shows that cadherins most often are constituents of cell-cell adhesive junctions (Fig. 3). This type of junctions involves autotypic junctions between the layers of the same glial cell in the axon myelin sheath [56]; adhesive junctions in synapses, where cadherins link pre- and postsynaptic membranes in the regions adjacent to the neurotransmitter secretion areas [57, 58]; the intermediate disks between the cardiomyocytes [59]; and some other. The best-known type of cell-cell adhesive junctions is zonula adherens located at the apico-lateral border of the epithelial layer a little lower than the tight junctions. Actin bunches attached to the adhesive junctions girding the cell on the cytoplasmic side are located parallel to the membrane surface and form a united contracting network in the epithelial layer. Assembling of the belt-like zonula adherens is apparently the basis for the occurrence of the epithelial morphology of the cell layer [60-63]. During morphogenesis, folding of the epithelial layers into tubes is often attained by contraction of actin filaments contained in the zonula adherens, which is associated with narrowing the apical end of each cell in the apical layer and results in the cell layer bending [64, 65].
Besides cadherins and catenins, adhesive junctions contain numerous proteins (such as vinculin, ezrin, moesin, and radixin), protein components of the actin cytoskeleton, and integral membrane proteins (e.g., epidermal growth factor receptor, EGF) [66]. Genetic studies on Drosophila revealed other components required for adhesive junction assembly. In particular, the genes whose mutations lead to disruptions in the course of zonula adherens assembling were identified in studies on Drosophila embryos. They involve the gene of beta-catenin homolog, armadillo, which is completely consistent with the view on the key role of this protein in cadherin-mediated adhesion [30, 67], as well as the genes crumb and stardust [67-69]. It was shown that gene crumb encodes the integral membrane protein that is required for epithelization of the ectodermic cells. In mutant individuals with inactive crumb gene normal cadherin-catenin complexes are expressed on the cell surface; however, their distribution is chaotic, leading to disruption in formation of mature zonula adherens in the epithelium [68-70].Fig. 3. Cell-cell junctions formed by cadherins: a) epithelial zonula adherens; b) intermediate disks between the cardiomyocytes; c) adhesive junctions restricting the area of neurotransmitter secretion in the synapse; d) autotypic junctions between the glial cell layers in axon myelin sheath [7].
It should be noted that in many cells cadherins can mediate adhesion without formation of morphologically pronounced adhesive junctions. Even in the epithelium of some organs, where cell-cell adhesion depends on E-cadherin, zonula adherens is absent [7]. Cadherin-mediated adhesion without cadherin accumulation in the adhesive junctions was also described for blastomeres [106], nerve ridge cells [71], and fibroblasts transfected with different types of cadherins [13].
REGULATION OF CADHERIN ACTIVITY
Cadherin-mediated adhesion can be regulated by a variety of extracellular signals, including growth factors [72-74], peptide hormones [75, 76], signals from gap junctions [77], and cholinergic receptor agonists [78]. In response to these external stimuli, different signals are generated in the cell, of which protein phosphorylation is, apparently, the most important for the regulation of cadherin function [7].
Protein kinase C (PKC) participates in the activation of E-cadherin-dependent mouse embryo cell compacting, which was demonstrated with the use of a combination of pharmacological agonists and antagonists. Embryo compacting is accelerated by the addition of PKC-stimulating agents (e.g., phorbol ester and diacylglycerol) and inhibited by PKC-blocking agents [79], the PKC effect being blocked by the addition of anti-E-cadherin antibodies. However, it was not determined which PKC-mediated way is activated in this case.
Using a similar experimental approach, a potential inhibitory effect of tyrosine phosphorylation on cadherin function was shown. Several scientific groups discovered that enhancement of tyrosine phosphorylation (transfection with v-src or incubation of the cells with pervanadate) weakens cadherin-mediated cell-cell adhesion. Components of the cadherin-catenin complex (primarily beta-catenin) undergo tyrosine phosphorylation in response to v-src transfection and incubation with pervanadate [80-82]. Attenuation of adhesion in these experiments was blocked by herbimicin, which is also indicative of participation of tyrosine phosphorylation in the regulation of cadherin activity. It was also shown that v-src can affect cadherin-mediated adhesion irrespective of beta-catenin [83]. The authors of this work used mutant E-cadherin that could directly bind with the C-terminal fragment of alpha-catenin and induce adhesion without the participation of beta-catenin. However, in this case transfection with v-src also significantly inhibited cell-cell adhesion.
Other data confirming the effect of tyrosine phosphorylation on cadherin-dependent adhesion are known. Tyrosine phosphorylation of beta-catenin is observed when cells are treated with hepatocyte growth factor (HGF) and EGF (agents that can induce dissociation of epithelial cells) [74]. Tyrosine kinases or their substrates can associate with the cadherin-catenin complex. It is known that p120cas, a member of the armadillo protein family, is a substrate for both src kinases and receptor tyrosine kinases [84]. It was shown that p120cas directly binds to the distal part of the cytoplasmic domain of E-cadherin, forming a whole complex with cadherin and beta-catenin or plakoglobin [52, 53, 84, 85]. Activation of the Erb-2/Neu receptor tyrosine kinase in the epithelial cells causes disassembling of the cell-cell junctions formed by E-cadherin, which results in the loss of the epithelial phenotype by the cells [86]. EGF receptor tyrosine kinase also can bind to the cadherin-catenin complex [87]. In addition, it was shown that cadherin-catenin complex can interact with receptor-dependent tyrosine phosphatases [49, 50, 88].
Cadherin function may also be affected by cell-cell communication via gap junctions. Inhibition of cell-cell communication by expression of the chimerical protein connexin 32/connexin 43 inhibitor (a protein that forms gap junctions) in Xenopus embryo cells leads to blastomere separation. A similar effect is observed when mutant cadherin is expressed in the embryo cells. This phenotype can be corrected by coexpression of connexin 37 that is insensitive to the inhibitor [77]. Similarly, cell-cell junction assembling in Novikov hepatoma cells is suppressed by anti-connexin and anti-cadherin antibodies [89]. The mechanism of signal transduction mediated by the gap junctions remains obscure. It is assumed that in this case cadherin-dependent adhesion and cell-cell junction assembling may be regulated via temporal increase in the concentration of Ca2+ and other small signal molecules (such as cyclic nucleotides or inositol phosphate) penetrating through the gap junctions and activating the intracellular processes that affect cadherin activity.
The strength of cell-cell interactions can be affected both by modulating cadherin activity and changing their expression level in the cell. It was demonstrated that an increase in cadherin content enhances cell adhesion [7, 90, 91]. It was also shown that cadherin expression in cultured cells is regulated by growth factors and peptide hormones [72, 73, 75, 76]. Another mechanism of regulation of cadherin activity is changing the extent of clustering of cadherin molecules in the junction area. As was mentioned above, lateral clustering of cadherin molecules can significantly affect the strength of cell-cell interaction. Changes in the extent of clustering can mediate rapid changes in cell adhesion strength. For example, mouse embryo blastomere compacting is associated with E-cadherin redistribution in the region of cell-cell junctions without any change in protein expression [92].
CADHERINS AND SIGNALING
To date, numerous data indicate that cell adhesion receptors can affect cell form, motility, and growth not only due to mechanical attachment of the cells to each other or to the substrate, but also by activating internal signaling [93]. Some papers report that many effects of cadherin on cell behavior are rapid and apparently caused by a series of short-term signals rather than by assembling stable long-term cell-cell junctions [4, 6, 94]. However, until recently only indirect evidence of cadherin ability to induce the production of secondary messengers in the cell have been known. For instance, it was shown that axon outgrowth stimulated by N-cadherin is associated with changes in the cytoplasmic Ca2+ concentration and activation of G-proteins and tyrosine kinases. However, it was not clear whether these signals result from the direct interaction of N-cadherin molecules [95, 96]. Because different signal molecules (such as proteins belonging to the non-receptor src kinase family as well as some membrane receptors and phosphatases) were found in the cell-cell junctions of epithelial cells [49, 97, 98], it was suggested that these molecules can mediate cadherin-dependent signaling. Data on the direct effect of cadherins on the signal processes appeared only during the last two years. It was shown that inter-cadherin junctions in cultured fibroblasts induced oscillations in the cytoplasmic Ca2+ concentration, antibodies raised against the first domain EC1 mimicking this effect. The oscillations occurred in the regions of cell-cell interactions and coincided in time with translocation of actin and other cytoplasmic proteins into the adhesive complexes [99]. N-Cadherin can regulate axon outgrowth by direct interaction with the EGF receptor, thereby activating the cascade of mitogen-activated protein kinases (MAPK) [21]. The experiments on cultured keratinocytes showed that adhesive junction formation leads to a rapid activation of MAPK-dependent signaling and that this effect is mediated by E-cadherin. In addition, E-cadherin can stimulate MAPK by ligand-independent activation of EGF receptors [100]. It also activates Cdc42, a low-molecular-weight GTPase belonging to the Rho family, which regulates the cytoskeleton structure [101].
For a long time beta-catenin, whose signal activity is well known, was considered as a candidate for the role of a messenger of signal transduction from cadherins, with which it is associated. beta-Catenin and its homolog armadillo from Drosophila are components of Wnt/wingless signal pathway that plays a key role in embryogenesis [102-104]. Recent data, however, indicate that the interaction between beta-catenin and cadherins is not prerequisite for manifestation of its signal activity. It was also shown that beta-catenin function as a cadherin partner during adhesive junction formation is not directly associated with its signal function in the cytoplasm and/or nucleus, where it affects transcription of genes by interacting with specific transcription factors [36, 105-111]. Free beta-catenin content in the cytoplasm is regulated by the protein product of the APC (adenomatous polyposis coli) gene. Formation of the complex between these two proteins is a signal for beta-catenin degradation. Conversely, triggering the Wnt signal pathway results in beta-catenin stabilization, its accumulation in the cytoplasm, and binding to the transcriptional factor Tcf, which, in turn, stimulates transcription of some genes. On the other hand, although the adhesive and signal functions of beta-catenin are separated, the formation of cell-cell junctions can apparently indirectly affect beta-catenin-dependent signaling. It was shown that overexpression of cadherins in the embryos of Xenopus laevis and Drosophila inhibited signal transduction via beta-catenin/armadillo [106, 112]. In Xenopus embryos, the inhibition is due to beta-catenin binding with C-cadherin on the inner surface of the cell membrane. As this takes places, beta-catenin is removed from its cytoplasmic pool, becoming inaccessible for participating in signaling. Thus, cadherins can regulate beta-catenin signaling activity by changing its distribution in the cell.
It cannot be ruled out, however, that cadherins indirectly contribute to signaling regulation. Approaching of the membranes of the neighboring cells during adhesive junction formation may enable the interaction of membrane receptors and their membrane-bound ligands on the neighboring cells and activate juxtacrine signaling. This hypothesis is corroborated by data on association of some signal molecules with the cadherin-catenin complexes and on high concentration of tyrosine kinase substrates in the regions of adhesive junctions. The group of juxtacrine receptors described to date involves notch, delta, sevenless, and bride-of-sevenless (boss) receptors participating in Drosophila embryogenesis [113, 114] and associated with the membrane form of tumor necrosis factor (TNF) and transforming growth factor (TGFalpha) [115]. It is tenable to assume that signaling via such receptors depends on the proximity of the surface of adjacent cells and, respectively, on formation of inter-cadherin junctions. This hypothesis is confirmed by the fact that expression of cadherins in the fibroblasts entails communication enhancement via gap junctions [116].
THE ROLE OF CADHERINS IN MORPHOGENESIS
The formation of tissues and organs during embryogenesis is determined by a number of processes coordinated in time and space, such as cell aggregation, polarization, differentiation, and migration. Because cell-cell and cell-nuclear matrix adhesive junctions play a key role in all these events, adhesion receptors are often called morphoregulatory molecules. One such adhesion-dependent processes is selective cell segregation. This phenomenon was discovered as early as in the 1950s. In classical works in embryology, it was shown that suspended cells from different amphibian blastophylla are capable of homotypic reaggregation to form junctions only with similar cells in correspondence with their histogenetic origin [117]. Later it was discovered that this homotypic aggregation is based on selective expression of specific adhesion molecules on different subpopulations of cells. The role of cadherins in this process was revealed by Nose et al. [15]. The L-cells were transfected with cDNA of either E- or P-cadherin. The suspensions were then mixed in vitro and analyzed for cluster formation. Under these conditions, highly selective adhesion between cells expressing cadherins of the same type was observed.
Another morphogenetic process in which cadherins play a key role is cell condensing (i.e., transition of cell population from dispersed state to condensed solid formation). An example of such condensing is blastomere assembling at the early stages of embryogenesis. E-Cadherin plays a crucial role in cell condensing in mouse embryo morula: the embryo structure is disrupted as a result of treatment of the cells with blocking anti-E-cadherin antibodies, introduction of antisense nucleotides to E-cadherin mRNA into the cell, and in transgenic mice defective by the gene encoding this protein [118-123]. Injection of antisense nucleotides into Xenopus laevis oocytes, which decreases expression of EP-cadherin (a Xenopus laevis protein homologous to E- and P-cadherins), significantly attenuates adhesion between the blastomeres and entails disruption of the embryo structure [124].
Numerous studies performed both in intact embryos and cultured cells revealed significant correlation between epithelization of mesenchymal cells and expression of specific cadherins in them. During somite development, mesenchymal cells comprising its future wall are polarized and temporarily form epithelium-like structures, the expression of N-cadherin in them significantly increasing [64, 65, 125]. Transfection of cultured mesenchymal cells with cDNA of different cadherins results in their epithelization [13, 126-128], whereas inhibition of cell-cell interactions by anti-cadherin antibodies leads to the loss of epithelial phenotype by the cells and stimulates cell motility and invasiveness [129-131].
One of the most vivid examples of participation of cadherins in morphogenesis is their role in the central nervous system development. At different stages of embryogenesis and in different structural layers, neuroepithelial tissues express more than 20 different cadherins involved in all key events of neurogenesis, beginning from selective aggregation of the cells at the earliest stages of embryo development and finishing with the formation of synapses [132-137]. Cadherins play a key role during neuroectoderm sorting and neural tube formation. It was also shown that before segregation of the neuroectoderm from the ectoblast in neurula, a coordinated decrease in the expression of E-cadherin and increase in that of N-cadherin occurs in the cells of future neuroectoderm. It is believed that it is N-cadherin that is responsible for selective cell uniting in the neural plate [64, 65, 138]. Interestingly, normal neural tube is formed in transgenic mice defective in the N-cadherin gene. Apparently, in this case N-cadherin is functionally substituted with other adhesion molecules (presumably, cadherin-6) [139]. It is known that the coordinated change in the cell shape induced by microfilament contraction in zonula adherens underlies neural tube folding and other morphogenetic processes that require change in the shape of the epithelial layers [64, 65]. Further development of the neural tube involves its segregation to separate regions due to local expression of different cadherins. For example, selective distribution of E- and R-cadherins, cadherin-6, and cadherin-8 in different regions of embryonic brain is observed [135, 136, 140, 141]. The possibility of selective segregation of nerve cells that express cadherins of the same type was demonstrated in vitro. In vivo such segregation of the neural tube to segments apparently prevents the migration of nerve cells between adjacent regions of the developing brain [10].
Normal expression of cadherins is required for neurite outgrowth activating and regulating. The expression of dominant-negative functionally inactive N-cadherin in developing frog retina blocks the axon and dendrite outgrowth. Those axons that are still formed are usually shorter and often do not have growth cones [142]. In the experiments in vitro, it was shown that growth and migration of axons change during neuron culturing on the substrates containing recombinant cadherins. In particular, recombinant N-cadherin enhances adhesion of axons to the substrate and enables their projecting in the direction of higher concentrations of the recombinant protein. Similar growth stimulation is observed when the neurons are cultured on the monolayer of cells transfected with N-cadherin cDNA [8, 95, 143-146]. Such effect on axon projecting is apparently due to the interaction of N-cadherin with FGF receptor with subsequent MAPK activation [21]. By contrast, another member of the cadherin family, T-cadherin, inhibits axon outgrowth [137, 147]. Thus, the coordinated action of different cadherins and other adhesion receptors expressed on the axon membrane and surrounding tissues ensure navigation of axon projecting to the peripheral targets.
Cadherins also play a key role in setting and stabilization of junctions between the neurons and formation of neural nets and neuromuscular junctions [148-153]. In mouse postnatal brain, cadherins of the same type are expressed in functionally related regions (e.g., in the thalamus nucleus and related cortex regions [154]). During chick eye development N-cadherin stabilizes the junctions between the axon termini and their targets. Formation of a branched net of neural termini in the retina may be blocked by injecting anti-N-cadherin antibodies [155]. It was shown that in synapses cadherins anchor the pre- and postsynaptic membranes, bordering the area of neurotransmitter secretion [57, 58]. E-Cadherin is also present in the myelin sheath of nerves, where it forms autotypic adhesion junctions between the plasma membrane layers of the same Schwann cell [56] (Fig. 3).
CADHERINS AND ONCOGENESIS
The ability of tumor cells for uncontrolled growth, migration, invasion into surrounding tissues, and metastasizing is often associated with disruption of cell-cell and cell-extracellular matrix junctions [156, 157]. For this reason, special attention is currently paid to identification and characterization of cell adhesion receptors involved in tumor development. With regard for the role of cadherins in cell-cell adhesion, maintenance of tissue structure, and regulation of epithelial cell phenotype, it was assumed that the disruption of cadherin-dependent cell-cell interactions in the epithelium may cause attenuation of cell-cell junctions, loss of epithelial phenotype, enhancement of cell motility, removal of contact suppression of growth, and, as a result, uncontrolled proliferation and invasion of tumor cells [158]. The majority of studies in this area focus on the role of E-cadherin in malignant cell transformation. In many works, it has been shown that E-cadherin expression is decreased or absent from different carcinomas (esophagus, stomach, or breast) [159-162]. Abnormal distribution of E-cadherin in tumor cells was often observed (it was absent from the regions of adhesion junctions). It should be noted that E-cadherin expression was most often decreased in the undifferentiated “aggressive” carcinomas that have high invasive potential [160].
Similar results were obtained on cultured cells. Frixen et al. [130] also reported that carcinoma cell lines with the epithelial noninvasive phenotype expressed E-cadherin, whereas the latter was absent from the cells with the fibroblastoid phenotype. Navarro et al. [163] revealed reciprocal dependence between the amount of E-cadherin expressed on the cell surface of different carcinomas and the ability of these cells for invasion. Malignant transformation of MDCK epithelial cells (which are noninvasive in normal state) as a result of injection of Harvey and Maloney sarcoma virus to the cell culture is accompanied by a decrease in E-cadherin expression on the cell surface. A similar change of MDCK cell phenotype from noninvasive for invasive is also observed after disruption of cell-cell junctions in the presence of anti-E-cadherin antibodies [129]. Conversely, transfection of the carcinoma cells with E-cadherin cDNA restores normal cell phenotype, decreases invasiveness and migration, and suppresses tumor growth [163-165].
The few studies on the role of P-cadherin in oncogenesis also revealed a correlation between decreased expression of this protein and the invasiveness of lung carcinomas [166] and melanomas [167]. Unexpected results were obtained when studying the effect of N-cadherin on tumor cells. It was discovered that expression of this protein is significantly enhanced in invasive undifferentiated breast carcinoma cells [168]. It was also shown that an increase in N-cadherin expression in the carcinoma cells simultaneously with the decrease in E- and P-cadherin expression changes the phenotype from epithelial to mesenchymal [169]. Transfection of MCF-7 carcinoma cells with N-cadherin cDNA significantly enhances the invasiveness and stimulates metastasis development despite the presence of E-cadherin in these cells [170]. Such an opposite effect of cell-cell adhesion mediated by E- and N-cadherins on cell behavior may be due to the ability of E-cadherin to form stable cell-cell junctions that prevent cell migration, whereas N-cadherin can form labile junctions required for such dynamic processes as axon projecting or migration and invasion of tumor cells [170].
Other components of cadherin complexes (primarily catenins) can also affect the growth and migration of transformed cells. As was mentioned above (see “Cadherins and Signaling”), normal expression of free beta-catenin in the cytoplasm is maintained by the oncosuppressing protein APC that binds with excessive beta-catenin and activates its degradation [105]. In patients with hereditary polyposis, who are predisposed to intestine cancer, mutations in the APC gene [171] or directly in beta-catenin gene [172] are often observed. As a result of these mutations, the APC protein lacks its ability to regulate beta-catenin level in the cytoplasm, which leads to uncontrolled activation of the Tcf transcription factor by beta-catenin and development of intestine tumors [105].
T-CADHERIN IS AN ATYPICAL MEMBER OF THE CADHERIN FAMILY
T-Cadherin (truncated) (or H-cadherin (heart), or cadherin-13) is one of the most unusual members of the cadherin superfamily. Although its N-terminal domain EC1 does not contain the His-Ala-Val sequence, its extracellular part comprised of five cadherin repeats is very similar in structure to the classical cadherins. A unique feature of this protein is the absence of both the transmembrane and cytoplasmic domains. It is anchored in the membrane via glycosylphosphatidylinositol (GPI) that attaches to the mature protein after cleavage its C-terminal sequence during processing in the endoplasmic reticulum [173]. Despite the absence of the cytoplasmic domain, T-cadherin can mediate any weak homophilic adhesion of the suspended cells [174]. The mechanisms of formation of cell-cell junctions via T-cadherin and classical cadherins are apparently significantly different because the majority of cadherins ensure adhesion only when they contain the cytoplasmic domain that mediates their binding with the cytoskeleton [11, 13]. Another unusual property of T-cadherin is simultaneous expression on the cell surface of its two forms (the mature protein and partially processed precursor containing an uncleaved propeptide, whose function remains obscure) [174].
T-Cadherin was first discovered in chick nervous system [137, 175]. Later its human homolog called cadherin-13 was identified [176]. The only physiological function of T-cadherin established so far is its participation in the regulation of neuron growth during embryogenesis. During formation of chick embryo hind limbs, the outgrowing axons avoid those regions where T-cadherin is expressed [137]. Neuron culturing on substrate containing recombinant T-cadherin significantly inhibits axon growth [147]. Contact suppression of axon growth as a result of homophilic binding between T-cadherin molecules located on the axon membrane and surrounding mesenchymal tissues is apparently a navigating mechanism whereby the direction of nerve fiber growth is determined.
Numerous recent data indicate that malignant tumor development is associated with the changes in T-cadherin expression. The loss of chromosome 16q24 locus containing T-cadherin gene correlates with the development of pancreas, lung, stomach, and ovary cancers [177-182]. The transfection of tumor cells with T-cadherin cDNA entails a decrease in the proliferative and invasive activities both in vitro [182] and in vivo as a result of challenging the mice with tumorigenic cell lines [183] as well as the loss of cancer cell sensitivity to the action of growth factors [184].
The mechanisms of T-cadherin effect on cell adhesion and proliferative activity are still unknown. It cannot be ruled out that the maintenance of mechanical junctions between the cells is not the main function of this protein. It is most likely that it serves as a signal receptor, a sensor that allows the cell to “sense” its environment. This hypothesis is corroborated by the data on T-cadherin distribution in the membrane: in the polarized intestinal cells it is located on the apical part of the cell rather than in the adhesive junctions on the basolateral cell surface [185]. It has long being known that many other GPI proteins may activate intracellular signaling [186-188]. The absence of the cytoplasmic domain in these proteins implies the presence of a membrane adapter protein. Owing to the interaction with the latter, the signal can be relayed across the membrane from the GPI proteins into the cell. We showed that, similar to other GPI proteins, T-cadherin is located on the cell surface in special plasma membrane domains (caveolae and lipid rafts) [189], which also contain other signal molecules (such as G-proteins, src kinases, rasproteins, and transmembrane receptors of growth factors [190]). It cannot be excluded that some of these molecules may serve as messengers during activation of T-cadherin-dependent signaling.
In our laboratory, the main attention is focused on investigation of the role that T-cadherin plays in the cardiovascular system function. Cell adhesion molecules play a crucial role in the maintenance of normal structure of vascular walls. The development of different pathologies (such as atherosclerosis and restenosis after balloon angioplasty and atherectomy) is characterized by enhanced migration, proliferation, and phenotypic modulation of the endothelial cells, which is often associated with disruption of cell-cell and cell-extracellular matrix junctions [191, 192]. The expression and functions of T-cadherin in the cardiovascular system have not been studied before. We performed a comparative study of T-cadherin expression in different human organs and tissues. The results show that T-cadherin content is maximal in the aorta, carotid, iliac, and kidney arteries, and in heart. In aorta wall, T-cadherin is contained in the endothelial and smooth muscle cells and pericytes. Its expression in the smooth muscle cells depends on the cell phenotype and proliferative activity [193-195] and increases in sclerotic lesion of vascular walls [193]. Preliminary studies performed on the model of balloon catheterization of rat carotid artery indicate that T-cadherin expression in smooth muscle cells increases in restenosis. The content of this protein is also elevated in the endothelium isolated from tumor vasculature [196]. In addition, anti-T-cadherin antibodies can affect the phenotype, adhesion, and motility of the endothelial cells in vitro (our unpublished data). With regard for these data, it is likely that T-cadherin plays a key role in the regulation of cell phenotype, migration, and growth, as well as in maintenance of vascular wall structure.
Study of the interaction of heterophilic interactions between T-cadherin and blood plasma lipoproteins is also of great interest. Originally, the work of our group was aimed toward searching for the receptors that mediate the hormone-like effect of low-density lipoproteins on the systems of intracellular signaling in smooth muscle cells in human vasculature [197, 198]. On the surface of membranes of aorta smooth muscle cells, we discovered two unusual lipoprotein-binding proteins with molecular weight of 105 and 130 kD. The characteristics of these proteins are indicative of their participation in lipoprotein-dependent signaling [199, 200]. After isolation of these receptors from human aorta medium and determination of their amino acid sequence, we discovered that the 105-kD protein is mature T-cadherin [201], whereas the 130-kD protein is its partially processed precursor [202]. It is known that increased lipoprotein content in blood plasma is a risk factor of development of vascular pathologies based on increased proliferation and migration of smooth muscle cells. A distinct relationship between atherosclerosis and restenosis pathogenesis and low-density lipoprotein content in blood was demonstrated [203]. It cannot be ruled out that lipoprotein binding to T-cadherin may affect T-cadherin-dependent regulation of growth and motility of vascular cells, thereby contributing to development of cardiovascular diseases.
REFERENCES
1.Grunwald, G. B. (1993) Curr. Opin. Cell
Biol., 5, 797-805.
2.Burridge, K., Fath, K., Kelly, T., Nuckolls, G.,
and Tutner, C. (1988) Annu. Rev. Cell Biol., 4,
487-525.
3.Garrode, D. R. (1993) Curr. Opin. Cell
Biol., 5, 30-40.
4.Gumbiner, B. M. (1996) Cell, 84,
345-357.
5.Burridge, K., and Chrzanovska-Wodnichka, M. (1996)
Annu. Rev. Cell Dev. Biol., 12, 463-519.
6.Geiger, B., and Ayalon, O. (1992) Ann. Rev. Cell
Biol., 8, 307-332.
7.Yap, A. S., Brieher, W. M., and Gumbiner, B. M.
(1997) Ann. Rev. Cell Dev. Biol., 13, 119-46.
8.Vleminckx, K., and Kemler, R. (1999)
BioEssays, 21, 211-220.
9.Angst, B. D., Marcozzi, C., and Magee, A. I. (2001)
J. Cell Sci., 114, 629-641.
10.Takeichi, M. (1995) Curr. Opin. Cell
Biol., 7, 619-627.
11.Ozawa, T., Ringwald, M., and Kemler, R. (1990)
Proc. Natl. Acad. Sci. USA, 87, 4246-4250.
12.Rimm, D. L., Koslov, E. R., Kebriaei, P., Cianci,
C. D., and Morrow, J. S. (1995) Proc. Natl. Acad. Sci. USA,
92, 8813-8817.
13.Nagafuchi, A., and Takeichi, M. (1988) EMBO
J., 7, 3679-3684.
14.Brieher, W. M., Yap, A. S., and Gumbiner, B. M.
(1996) J. Cell Biol., 135, 487-496.
15.Nose, A., Nagafuchi, A., and Takeichi, M. (1988)
Cell, 54, 993-1001.
16.Takeichi, M. (1991) Science, 251,
1451-1455.
17.Nose, A., Tsui, K., and Takeichi, M. (1990)
Cell, 61, 147-155.
18.Blaschuk, O. W., Sullivan, R., David, S., and
Pouliot, Y. (1990) Dev. Biol., 139, 227-229.
19.Cepek, K. L., Shaw, S. K., Parker, C. M.,
Russell, G. J., Morrow, J. S., Rimm, D. L., and Brenner, M. B. (1994)
Nature, 372, 190-193.
20.Williams, E. J., Furness, J., Walsh, F. S., and
Doherty, P. (1994) Neuron, 13, 583-594.
21.Doherty, P., Williams, G., and Williams, E. J.
(2000) Mol. Cell. Neurosci., 16, 283-95.
22.Shapiro, L., Fannon, A. M., and Kwong, P. D.
(1995) Nature, 374, 327-337.
23.Nagar, B., Overduin, M., Ikura, M., and Rini, J.
M. (1996) Nature, 380, 360-364.
24.Hirokawa, N., and Heuser, J. E. (1981) J. Cell
Biol., 91, 399-409.
25.Ozawa, M., Baribault, H., and Kemler, R. (1989)
EMBO J., 8, 1711-1717.
26.McCrea, P., and Gumbiner, B. (1991) J. Biol.
Chem., 266, 4514-4520.
27.Hirano, S., Kimoto, N., Shimoyama, Y., Hirohashi,
S., and Takeichi, M. (1992) Cell, 70, 293-301.
28.Kintner, C. (1992) Cell, 69,
225-236.
29.Watabe, M., Nagafuchi, A., Tsukita, S., and
Takeichi, M. (1994) J. Cell Biol., 127, 247-256.
30.Cox, R. T., Kirkpatrick, C., and Peifer, M.
(1996) J. Cell Biol., 134, 133-148.
31.Stappert, J., and Kemler, R. (1994) Cell
Adhes. Commun., 130, 369-381.
32.Aberle, H., Butz, S., Stappert, J., Weissig, H.,
Kemler, R., and Hoschuetzky, H. (1994) J. Cell Sci., 107,
3655-3663.
33.Jou, T-S., Stewart, D. B., Stappert, J., Nelson,
W. J., and Marrs, J. A. (1995) Proc. Natl. Acad. Sci. USA,
92, 5067-5071.
34.Hulsken, J., Birchmeier, W., and Behrens, J.
(1994) J. Cell Biol., 127, 2061-2069.
35.Oyama, T., Kanai, Y., Ochiai, A., Akimoto, S.,
and Oda, T. (1994) Cancer Res., 54, 6282-6287.
36.Funayama, N., Fagotto, F., McCrea, P., and
Gumbiner, B. M. (1995) J. Cell Biol., 128, 959-968.
37.Fujimori, T., and Takeichi, M. (1993) Mol.
Biol. Cell, 4, 37-47.
38.Hermiston, M. L., and Gordon, J. L. (1995) J.
Cell Biol., 129, 489-506.
39.Knudsen, K. A., Soler, A. P., Johnson, K. R., and
Wheelok, M. J. (1995) J. Cell Biol., 130, 67-77.
40.Peifer, M., and Wieschaus, E. (1990) Cell,
63, 1167-1178.
41.Peifer, M., Berg, S., and Reynolds, A. B. (1994)
Cell, 76, 789-791.
42.Nagafuchi, A., Ishihara, S., and Tsukita, S.
(1994) J. Cell Biol., 127, 235-245.
43.Cowin, P., Kapprell, H.-P., Franke, W. W.,
Tamkun, J., and Hynes, R. O. (1986) Cell, 46,
1063-1073.
44.Korman, N. J., Eyre, R. W., Klaus-Kowtun, V., and
Stanley, J. R. (1989) N. Engl. J. Med., 321, 631-635.
45.Witcher, L. L., Collins, R., Puttagunta, S.,
Mechanic, S. E., Munson, M., Gumbiner, B., and Cowin, P. (1996) J.
Biol. Chem., 271, 10904-10909.
46.Franke, W. W., Goldschmidt, M. A., Zimbelmann,
R., Mueller, H. M., Schiller, D. L., and Cowin, P. (1989) Proc.
Natl. Acad. Sci. USA, 86, 4027-4031.
47.Haegel, H., Larue, L., Ohsugi, M., Fedorov, L.,
Herrenknecht, K., and Kemler, R. (1995) Development, 121,
3529-3537.
48.Bierkamp, C., Mclaughlin, K. J., Schwarz, H.,
Huber, O., and Kemler, R. (1996) Dev. Biol., 180,
780-785.
49.Brady-Kalnay, S. M., Rimm, D. L., and Tonks, N.
K. (1995) J. Cell Biol., 130, 977-986.
50.Kypta, R. M., Su, H., and Reichardt, L. F. (1996)
J. Cell Biol., 134, 1519-1529.
51.Reynolds, A. B., Daniel, J., McCrea, P.,
Wheelock, M. J., Wu, J., and Zhang, Z. (1994) Mol. Cell. Biol.,
14, 8333-8342.
52.Shibamoto, S., Hayakawa, M., Takeuchi, K., Hori,
T., and Miyazawa, K. (1995) J. Cell Biol., 128,
949-957.
53.Staddon, J. M., Smales, C., Schulze, C., Esch, F.
S., and Rubin, L. L. (1995) J. Cell Biol., 130,
369-381.
54.Nagafuchi, A., Takeichi, M., and Tsukita, S.
(1991) Cell, 65, 849-857.
55.Roh, J.-Y., and Stanley, J. R. (1995) J. Cell
Biol., 128, 939-947.
56.Fannon, A. M., Sherman, D. L., Ilyina-Gragerova,
G., Brophy, P. J., Friedrich, V. L. J., and Colman, D. R. (1995) J.
Cell Biol., 129, 189-202.
57.Fannon, A. M., and Colman, D. R. (1996)
Neuron, 17, 423-434.
58.Uchida, N., Honjo, Y., Johnson, K. R., Wheelock,
M. J., and Takeichi, M. (1996) J. Cell Biol., 135,
767-797.
59.Volk, T., and Geiger, B. (1986) J. Cell
Biol., 103, 1441-1450.
60.Mooseker, M. S., Bonder, E. M., Conzelman, K. A.,
Fishkind, D. J., Howe, C. L., and Keller, T. C. S. I. (1984) J. Cell
Biol., 99, 104-112s.
61.Madara, J. L. (1987) Am. J. Physiol.,
253, C171-175.
62.Madara, J. L., Moore, R., and Carlson, S. (1987)
Am. J. Physiol., 253, 854-861.
63.Madara, J. L., Stafford, J., Barenberg, D., and
Carlson, S. (1988) Am. J. Physiol., 254, G416-423.
64.Duband, J.-L., Dufour, S., Hatta, K., Takeichi,
M., and Edelman, G. M. (1987) J. Cell Biol., 104,
1361-1374.
65.Duband, J.-L., Volberg, T., Sabanay, I., Thiery,
J.-P., and Geiger, B. (1988) Development, 103,
325-344.
66.Tsukita, S., Tsukita, S., Nagafuchi, A., and
Yonemura, S. (1992) Curr. Opin. Cell Biol., 4,
834-839.
67.Muller, J.-A. J., and Wieschaus, E. (1996) J.
Cell Biol., 134, 149-163.
68.Grawe, F., Wodarz, A., Lee, B., Knust, E., and
Skaer, H. (1996) Development, 122, 951-959.
69.Tepass, U. (1996) Dev. Biol., 177,
217-225.
70.Wodarz, A., Hinz, U., Engelbert, N., and Knust,
E. (1995) Cell, 82, 67-76.
71.Nakagawa, S., and Takeichi, M. (1995)
Development, 121, 1321-1332.
72.Bradley, R. S., Cowin, P., and Brown, A. M. C.
(1993) J. Cell Biol., 123, 1857-1865.
73.Hinck, L., Nelson, W. J., and Papkoff, J. (1994)
J. Cell Biol., 124, 729-741.
74.Shibamoto, S., Hayakawa, M., Takeuchi, K., Hori,
T., and Oku, N. (1994) Cell Adhes. Commun., 1,
295-305.
75.Brabant, G., Hoang-Vu, C., Behrends, J., Cetin,
Y., and Potter, E. (1995) Endocrinology, 136,
3113-3119.
76.Bracke, M. E., Vyncke, B. M., Bruyneel, E. A.,
Vermeulen, S. J., and De Bruyne, G. K. (1995) Br. J. Cancer,
68, 282-289.
77.Paul, D. L., Yu, K., Bruzzone, R., Gimlich, R.
L., and Goodenough, D. A. (1995) Development, 121,
371-381.
78.Williams, S. L., Hayes, V. Y., Hummel, A. M.,
Tarara, J. E., and Healsey, T. J. (1993) J. Cell Biol.,
121, 643-654.
79.Winkel, G. K., Ferguson, J. E., Takeichi, M., and
Nuccitelli, R. (1990) Dev. Biol., 138, 1-15.
80.Matsuyoshi, N., Hamaguchi, M., Taniguchi, S.,
Nagafuchi, A., Tsukita, S., and Takeichi, M. (1992) J. Cell
Biol., 118, 703-714.
81.Hamaguchi, M., Matsuyoshi, N., Ohnishi, Y.,
Gotoh, B., Takeichi, M., and Nagai, Y. (1993) Dev. Suppl.,
41-46.
82.Behrens, J., Vakaet, L., Friis, R., Winterhager,
E., and Roy, F. V. (1993) J. Cell Biol., 120,
757-766.
83.Takeda, H., Nagafuchi, A., Yonemura, S., Tsukita,
S., and Behrens, J. (1995) J. Cell Biol., 131,
1839-1847.
84.Reynolds, A. B., Herbert, L., Cleveland, J. L.,
Berg, S. T., and Gaut, J. R. (1992) Oncogene, 7,
2439-2445.
85.Daniel, J. M., and Reynolds, A. B. (1995) Mol.
Cell. Biol., 15, 4819-4824.
86.Khoury, H., Dankort, D. L., Sadekova, S.,
Naujokas, M. A., Muller, W. S., and Park, M. (2001) Oncogene,
20, 788-799.
87.Hoschuetzky, H., Aberle, H., and Kemler, R.
(1994) J. Cell Biol., 127, 1375-1380.
88.Zondag, G. C., Moolenaar, W. H., and Gebbink, M.
F. (1996) J. Cell Biol., 134, 1513-1517.
89.Meyer, R. A., Laird, D. W., Revel, J.-P. , and
Johnson, R. G. (1992) J. Cell Biol., 119, 179-189.
90.Steinberg, M. S., and Takeichi, M. (1994)
Proc. Natl. Acad. Sci. USA, 91, 206-209.
91.Angres, B., Barth, A., and Nelson, W. J. (1996)
J. Cell Biol., 134, 549-557.
92.Vestweber, D., Gossler, A., Boller, K., and
Kemler, R. (1987) Dev. Biol., 124, 451-456.
93.Rosales, C., O'Brien, V., Kornberg, L., and
Juliano, R. (1995) Biochim. Biophys. Acta, 1242,
77-98.
94.Gumbiner, B. (1990) Curr. Opin. Cell
Biol., 2, 881-887.
95.Doherty, P., Ashton, S. V., Moore, S. E., and
Walsh, F. S. (1991) Cell, 67, 21-33.
96.Bixby, J. L., Grunwald, G. B., and Bookman, R. J.
(1994) J. Cell Biol., 127, 1461-1475.
97.Tsukita, S., Oishi, K., Akiyama, T., Yamanashi,
Y., Tamamoto, T., and Tsukita, S. (1991) J. Cell Biol.,
113, 867-879.
98.Woods, D. F., and Bryant, L. (1993) J. Cell
Sci. Suppl., 17, 171-181.
99.Ko, K. S., Arora, P. D., Bhide, V. V., Chen, A.,
and McCulloch, C. A. (2001) J. Cell Sci., 114 (Pt. 6),
1155-1167.
100.Pece, S., and Gutkind, J. S. (2000) J. Biol.
Chem., 275, 41227-41233.
101.Kim, S. H., Li, Z., and Sacks, D. B. (2000)
J. Biol. Chem., 275, 36999-37005.
102.Gumbiner, B. (1995) Curr. Opin. Cell
Biol., 7, 634-640.
103.Peifer, M. (1995) Trends Cell Biol.,
5, 224-229.
104.Miller, J. R., and Moon, R. T. (1996) Genes
Dev., 10, 2527-2539.
105.Resnik, E. (1997) Nat. Genet.,
16, 9-11.
106.Fagotto, F., and Gumbiner, B. M. (1996) Dev.
Biol., 180, 445-454.
107.Orsulic, S., and Peifer, M. (1996) J. Cell
Biol., 134, 1283-1300.
108.Behrens, J., von Kries, J. P., Kuhl, M., Bruhn,
L., and Wedlich, D. (1996) Nature, 382, 638-642.
109.Molenaar, M., van de Wetering, M., Oosterwegel,
M., Peterson-Maduro, J., and Godsave, S. (1996) Cell, 86,
391-399.
110.Novak, A., and Dedhar, S. (1999) Cell. Mol.
Life Sci., 56, 523-537.
111.Ling, Y., Zhong, Y., and Perez-Soler, R. (2001)
Mol. Pharmacol., 59, 593-603.
112.Sanson, B., White, P., and Vincent, J.-P.
(1996) Nature, 383, 627-630.
113.Hafen, E., Dickson, B., Raabe, T., Brunner, D.,
Oellers, N., and van der Straten, A. (1993) Dev. Suppl.,
41-46.
114.Artavanis-Tsakonas, S., Matsuno, K., and
Fortini, M. E. (1995) Science, 268, 225-232.
115.Bosenberg, M. W., and Massague, J. (1993)
Curr. Opin. Cell Biol., 5, 832-838.
116.Musil, L. S., Cunningham, B. A., Edelman, G.
M., and Goodenough, D. A. (1990) J. Cell Biol., 111,
2077-2088.
117.Townes, P., and Holtfretter, J. (1955) J.
Exp. Zool., 128, 53-120.
118.Hyafil, F., Morello, D., Babinet, C., and
Jacob, F. (1980) Cell, 21, 927-934.
119.Hyafil, F., Babinet, C., and Jacob, F. (1981)
Cell, 26, 447-454.
120.Reima, I. (1990) Cell Differ. Dev.,
29, 143-153.
121.Ao, A., and Erickson, R. P. (1992) Antisense
Res. Dev., 2, 153-163.
122.Larue, L., Ohsugi, M., Hirchenhain, J., and
Kemler, R. (1994) Proc. Natl. Acad. Sci. USA, 91,
8263-8267.
123.Riethmacher, D., Brinkmann, V., and Birchmeier,
C. (1995) Proc. Natl. Acad. Sci. USA, 92, 855-859.
124.Heasman, J., Ginsberg, D., Geiger, B.,
Goldstone, K., Pratt, T., Yoshida-Noro, C., and Wylie, C. (1994)
Development, 120, 49-57.
125.Hatta, K., Takagi, S., Fujisawa, H., and
Takeichi, M. (1987) Dev. Biol., 120, 215-227.
126.Hatta, K., Nose, A., Nagafuchi, A., and
Takeichi, M. (1988) J. Cell Biol., 106, 873-881.
127.Mege, R. M., Matsuzaki, F., Gallin, W. J.,
Goldberg, J. I., and Cunningham, B. A. (1988) Proc. Natl. Acad. Sci.
USA, 85, 7274-7278.
128.McNeili, H., Ozawa, M., Kemler, R., and Nelson,
W. J. (1990) Cell, 62, 309-316.
129.Behrens, J., Mareel, M. M., Van Roy, F. M., and
Birchmeier, W. (1989) J. Cell Biol., 108, 2435-2447.
130.Frixen, U. H., Behrens, J., and Sachs, M.
(1991) J. Cell Biol., 113, 173-185.
131.Thiery, J. P., Boyer, B., Tucker, G.,
Gavrilovic, J., and Valles, A. M. (1988) Ciba Found. Symp.,
141, 48-74.
132.Redies, C., Engelhart, K., and Takeichi, M.
(1993) J. Comp. Neurosci., 333, 398-416.
133.Redies, C., and Muller, H.-A. (1994) Cell
Adhes. Commun., 2, 511-520.
134.Murphy-Erdoch, C., Napolitano, E. W., and
Reichardt, L. F. (1994) Dev. Biol., 161, 107-125.
135.Shimamura, K., and Takeichi, M. (1992)
Development, 116, 1011-1019.
136.Ganzler, S., and Redies, C. (1995) J.
Neurosci., 15, 4157-4172.
137.Fredette, B. J., and Ranscht, B. (1994) J.
Neurosci., 14, 7331-7346.
138.Friedlander, D. R., Mege, R. M., Cunningham, B.
A., and Edelman, G. M. (1989) Proc. Natl. Acad. Sci. USA,
86, 7043-7047.
139.Radice, J. L., Rayburn, H., Matsunami, H.,
Knudsen, K. A., Takeichi, M., and Hynes, R. O. (1997) Dev.
Biol., 181, 64-78.
140.Redies, C., and Takeichi, M. (1996) Dev.
Biol., 180, 413-423.
141.Redies, C. (1997) Cell Tissue Res.,
290, 405-413.
142.Riehl, R., Johnson, K., Bradley, R., Grunwald,
G. B., Cornel, E., Lilienbaum, A., and Holt, C. E. (1996)
Neuron, 17, 837-848.
143.Matsunaga, M., Hatta, K., Nagafuchi, A., and
Takeichi, M. (1988) Nature, 334, 62-64.
144.Doherty, P., Cohen, J., and Walsh, F. S. (1990)
Neuron, 5, 209-219.
145.Doherty, P., Fruns, M., Seaton, P., Dickson,
G., Barton, C. H., Seras, T. A., and Walsh, F. S. (1990) Nature,
343, 464-466.
146.Doherty, P., Rowett, L. H., Moore, S. E., Mann,
D. A., and Walsh, F. S. (1991) Neuron, 6, 247-258.
147.Fredette, B. J., Miller, J., and Ranscht, B.
(1996) Development, 122, 3163-3171.
148.Cifuentes-Diaz, C., Nicolet, M., Goudou, D.,
Rieger, F., and Mege, R. M. (1994) Development, 120,
1-11.
149.Chifuentes-Diaz, C., Goudou, D., Padilla, F.,
Facchinetti, P., Nicolet, M., Mege, P. M., and Rieger, F. (1996)
Eur. J. Neurosci., 8, 1666-1676.
150.Tanaka, H., Shan, W., Phillips, G. R., Arndt,
K., Bozdagi, O., Shapiro, L., Huntley, G. W., Benson, D. L., and
Colman, D. R. (2000) Neuron, 25, 93-107.
151.Uemura, T. (1998) Cell, 93,
1095-1098.
152.Ranscht, B. (2000) Int. J. Dev.
Neurosci., 18, 643-651.
153.Bruses, J. L. (2000) Curr. Opin. Cell
Biol., 12, 593-597.
154.Suzuki, S. C., Inoue, T., Kimura, Y., Tanaka,
T., and Takeichi, M. (1997) Mol. Cell. Neurosci., 9,
433-447.
155.Inoue, A., and Sanes, J. R. (1997)
Science, 276, 1428-1431.
156.Albeda, S. M. (1993) Lab. Invest.,
68, 4-17.
157.Vasiliev, J. M., and Gelfand, I. M. (1981)
Neoplastic and Normal Cells in Culture, Cambridge University
Press, London.
158.Takeichi, M. (1993) Curr. Opin. Cell
Biol., 5, 806-811.
159.Shiozaki, H., Tahara, H., and Oka, H. (1991)
Am. J. Pathol., 139, 17-23.
160.Schipper, J. H., Frixen, U. H., Behrens, J.,
Unger, A., Jahnke, K., and Birchmeier, W. (1991) Cancer Res.,
51, 6328-6337.
161.Perl, A. K., Wilgenbus, P., Dahl, U., Semb, H.,
and Christofori, G. (1998) Nature, 392, 190-193.
162.Christofori, G., and Semb, H. (1999) Trends
Biochem. Sci., 24, 73-76.
163.Navarro, P., Gomez, M., Pizarro, A., Gamallo,
C., Quintanilla, M., and Cano, A. (1991) J. Cell Biol.,
115, 517-533.
164.Vleminckx, K., Vakaet, L., Mareel, M., Fiers,
W., and Van Roy, F. (1991) Cell, 66, 107-119.
165.Chen, W., and Obrink, B. (1991) J. Cell
Biol., 114, 319-327.
166.Smythe, W. R., Williams, J. P., Wheelock, M.
J., Johnson, K. R., Kaiser, L. R., and Albelda, S. M. (1999) Lung
Cancer, 24, 157-168.
167.Seline, P. C., Norris, D. A., Horikawa, T.,
Fujita, M., and Middleton, M. H. (1996) J. Invest. Dermatol.,
106, 1320-1324.
168.Hazan, R. B., Kang, L., Whooley, B. P., and
Borgen, P. I. (1997) Cell Adhes. Commun., 4, 399-411.
169.Islam, S., Carey, G. T., Wolf, M. G., Wheelock,
M. J., and Johnson, K. R. (1996) J. Cell Biol., 135,
1643-1654.
170.Hazan, R. B., Phillips, G. R., Qiao, R. F.,
Norton, L., and Aaronson, S. A. (2000) J. Cell Biol.,
148, 779-790.
171.Bodmer, W. (1999) Cytogenet. Cell
Genet., 86, 99-104.
172.Morin, P. J., Sparks, A. B., Korinek, V.,
Barker, N., Clevers, H., Vogelstein, B., and Kinzler, K. W. (1997)
Science, 275, 1787-1790.
173.Ranscht, B., and Dours-Zimmermann, M. T. (1991)
Neuron, 7, 391-402.
174.Vestal, D. J., and Ranscht, B. (1992) J.
Cell Biol., 119, 451-461.
175.Ranscht, B., and Bronner-Fraser, M. (1991)
Development, 111, 15-22.
176.Tanihara, H., Sano, K., Heimark, R. L., St.
John, T., and Suzuki, S. (1994) Cell Adhes. Commun., 2,
15-26.
177.Takeuchi, T., Misaki, A., Chen, B. K., and
Ohtsuki, Y. (1999) Histopathology, 35, 87-8.
178.Kawakami, M., Staub, J., Cliby, W., Hartmann,
L., Smith, D. I., and Shridar, V. (1999) Int. J. Oncol.,
15, 715-720.
179.Sato, M., Mori, Y., Sakurada, A., Fujimura, S.,
and Horii, A. (1998) J. Hum. Genet., 43, 285-286.
180.Sato, M., Mori, Y., Sakurada, A., Fujimura, S.,
and Horii, A. (1998) Hum. Genet., 103, 96-101.
181.Mori, Y., Matsunaga, M., Abe, T., Fukushige,
S., Miura, K., Sunamura, M., Shiiba, K., Sato, M., Nukiwa, T., and
Horii, A. (1999) Br. J. Cancer, 80, 556-562.
182.Lee, S. W. (1996) Nat. Med., 2,
776-782.
183.Lee, S. W. (1998) Carcinogenesis,
19, 1157-1159.
184.Takeuchi, T., Misaki, A., Liang, S. B.,
Tachibana, A., Hayashi, N., Sonobe, H., and Ohtsuki, Y. (2000) J.
Neurochem., 74, 1489-1497.
185.Koller, E., and Ranscht, B. (1996) J. Biol.
Chem., 22, 30061-30067.
186.Stefanova, I., Horeishi, V., Ansotegui, I. J.,
Knapp, W., and Stockinger, H. (1991) Science, 254,
1016-1019.
187.Shenoy-Scaria, A. M., Kwong, J., Fujita, T.,
Olszowy, M. W., Shaw, A. S., and Lublin, D. M. (1992) J.
Immunol., 149, 3535-3541.
188.Solomon, K. R., Rudd, C. E., and Finberg, R. W.
(1996) Proc. Natl. Acad. Sci. USA, 93, 6053-6058.
189.Philippova, M. P., Bochkov, V. N., Stambolsky,
D. V., Tkachuk, V. A., and Resink, T. J. (1998) FEBS Lett.,
429, 207-210.
190.Anderson, R. G. W. (1998) Annu. Rev.
Biochem., 67, 199-225.
191.Jang, Y., Lincoff, A. M., Plow, E. F., and
Topol, E. J. (1994) J. Am. Coll. Cardiol., 24,
1591-1601.
192.Hamon, M., Bauters, C., McFadden, E. P.,
Wernert, N., Lablanche, J. M., Dupuis, B., and Bertand, M. E. (1995)
Eur. Heart J., 16 (Suppl. 1), 33-48.
193.Ivanov, D., Philippova, M., Antropova, J.,
Gubaeva, F., Iljinskaya, O., Tararak, E., Bochkov, V., Erne, P.,
Resink, T., and Tkachuk, V. (2001) Histochem. Cell Biol.,
115, 231-242.
194.Kuzmenko, Y. S., Stambolsky, D., Kern, F.,
Bochkov, V. N., Tkachuk, V. A., and Resink, T. J. (1998) Biochem.
Biophys. Res. Commun., 246, 489-494.
195.Kuzmenko, Y. S., Kern, F., Bochkov, V. N.,
Tkachuk, V. A., and Resink, T. J. (1998) FEBS Lett., 434,
183-187.
196.Wyder, L., Vitaliti, A., Schneider, H.,
Hebbard, L. W., Moritz, D. R., Wittmer, M., Ajmo, M., and Klemenz, R.
(2000) Cancer Res., 60, 4682-4688.
197.Tkachuk, V. A., Kuzmenko, Y. S., Resink, T. J.,
Stambolsky, D. V., and Bochkov, V. N. (1994) Mol. Pharmacol.,
46, 1129-1137.
198.Bochkov, V. N., Tkachuk, V. A., Kuzmenko, Y.
S., Borisova, Y. L., Buhler, F. R., and Resink, T. J. (1994) Mol.
Pharmacol., 45, 262-270.
199.Kuzmenko, Y. S., Bochkov, V. N., Philippova, M.
P., Tkachuk, V. A., and Resink, T. J. (1994) Biochem. J.,
303, 281-287.
200.Bochkov, V. N., Tkachuk, V. A., Philippova, M.
P., Stambolsky, D. V., Buhler, F. R., and Resink, T. J. (1996)
Biochem. J., 317, 297-304.
201.Tkachuk, V. A., Bochkov, V. N., Philippova, M.
P., Stambolsky, D. V., Kuzmenko, Y. S., Sidorova, M. V., Molokoedov, A.
S., Spirov, V. G., and Resink, T. J. (1998) FEBS Lett.,
421, 208-212.
202.Stambolsky, D. V., Kuzmenko, Y. S., Philippova,
M. P., Bochkov, V. N., Bespalova, Z. D., Azmuko, A. A., Kashirina, N.
M., Vlasik, T. N., Tkachuk, V. A., and Resink, T. J. (1999) Biochim.
Biophys. Acta, 1416, 155-160.
203.Ross, R. (1995) Annu. Rev. Physiol.,
57, 791-804.