Kinetics and pH Dependence of Light-Induced Deprotonation of the Schiff Base of Rhodopsin: Possible Coupling to Proton Uptake and Formation of the Active Form of Meta II
O. Kuwata1,2, C. Yuan1, S. Misra3,4, R. Govindjee1, and T. G. Ebrey3*
1Center for Biophysics and Computational Biology, and Department of Biochemistry, 600 S. Mathews, University of Illinois, Urbana, Illinois, 61801 USA2Present address: Institute of Biological Science, University of Tsukuba, Tsukuba, Ibaraki, 305-8852, Japan
3Departments of Botany and Zoology, Box 351330, University of Washington, Seattle, WA 98195, USA; E-mail: tebrey@u.washington.edu
4Present address: Laboratory of Molecular Biology, National Institute of Diabetes and Digestive and Kidney Disorders, National Institutes of Health, Bethesda, Maryland, 20892 USA
* To whom correspondence should be addressed.
Received June 8, 2001; Revision received July 12, 2001
In this paper we first review what is known about the kinetics of Meta II formation, the role and stoichiometry of protons in Meta II formation, the kinetics of the light-induced changes of proton concentration, and the site of proton uptake. We then go on to compare the processes that lead to the deprotonation of the Schiff base in bacteriorhodopsin with rhodopsin. We point out that the similarity of the signs of the light-induced electrical signals from the two kinds of oriented pigment molecules could be explained by bacteriorhodopsin releasing a proton from its extracellular side while rhodopsin taking up a proton on its cytoplasmic side. We then examined the pH dependence of both the absorption spectrum of the unphotolyzed state and the amplitude and kinetics of Meta II formation in bovine rhodopsin. We also measured the effect of deuteration and azide on Meta II formation. We concluded that the pKa of the counter-ion to the Schiff base of bovine rhodopsin and of a surface residue that takes up a proton upon photolysis are both less than 4 in the unphotolyzed state. The data on pH dependence of Meta II formation indicated that the mechanisms involved are more complicated than just two sequential, isospectral forms of Meta II in the bleaching sequence. Finally we examined the evidence that, like in bacteriorhodopsin, the protonation of the Schiff bases's counter-ion (Glu113) is coupled to the changing of the pKa of a protonatable surface group, called Z for rhodopsin and tentatively assigned to Glu134. We conclude that there probably is such a coupling, leading to the formation of the active form of Meta II.
KEY WORDS: rhodopsin, light-induced deprotonation, Schiff base, pH dependence
This paper is on the intersection of two topics that were favorites of the late Andrei Kaulen. The first is the processes involved in the light-induced deprotonation of the Schiff base in retinal pigments, especially bacteriorhodopsin (see his masterful review [1]) and thus the link between the absorption of light and the physiological result for the pigment, the pumping of a proton. The second involves the intriguing similarities between the bacteriorhodopsin and rhodopsin systems and in particular the possibility that light-driven proton movements in rhodopsin may be as important as such changes in bacteriorhodopsin (see, in particular, his papers with Drachev and Skulachev [2, 3]). Future researchers will continue to build upon Kaulen's important contributions to these and other areas of research.
Light initiates a sequence of spectral changes in the visual pigment rhodopsin (RH), which lead to electrical excitation of a photoreceptor cell. Within a few milliseconds, the primary photoproduct, Bathorhodopsin, decays through the Blue Shifted Intermediate (BSI) through Lumirhodopsin (Lumi) to Metarhodopsin I (Meta I). Meta I then decays to Metarhodopsin II (Meta II) (reviewed in [4-7]). Lumi may also have alternative pathways of decay (discussed below and in [8, 9]). Meta II is perhaps the key intermediate in the photolysis of the rhodopsin because it is this intermediate that interacts with the G-protein transducin, initiating the enzyme cascade leading to visual excitation. It is therefore of great interest to investigate the molecular events that take place during the formation of the Meta II intermediate in order to understand the mechanism of the activation of transducing. We will first give an in depth review of what is known about Meta II.
The retinal chromophore of rhodopsin is attached to Lys296 by a Schiff base linkage. It was cogently argued at the time of the discovery of Metarhodopsin II [10] that the Schiff base is protonated in Meta I but deprotonated in Meta II. This was later shown more directly using resonance Raman spectroscopy to observe the C=N and C=NH+ vibrations of Meta II and Meta I, respectively [11]. Many other aspects of the excitation process are not clear. While the pigment has to be in the Meta II state to activate transducin, being in the Meta II state is by itself not sufficient for activation. There is good evidence that Meta II is quickly phosphorylated and then binds arrestin, disabling the ability of Meta II to activate transducin (see reviews, cited above). There is no change in the spectrum of Meta II upon phosphorylation or arrestin binding. Another area of uncertainty involves the exact sequence of photointermediates after photoexcitation. The sequence from Batho to Meta I has been fairly well agreed upon, once it was realized that one of the intermediates, BSI, is “frozen out” by low temperatures and can only be observed easily in flash photolysis experiments (reviewed in [6]). However, there is uncertainty about the lifetimes and processes taking place in the later stages of bleaching, starting with the formation and especially the decay of Meta I. There are several reasons for this. One is that different investigators have used different preparations that seem to have quite different behaviors. From the most physiological to the most disrupted, common preparations are the following. 1) Intact photoreceptor cells, in which transducin and rhodopsin kinase are present. Transducin and rhodopsin kinase can interact with rhodopsin's bleaching intermediates and shift the equilibrium between them (transducin, especially, can shift the Meta I<-->Meta II equilibrium). 2) Purified membrane suspension of rod outer segment (ROS) disk membranes, in which most of the extrinsic peripheral membrane proteins have been removed and rhodopsin itself is in its native lipid environment. These two preparations, being particulate, scatter transmitted light badly and so it is more difficult to obtain high quality spectral data from them. 3) Rhodopsin solubilized in digitonin, a detergent in which rhodopsin can be regenerated after bleaching. Relative to rhodopsin in other detergents, the thermal stability of rhodopsin in digitonin is more similar to that of rhodopsin in ROS disk membranes [12, 13]. 4) Rhodopsin solubilized in dodecyl maltoside or a similar detergent where regeneration is still possible but thermal stability is further decreased (see references above). 5) Rhodopsin solubilized in a detergent that does not allow regeneration and therefore is deemed harsher than those that do allow it.
As an example of the effects of using different sample preparations, we consider the experimental evidence for alternative pathways for the decay of Lumi. The data is complex but the simplest interpretation is that part of the Lumi decays to this new species, called by Thorgeirsson et al. [8, 9], the particularly unfortunate name of Meta I380 (unfortunate because it probably would be best to reserve the name Meta I for protonated Schiff base species and it is very likely that the new short wavelength species is an unprotonated Schiff base). We propose calling it “T” in honor of Thorgeirsson. The experiments of Thorgeirsson et al. were performed in mildly sonicated ROS. It is most prominent at higher temperatures and so, at room temperature, like in the experiments presented below [14], the preferred pathway for the decay of Lumi seems to be through Meta I to Meta II. We tentatively ignore this new species (T380) in this paper as not being a major participant in most experimental situations, including the one examined here. However, at higher temperature, significant amounts of the pigment can flow through this pathway from Lumi.
Equilibration between Meta I and Meta II and complex formation kinetics for Meta II. The original experiments of Matthews et al. (1963) on rhodopsin in digitonin showed that Meta I was in pH- and temperature-dependent equilibrium with Meta II. The observed pH dependence was at first surprising in that one might expect that the equilibrium between a protonated and unprotonated Schiff base would favor the protonated Schiff base with increasing proton concentration. Instead, the equilibrium is shifted towards Meta II with increasing proton concentration. This suggests that the protonation state of one or more groups, other than the Schiff base itself, can shift the equilibrium from Meta I to Meta II. Later, it was shown that when light initiates events that lead to Schiff base deprotonation, the proton is transferred to a glutamic acid buried in the center of the membrane, Glu113 [15]. The Meta I-->Meta II transition is identified with this proton transfer.
In early reports, the formation of Meta II from Meta I, monitored spectroscopically at 380 nm, was fitted as a single exponential. Others found the process to be multiexponential even in ROS (both sets of data are summarized in [16]). Most recent reports find two or three components of Meta II formation in ROS or digitonin at 15-25°C [14, 17-20]. To explain these kinetics, Straume et al. [19] favored a model in which two Meta IIs were each independently in equilibrium with Meta I. Thorgeirsson et al. [8] explained their data in part by a Meta II like species formed directly from Lumi. Hofmann and co-workers [21, 22] favored two Meta IIs in series. Although these models do not exclude each other [14], with the available evidence the series model is attractive because: 1) it parallels a model for the formation of the key M intermediate in bacteriorhodopsin (see below), and 2) it is compatible with experimental results that suggest that proton uptake is required for the stabilization and maintenance of Meta II. We consider the latter idea first.
The role of protons in forming Meta II. The equilibrium between Meta I and Meta II is pH-dependent with the higher concentration of protons favoring the deprotonated form of the Schiff base (Meta II). Since the formation of Meta II reflects the intra-protein proton transfer between the Schiff base and Glu113 [15], the origin of the pH dependence of this equilibrium is not obvious; it must reflect the role of another protonatable group. The Meta I/Meta II equilibrium was studied in some detail by Parkes and Liebman [23] who made careful measurements of the thermodynamics and kinetics of the Meta I to Meta II transition between pH 5.9 and 8.1; these experiments were done at lower temperatures (-1 to 5°C) so that they could follow the kinetics accurately even with limited time resolution (~60 msec). Their results suggested that a proton is taken up during the formation of Meta II. They initially found that n, the number of protons taken up was 0.70 ± 0.04. Gibson, Parkes and Liebman [24] later showed that because of the surface potential affecting the local membrane surface pH, the apparent number of protons taken up was distorted, and that when the surface potential effects are taken into account, n equals 1. Miller and Oesterhelt [25] had earlier reported that the surface potential of the purple membrane distorted some proton stoichiometries associated with bacteriorhodopsin. Thus, examining the pH dependence of the equilibrium between Meta I and Meta II suggests that one proton is taken up in going from Meta I to Meta II.
Note that with this technique only protons taken up/released that control the Meta I/Meta II equilibrium are measured. More recently, Arnis and Hofmann [21] examined the kinetics of Meta II formation at different pH values and formulated a model in which Meta II formation is divided into two steps, Schiff base deprotonation and then proton uptake (see below). They suggested the existence of two Meta IIs in series, Meta I<-->Meta IIa<-->Meta IIb, with the proton being taken up in the second step. Thus, of the three exponential phases reported in the 1 µsec to 500 msec time range, the fastest can be assigned to the Lumi to Meta I transition while the other two are assigned to the Meta I<-->Meta IIa<-->Meta IIb. According to Hofmann et al. [5, 26] the rate of the transition from Meta IIa to Meta IIb at 5°C is 10 msec in dodecyl maltoside; at 20°C in ROS membranes, Meta I-->Meta IIa is ~5 msec while Meta IIa<-->Meta IIb is now 1 msec. Meta II takes minutes to decay.
Stoichiometry of the protonation changes associated with Meta II formation. The pH dependence of the Meta I/Meta II equilibrium provides evidence of transient protonation changes upon light activation of rhodopsin. Accurate determination of the stoichiometry and the kinetics of proton binding/release from light-activated rhodopsin is complicated by many seemingly contradictory results that may depend on the experimental preparation used. Below we attempt to summarize and evaluate these results. As with experiments on the photochemistry, several different preparations of rhodopsin have been used: intact cells, ROS membranes, washed ROS membranes, rhodopsin reconstituted in lipid vesicles, rhodopsin in mild (regenerating) detergents and rhodopsin in stronger detergents.
The first report of light causing protonation changes of rhodopsin was by Radding and Wald [27]. They showed that bleaching bovine rhodopsin caused the pH of the suspending medium to increase, and they surmised that a proton was taken up for every rhodopsin bleached. Soon thereafter Matthews et al. [10] reported that Metarhodopsin existed in two interconvertible forms, Meta I and Meta II. They found that the pKa of the group that controlled the equilibrium between Meta I and Meta II was ~6.4 at 170 mM salt. Several paths of research were then pursued. One line of investigation was to measure near room temperature the number of protons taken up or released from light-activated rhodopsin. Falk and Fatt [28] found that in frog ROS near neutral pH, one proton/bleached rhodopsin is taken up. These workers used pH electrodes to measure protons and showed that the changes occurred within 500 msec. Emrich [29] was the first to report results with pH sensitive dyes and found a light-initiated proton uptake for ROS membranes at 20°C (7 to 31°C) using bromocresol purple as an indicator.
Similarly, Wong and Ostroy [30] used a pH indicator dye, bromocresol green, to measure light-induced changes for rhodopsin in digitonin at low temperatures, e.g., -10°C. In this way they could measure proton changes with some kinetic resolution and found that one proton was taken up during Meta II formation and released later, in times of the order of minutes at 25°C, concurrent with the decay of Meta II. Later Emrich and Reich [31] reported a variable proton/Meta II stoichiometry at low bleached fractions of RH converted to Meta II, which converged to one proton/Meta II at higher bleaches. It seems likely that the presence of transducin contributed to the variable stoichiometry [32]. In 1978 Bennett [33] reported that over the pH range 6-8 two protons are taken up per Meta II formed in ROS membranes at 3°C. At warmer temperatures, the Meta II decays but also loses protons itself so that now an “unprotonated” Meta II is formed. The pH changes were measured with a pH electrode. Using the pH sensitive dye bromocresol purple, washed ROS membranes, and reasonable time resolution (milliseconds at 20°C), Bennett [34] reported that two protons were taken up as Meta II was formed, but that at pH values greater than 5, one of the protons was released before Meta II decayed, so that the H+/Meta II stoichiometry equals 2 only at low pH values. It is not clear why this pH dependence differed from that reported earlier. Schleicher and Hofmann [32] found that for low bleaches in unwashed ROS, two protons were taken up per Meta II but that at higher bleaches, where there is much more rhodopsin photolyzed than transducin present, the stoichiometry went down to 1 proton/Meta II. Later, working with rhodopsin solubilized in octyl glucoside or dodecyl maltoside, Arnis and Hofmann [21] found two protons are taken up per Meta II at pH 6. Arnis et al. [22] reported stoichiometries of 2 for pH 6.0 and for pH 7.5, and a net release at higher pH values.
Another method used to determine the number of protons taken up/released upon Meta II formation is photocalorimetry. Cooper and Converse [35] measured the amount of heat released from a solution of ROS membranes after photolysis in several different buffers with differing heats of proton ionization. They found that there were no protonation changes during Meta II formation at pH 8.0, and only one proton was taken up during Meta II formation at pH 5.4.
To summarize, several experiments following light-induced protonation changes with pH sensitive dyes or pH electrodes [27, 28, 30, 32], pH effects on the Meta I/Meta II equilibrium [24] and photocalorimetric experiments [35] find that just one proton is taken up in forming Meta II at lower pH values. In contrast, Bennett [36] and Arnis and Hofmann [21] find that up to two protons are taken up per Meta II at lower pH values. While some experiments would only measure the protons that must be taken up to allow Meta II formation (“essential” protons [23]), others measure all the protons taken up, essential and non-essential. This could explain part of the discrepancy. However, in view of the difficulty of measuring proton stoichiometries, we tentatively assume that the simpler, more comprehensive set of data is accurate and that only one proton is taken up in going from Meta I to Meta II in ROS membranes. Arnis and Hofmann's experiments show that Meta II can be transiently formed without proton uptake (Meta IIa) but that a stabilized Meta II (Meta IIb) requires proton uptake [21, 37]. The question of the kinetics of the proton changes will now be addressed.
Kinetics of the protonation changes in photolyzed rhodopsin. Several of the techniques used to measure the stoichiometry of the protonation changes have also been used to measure the time course with which the proton is taken up during the photolysis of rhodopsin. Measurements in ROS membranes or rhodopsin in digitonin at lower temperatures (-10.5°C [30]) or at warmer temperatures (23°C [31]; 37°C [33]) found proton uptake fairly well matched the Meta I to Meta II transition rate, although resolution and precision were limited. Bennett [36] reported good time resolution data for proton uptake (bromocresol purple) and Meta II formation (absorbance at 365 nm), and found that they matched well. A small (10% of total), very slow phase (t1/2 of ~2 sec vs. 10 msec for the main phase at 20oC) of Meta II formation was observed. At pH values above 5 a proton was released with about the same lifetime as the slow phase, 2 sec. This transient change in protonation, if present, probably could not have been seen by Bennett [33], Schleicher and Hofmann [32], Wong and Ostroy [30] or Emrich [29] with the methods they used. Kaupp et al. [38] used the pH indicator bromocresol purple to show that in sonicated ROS membranes proton uptake and Meta II formation times were essentially identical at 20°C (both with halftimes of ~5 msec).
In 1993 Arnis and Hofmann [21] solubilized bovine rhodopsin in two different detergents, octyl glucoside or dodecyl maltoside (in these detergents Meta II formation is greatly accelerated compared to ROS or digitonin solutions) and showed that at some temperatures, the light-induced proton uptake signal from bromocresol purple lagged significantly behind Meta II formation. Their experiments were performed at such a low temperature (3°C) that their time base was fast enough (unlike before, when everyone's resolution was too slow) to detect any possible deprotonation associated with Meta II on the 1-10 sec time scale [Footnote: Even more recently Szundi et al. [39] and Meyer and Hofmann [40] have reported that there is a very rapid (less than a millisecond) light-induced proton release and reuptake, perhaps before Meta I decays, followed by a “slower” proton uptake matching Meta II formation. It is difficult to evaluate this putative very rapid protonation change without further study, to be sure it is not a reflection of membrane surface dipole potential changes]. So far, all the kinetics of proton uptake which have been reported have been plausibly represented as a single exponential phase, so if there were two different groups that took up a proton during Meta II formation, they would have to become protonated with about the same kinetics. This is an argument, albeit not a strong one, that only one proton is taken up in forming Meta II. This will be pursued below.
There is another way to potentially follow proton kinetics in intact ROS, and that is to use the Early Receptor Potential (ERP) or the Early Receptor Current (ERC). The ERP is a very fast potential/current evoked from oriented photoreceptors with bright flashes (reviewed in [41, 42]). A similar potential can be elicited from oriented bacteriorhodopsin. At room temperature, two phases can be easily observed for the ERP, a somewhat faster (sub-milliseconds) corneal negative R1 and a bit slower (1-10 msec) potential of opposite sign, R2. For rhodopsin, Cone [43, 44] showed that the kinetics of appearance of R2 and Meta II were identical within experimental error (this correlation was also reported by Spalink and Stieve [45] for bovine retinas at 37°C). This finding, along with R2's temperature dependence, led Cone [43, 44] to propose that R2 was due to charge movements associated with the Meta I to Meta II transition. (R1 was similarly linked to the RH to Meta I transitions.) Cafiso and Hubbell [46] used spin labels and Bennett et al. [36] used membrane absorbed dyes to detect changes in the electrical properties of the ROS disk membrane interface. The signals these groups detected could plausibly be explained as having the same origin as the R2 phase of the ERP as discussed above, that is, both are due to the cytoplasmic face becoming more positive; this could arise from the uptake of a proton from the medium at the cytoplasmic surface of the disk. The interfacial potential change measured by Cafiso and Hubbell titrated with a pKa of ~6.8 at 135 mM salt, similar to the pKa of the Meta I to Meta II equilibrium.
Cone [43, 44] argued that since R2 is caused by charge movement associated with Meta II formation, and since it is known that at least one proton is taken up when Meta II is formed, that such a proton uptake could contribute to the charge movement associated with Meta II formation. (Movement of charged groups or dipoles within the pigment could also contribute to the observed electrical signal, and indeed must be the source of R1.) Two recent studies seem to demonstrate this assignment. Dickopf et al. [20] studied oriented ROS membrane fragments incorporated into lipid vesicles. They found that the rise of the light-evoked electrical current could be resolved into two components with similar time constants, 1.1 and 3.0 msec at 22°C, pH 7.8. Together these probably correspond to R2; R1 was not observed, perhaps because the recording method used makes very fast events difficult to detect. The photocurrent's amplitude and time course were pH dependent, with the amplitude falling with increasing pH. These changes did seem to track the pH dependence of the amplitude of Meta II produced, although both determinations did not quite match what had been reported in other studies. However, this may be because of the unusual environment of the rhodopsin, in sonicated membrane fragments attached to a lipid support membrane. Sullivan [47] titrated the R2 component for rhodopsin expressed and then incorporated into HEK293S cells. He found that ¾ of the R2 amplitude disappeared upon raising the pH with a pKa of 6.3, again suggesting that a large part of R2 is due to proton uptake by the photolyzed rhodopsin in forming Meta II.
In summary, three kinds of techniques for measuring proton kinetics: pH electrode, pH sensitive dyes, and the shape of the R2 component of the ERP find that the formation of Meta II is intimately and kinetically associated with the uptake of a proton. Arnis and Hofmann [21] show that in some circumstances proton uptake can lag behind Meta II formation. Moreover, titration data for the equilibrium between Meta I and Meta II, along with the titration of the fast photocurrent due to proton uptake associated with Meta II formation, find that the pKa of the group binding the proton is approximately 6.4 in 150 mM salt.
Site of proton uptake. Arnis and co-workers [22] showed that the light-induced proton uptake by rhodopsin was completely abolished in a mutant in which Glu134 was changed to a neutral residue. The authors reasonably enough argued that this shows that Glu134 is the site of proton uptake. As noted below, the uptake of a proton by Glu134, located on the cytoplasmic surface of the pigment [48], would be of the right sign to contribute to the R2 photovoltage from rods. In addition, using FTIR spectroscopy Fahmy et al. [49] have recently shown that Glu134 is likely deprotonated in the dark, but is protonated in Meta II in the presence of transducin. Can we identify Glu134 as the site which, when it takes up a proton, shifts the equilibrium between Meta I and Meta II towards Meta II? This seems entirely plausible and the consequences of this mechanism will be taken up in the Discussion section of this paper. Since Arnis et al. worked in the context that there were two protons taken up in Meta II formation, they had to posit that the alteration of one locus for proton uptake, Glu134, affected the second locus. If we assume that there is only one proton taken up, then this awkward problem disappears. Although it appears the proton uptake site is Glu134, other residues may be involved and so to avoid making premature conclusions we will call the proton uptake group, Z.
Comparison of the formation of Meta II with the formation of the M intermediate of bacteriorhodopsin. It is useful to summarize briefly what is known about the photochemical sequence following light absorption by another retinal protein, bacteriorhodopsin (BR). At present there is no evidence that bacteriorhodopsin is connected evolutionarily to rhodopsin, though it cannot be excluded. However, although the chromophore binding sites and chromophore isomeric specificity of the two pigments differ, there are striking similarities in their photochemistry. In both pigments light acts by photoisomerizing a retinal chromophore, attached to the parent pigment by a Schiff base linkage with a lysine residue in the 7th transmembrane helix. As in rhodopsin, the primary photoproduct of bacteriorhodopsin is a red shifted species, here called K. It is followed by a species blue shifted from K which in bacteriorhodopsin seems to combine features of the Lumi and Meta I intermediates of rhodopsin; it is called L. L decays with complex kinetics to M, with an apparent lifetime of ~80 µsec at 20°C. The L to M transition is due to the transfer of a proton from the Schiff base to Asp85. This is directly analogous to the Meta I to Meta II transition. Conceptually there may be as many as three phases in M formation [50, 51] although rapid kinetics between two of the states could reduce this to two and possibly even one observable phase under some circumstances [52-54]. The first (fast) phase is due to the initial transfer of a proton from the protonated Schiff base to the primary proton acceptor, Asp85. Asp85, is part of the negative counter-ion to the positively charged protonated Schiff base of retinal similar to the role of Glu113 in rhodopsin. The next phase reflects a physical coupling event which links the protonation of the counter-ion (Asp85) in the interior of the pigment to the change in pKa of the Proton Release Complex (PRC) at the extracellular surface of BR. It is by this mechanism that the PRC can sense that the counter-ion is protonated. It is not known precisely what this physical coupling event is, but a reasonable hypothesis is that the positively charged Arg82 changes its position within the pigment as M is formed [55] and that several internal water molecules within bR move and rearrange their hydrogen bonds (Maeda, this volume). The movement of Arg82 away from the Schiff base towards the Proton Release Complex could cause the Complex to deprotonate [56-59]. Finally, as a consequence of the physical changes in M, a proton from the Proton Release Complex is released from the membrane, leading to a third chemically/structurally distinct form of the M intermediate (see Fig. 1a); all three Ms have a deprotonated Schiff base and very similar absorption spectra. Some consequences of these changes can be investigated by altering the external pH. If, for example, the pH is raised high enough so that the Proton Release Complex is already deprotonated in the dark, the pKa of Asp85 increases by about 5 pH units [60]. This in turn greatly alters the kinetics of light-induced Schiff base deprotonation so that M is formed with an apparent lifetime of less than 6 µsec, instead of ~80 µsec. Below, in our own experiments we try to find analogous reactions to these in the formation of Meta II. Note that the structural changes involved in going from one M to another are quite restrained and while large protein conformational changes may occur in the photocycle, they are not required up to this stage.
In bacteriorhodopsin, a photocurrent/photovoltage on the sub millisecond time scale is composed of two phases of the opposite sign, called B1 and B2. Liu [61] showed that the photocurrent kinetics of B2 match the kinetics of proton release and formation of the M intermediate. In both systems, RH and BR, light absorbed by the deprotonated Schiff base photointermediate, Meta II or M, gave a photovoltage of the opposite sign to R2 or B2, as expected if these signals reflect the photoreversed proton movement from the counter-ion back to the Schiff base.Fig. 1. Schematic of possible contributions by charge movements to the R2 component of the Early Receptor Potential (ERP) and the B2 component from oriented bacteriorhodopsin.
In addition, the kinetics of the associated photocurrent/photovoltage signals accelerate at alkaline pH in a manner similar to the kinetics of M formation [62]. As noted above, Misra [53] argued that for BR, theoretically there should be three phases to the rise of M and thus potentially three phases to the associated electrical signal. It is of course possible that one or more of the transitions are so fast or arranged in other ways that they are not rate limiting so that one could observe only two or even one of these components.
Comparison of the photoelectric responses of rhodopsin and bacteriorhodopsin.There are intriguing similarities between the flash photoelectric responses of visual pigments and bacteriorhodopsin. Figure 1 illustrates a hypothetical but plausible set of charge movements that RH and BR would go through after a flash. Note that in both cases, the main counter-ion, Glu113 (RH) (Fig. 1b) or Asp85 (BR) (Fig. 1a) is on the N-terminal side (extracellular or intradiskal) of the membrane-embedded pigment molecule. In both cases photoisomerization moves the Schiff base proton away from the counter-ion, toward the C-terminal side, giving rise to R1 (RH) and B1 (BR). In BR, the formation of M1 occurs when the Schiff base protons of some of the activated pigment molecules move in the opposite direction, towards the N terminal side of the membrane, as they are transferred from the Schiff base to Asp85. The movement of the Schiff base proton gives rise to a current with the positive sign of B2 and with the approximate initial kinetics of B2. Then the protonation of Asp85 allows a set of events (M1 to M2), which includes and may even be mostly restricted to a rearrangement of internal water molecules and the movement of a positively charged residue, Arg82, away from the now neutralized Asp85 and towards the N-terminal side of the membrane (Maeda, this volume, and [59]). This probably also contributes to B2. Finally in the M2 to M3 transition, as a consequence of these changes, a proton is now released from the interface into solution. This positive current may also contribute to B2. Thus, in generating B2 there almost certainly are contributions from a proton being released from the membrane as M3 is being formed. This sequence of events is shown in Fig. 1a. If we accept this sequence, then the correlation between the kinetics of Meta II formation and the R2 kinetics implies that proton uptake normally follows Meta II formation.
As noted above, Cone [43] proposed that the proton which is taken up in forming Meta II would substantially contribute to the R2 current. The only difference between rhodopsin and bacteriorhodopsin in this event is that in BR, the proton current in the direction from C-terminus (intracellular) to N-terminus (extracellular), is caused by proton release from the N-terminal side, while in rhodopsin, a proton current of the same sign arises when a proton is taken up on the C-terminal side of the membrane (Fig. 1b). Thus, the formation of Meta II in rhodopsin and the M intermediate in bacteriorhodopsin are analogous in several ways. The kinetics of formation of these photointermediates are complex in both cases. In both pigments, there are electrical signals that are intimately associated with the formation of these key photointermediates especially M and Meta II. Finally, a change in the protonation state of a surface residue appears to contribute to the complex kinetics of formation of M and Meta II.
Rationale for the experiments. In bacteriorhodopsin, the concept of “coupling of protonatable residues” has proven illuminating in analyzing the titration behavior of the pigment in the dark [60, 63, 64]. In essence, the model emphasizes that the pKas of important residues whose protonation states change during the photocycle mutually influence each other. Importantly, the coupling between these residues in the initial (dark) state of the pigment is also manifested during the photocycle. Specifically, in bacteriorhodopsin, the pKas of the Schiff base, the counter-ion Asp85, and the PRC are coupled to each other. These are the residues involved in the formation of M. Because of the parallels between bacteriorhodopsin and rhodopsin noted in this section, we surmise that a similar coupling may be present between the Schiff base, Glu113, and Glu134 in rhodopsin.
In this paper, building on these earlier results, we examined the pH dependence of Meta II formation over a broader pH range than had hitherto been studied. We determined both amplitude and kinetics of Meta II formation as a function of pH. We wanted to see if any anomalous changes occur in these processes that would suggest that the pKa of the group on the cytoplasmic surface of rhodopsin that controls the Meta I/Meta II equilibrium, is coupled to the pKa of the Schiff base (or the counter-ion to the Schiff base) in the Metarhodopsin state. The limits of our titration should also provide us with limits to the pKas of the group regulating the pKa of the Schiff base counter-ion. In addition, we also examined other agents known to inhibit or accelerate protonation changes in other systems to see if we can assign the steps involved in the Meta II transition to particular proton transfer steps.
MATERIALS AND METHODS
Sample preparation. All procedures involving rhodopsin samples were performed under dim red light (Kodak No. 1A safety lamp) and the samples were kept on ice unless otherwise indicated.
Digitonin was the detergent used to solubilize the pigments so as to avoid light scattering problems by the ROS membrane fragments. Purification of digitonin was done according to Bridges [65]. Digitonin purchased from several vendors (Sigma, Fluka, and Calbiochem, USA) was pooled and dissolved in boiling water. After being kept in a refrigerator for one week, the solution was centrifuged and the supernatant filtered through 0.45 µm nitrocellulose filter unit (Nalgene, Milwaukee, WI) to eliminate small particles. The filtrate was then freeze-dried for later use.
Frozen bovine retinas obtained from Hormel (Austin, MN) were thawed and suspended in 43% sucrose in R-buffer (120 mM KCl, 60 mM NaCl, 10 mM MgCl2, 2 mM DTT, 0.1 mM PMSF, 20 mM MOPS, pH 7.4). ROS membranes were isolated from the homogenate by sucrose floatation using 43% sucrose in R-buffer, and then 38% sucrose in R-buffer. Membranes were washed with R-buffer twice, then three times by water. The preparation of gecko ROS was as described in [66]. To avoid any possible effects of residual transducin on Meta II formation, the ROS membranes were washed extensively with a hypertonic solution (5 mM Tris-HCl, 5 mM KCl, pH 7.0). The washed membranes were solubilized to extract rhodopsin by gentle agitation with 2% digitonin in water at 4°C overnight. After centrifugation to remove undissolved material, the extract was kept frozen until used. Aliquots of detergent extracts of bovine rhodopsin were mixed with NaCl (final concentration: 140 mM) and a mixture of buffers (MES, MOPS, TRICINE, CHES; final concentration: 10 mM each) adjusted to pH values between 4 and 10. The absorption spectrum of each sample was measured prior to flash photolysis with a Model 14DS UV-VIS spectrophotometer (AVIV Associates Inc., Lakewood, NJ). The pH was also measured after mixing.
For the D2O experiments, the ROS membranes were washed extensively with buffer solutions made up in D2O and then incubated with 2% digitonin, also dissolved in D2O at 4°C for 24 h to 4 days.
Samples with added sodium azide at various concentrations were analyzed in the same manner by recording the light-induced absorbance changes at 480 nm to monitor the decay of Meta I in addition to at 380 nm for the formation of Meta II.
Kinetic measurements and data analysis. The kinetics of Metarhodopsin II formation were measured with a home made flash photolysis setup [67, 68]. Time-resolved absorbance changes at 380 nm were measured after a short flash (532 nm, 7 nsec, ~20 mJ/cm2) from a Nd-YAG laser (Quanta-Ray--> DCR-11, Spectra-Physics, Mountain View, CA). The signal from the photomultiplier tube was digitized and first stored on a Le Croy transient digitizer (TR8837F from Le Croy Research, Spring Valley, NY) and then transferred to a computer. The data were recorded at 20oC with full scale duration ranging from 100 msec to 5 sec. Absorption spectra of the flash-bleached samples were taken and their difference from the ones taken prior to the measurement were used to estimate the amount of bleached pigment in the sample.
The kinetic traces of the absorbance changes were normalized based on the amount of the bleached sample, then fitted to three exponential component(s) (two rising and one decaying) by using KaleidaGraph. software (Synergy Software, Reading, PA) and the time constants and amplitudes were plotted versus pH. For each set of experiments the spectral traces were recorded in two different time scales, 100 msec and 5 sec.
Several possible reaction schemes for the formation of Meta II subspecies were considered and the theoretical predictions based on those model schemes were compared to the observed titration curves of the amplitudes.
RESULTS
Titration of the initial (dark) state of rhodopsin. The purpose of the first set of experiments is to determine if there is a measurable coupling of the deprotonation of the Schiff base/protonation of the primary proton acceptor, Glu113, with the protonation change at the cytoplasmic surface of rhodopsin. For the coupling of the protonation of the primary proton acceptor (Asp85) in bacteriorhodopsin to the protonation change at the extracellular surface of the Proton Release Complex (PRC) of BR, four types of evidence were obtained [60, 64, 69]. 1) The presence of a complex titration curve for the counter-ion, Asp85 (monitored spectroscopically at 605 nm) could be most easily explained by two interacting protonatable groups, the counter-ion Asp85 and the Proton Release Complex. 2) A very small shift (1.5 nm) in the absorption spectrum of BR is observed when the Proton Release Complex deprotonates in the dark, suggesting that the PRC interacts with the chromophore. 3) A change in the kinetics of M formation at alkaline pH (specifically, a large increase in the amplitude of the fastest component) suggests that the counter-ion pKa is influenced by the protonation state of the PRC. In a complementary fashion, a large change in the pKa of the Proton Release Complex is predicted when the counter-ion is protonated; this is what is required for the proton release observed when light causes the Schiff base to be deprotonated and the counter-ion to be protonated. 4) The pKa of the Proton Release Complex [62] of ~9.2 is close to the high pKa in the complex titration curve of the counter-ion Asp85 (~9.5).
We looked for similar phenomena in bovine rhodopsin. First, is there any evidence for a complex titration curve for the counter-ion of the protonated Schiff base of rhodopsin, Glu113? We assume that rhodopsin with a protonated counter-ion would absorb near where a pigment with a neutral counter ion, e.g., the rhodopsin mutant Glu113Asn would absorb. There is some complexity because the mutants' absorption spectrum depends on the type of anion present in the salt used [70, 71]. Nevertheless, red shifts of the absorption spectrum compared to control (11-cis retinal regenerated WT) rhodopsin (498 nm) were found for Glu113Ala (506 nm) and Glu113Asn (520 nm) [70, 71] (however, Glu113Gln absorbs at 490 nm [72]). Moreover, the pKa of the Schiff base dropped from greater than 14 [73] to ~5.7 (Glu113Ala) or 6.7 (Glu113Gln). In BR, when the primary counter-ion, Asp85 is changed to Asn, the spectrum red shifts by ~30 nm. Similarly, the absorption spectrum of wild type BR shifts ~30 nm when Asp85 is protonated from the bulk under acidic conditions, and the pKa of the Schiff base is reduced [74, 75]. In summary, we expect that when Glu113 is protonated, the absorption spectrum of rhodopsin would shift ~20 nm to the red. There is no evidence for such a shift down to pH 4, at which rhodopsin begins to denature ([27, 76, 77] and Koutalos, Liang, and Ebrey, unpublished results). We conclude that Glu113 cannot be protonated from the bulk above pH 4. Without a way to measure the pH dependence of the protonation of Glu113, we cannot determine if there is a complex titration curve or not (but see “Discussion”).
Second, is there a change in the absorption spectrum when the putative protonatable group on the surface of rhodopsin, named Z, changes its protonation state? In BR, we showed that when the pH was raised so that the Proton Release Complex on the extracellular surface is deprotonated, there was a very small (~1.5 nm) shift in the absorption spectrum of the native unphotolyzed pigment [63]. We hypothesize (see below) that the protonatable group on cytoplasmic surface of rhodopsin, which we nominally call group Z, is normally in its anionic (e.g., COO-) form but protonation of Glu113 causes Z to protonate (e.g., COOH). If so, then lowering the pH below the pKa of Z when RH is in its initial set of protonation values (the Schiff base is protonated and the counter-ion is anionic) could lead to a small spectral change when Z becomes protonated. However, so far we have not observed a pH dependent change in the absorption spectrum of rhodopsin that could reflect this protonation. We suggest that the pKa of Z in the initial state of RH is also less than 4 or that protonation of Z causes only a very small shift of the absorption band of initial rhodopsin (less than 1 nm).
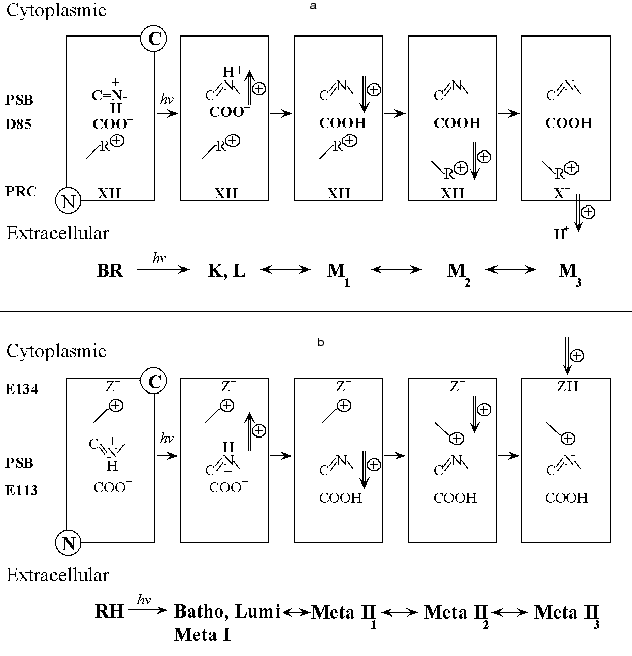
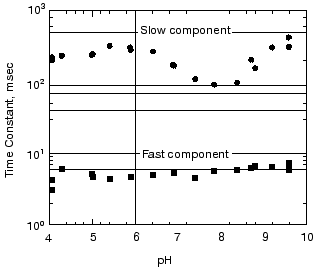
pH dependence of the kinetics of Meta II formation. The third set of experiments we conducted was to look for a dramatic effect of pH on the formation of Meta II, taken as the rise of absorption at 380 nm. In order to evaluate the pH dependency, the rising phase of each trace taken at a given pH was fit by two exponential curves. Initially each trace was fitted by a single exponential curve, which resulted in obvious deviation of the observed curve from the calculated one. Better fits were obtained with two successive exponential components with time constants separated by more than an order of magnitude (fast, ~3-5 msec; slow, ~100-400 msec) throughout the pH range we measured (Fig. 2). The rate constants based on these time constants and their relationship to the actual rate constants for a particular process depends on the model used to represent the observed changes (see “Discussion”). The amplitude of each component was also obtained in the curve fitting of each measurement (Fig. 3). In the lower pH range the amplitude of the slow component titrates with a pKa of ~6.0 and so is assigned to the titration of the surface group which controls the equilibrium between Meta I and Meta II, as initially observed by Matthews and co-workers [10].
As mentioned above, in BR, when the pH is raised so that the Proton Release Complex deprotonates in the dark, then the kinetics of M formation accelerate dramatically [63, 78] presumably because of the increased pKa of the primary proton acceptor, Asp85 [60]. Specifically, the amplitude of the fastest component of M formation increases dramatically, at the expense of the slower components [53, 62]. In rhodopsin, we might expect to see a dramatic change in the kinetics of Meta II formation, as the pH is lowered below the pKa of the protonatable group, Z. The amplitude of the fast component of Meta II, however, decreases slightly between pH 8 and 4, rather than increasing. Our data on the pH dependence of the kinetics of Meta II formation, for rhodopsin in digitonin (Fig. 3) shows that while there are some effects of proton (pH) changes, they are not dramatic. However, the amplitude of slow component is strongly pH dependent. A fivefold increase in the amplitude is observed at low pH with pKa close to 6. The pKa of the major transition in the acid range is presumably the Meta I to Meta II equilibrium with pKa of 6.3 observed by Matthews et al. [10]. These results are more or less consistent with those of other investigators who studied the pH dependence of Meta II formation although usually over much smaller pH ranges at either lower temperatures [23, 24, 79]; or in ROS membranes [14]. We conclude that the pKa of the group Z on the cytoplasmic surface is either quite low (less than 4) or that it is not titratable.Fig. 2. pH dependence of the rate constants of Meta II formation for bovine rhodopsin. The two time constants, fast and slow, were obtained by fitting the observed kinetics to two exponentials (see “Materials and Methods”).
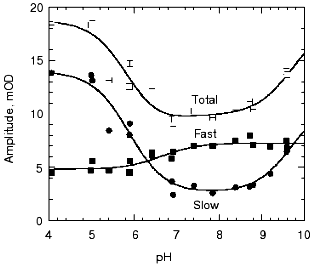
Effects of deuteration on the formation of Meta II. We next examined the effect of deuteration on the formation of Meta II. It is known that membrane proteins like rhodopsin can take very long times for deuterons to completely exchange with protons [80]. However, some of the protons in rhodopsin can also exchange extremely rapidly; Deng et al. [81] showed the Schiff base proton could exchange with a deuteron in less than 3 msec. Rhodopsin samples, as discussed in the methods section, were exposed to D2O for 1 to 4 days. Deuteration slows the overall kinetics of Meta II formation (Fig. 4) for the Gecko P521 pigment (a member of the Long Wavelength Sensitive (“red cone”) visual pigment family, reviewed in [82]). The size of the slowing effect was ~3.5 for the fast component of the rise of bovine Meta II (data not shown) and 4.3 for the fast component of the P521 pigment. After incubation for 1 day, the slow component of Meta II formation showed a smaller deuterium effect, but prolonged incubation of up to 4 days increased the size of the effect from 2.5- to 3.5-fold. The implications of these deuterium effects are considered below in the discussion. Briefly, the deuterium effect highlights the involvement of proton translocation in both of the observable components of Meta II formation.Fig. 3. pH dependence of the amplitudes of the fast and slow components of Meta II.
Effect of azide on the formation of Meta II. Azide added to the medium accelerated the decay of Meta I as well as the formation of Meta II (Fig. 5). The absorption at 480 nm was measured in order to monitor the amount of Meta I. The kinetics of Meta I decay, shown as the decrease in absorption at 480 nm, agree well with the Meta II formation kinetics measured at 380 nm.Fig. 4. Effect of D2O on Meta II formation. Comparison of the traces for fast components (100 msec full scale) in the formation of Meta II in Gecko P521 in digitonin in H2O and D2O.
At longer time scales Meta II started to decay at high azide concentration concomitant with a rise in absorption at 480 nm, presumably due to the formation of Meta III.Fig. 5. Effect of azide on Meta II formation. Rhodopsin samples in the concentrations of azide indicated were photolyzed and the decay of Meta I and the formation of Meta II analyzed as a sum of three exponentials. The third exponential has a small amplitude and so was ignored in the other analyses but in this case we followed the changes to very long times.
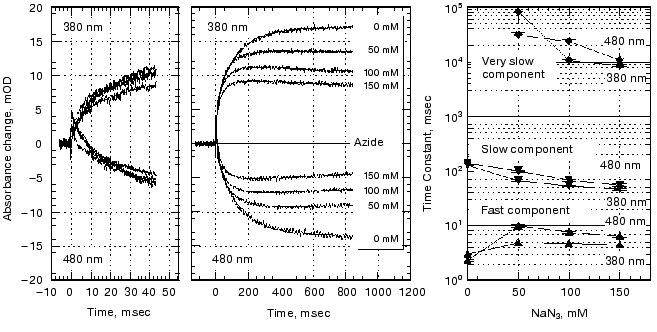
The difference of the effect between the fast and slow component should be attributed to the difference on the way proton(s) are involved in those steps. The azide effect is not the result of elevated sodium concentration, as we have examined the kinetics at higher NaCl concentrations and found no change (data not shown). Azide not only accelerated the formation of the slow component but also reduced the total apparent amount of Meta II formed. This may also be due to the fact that azide facilitated the formation of Meta III and thus depleted the Meta II on a timescale observable in our experiments.
DISCUSSION
The earlier data reviewed in the introduction to this paper provided strong evidence that a proton is taken up in forming Meta II, that the pKa of the group taking up the proton is 6.4 (in 150 mM salt), that Glu134 is the group (or a part of the group) taking up the proton, and that the proton is taken up after Meta II starts to form, and in some cases [21] can be temporally separated from the formation of the 380 nm absorbing species.
Proton uptake is crucial in forming the activated form of RH. The finding that Glu134Asn can constitutively activate transducin in the absence of 11-cis retinal [83] suggests that a neutral, polar residue at the 134 position (i.e., protonated E134 in the WT) is required for activation. Moreover, the pH dependence of transducin activation [84] and the effect of photoreversal on two of the forms of Meta II [37] also suggest that the protonated form of Meta II (i.e., with protonated Glu134) is required for activation. Finally, there is accumulating direct evidence that Glu134 is protonated when transducin forms a complex with Meta II [49].
Evidence for the coupling of the pKs of Glu113 and Z (Glu134). As noted in the results, we found no evidence of a red shifted form of RH with a protonated counter-ion (Glu113) down to pH 4. Moreover, the extraordinarily high pKa of the protonated Schiff base of bovine rhodopsin (greater than 15 [73]) probably requires a very low pKa for its counter-ion [85]. Thus a very low pKa for Glu113 in RH, say 1.0, is quite likely. This very low pKa would plausibly explain why we can neither follow the pH titration of the counter-ion Glu113 or even determine its main pKa. In addition, we (Figs. 2 and 3) and others (see introductory section) observe no pH dependence of Meta II formation kinetics that can be explained as being due to the protonation of Glu134 at acidic pH in the dark, suggesting that its pKa is also quite low.
Despite our present inability to follow the titration of the counter-ion, there is some indirect evidence that the pKas of Glu113 and Glu134 are coupled in Metarhodopsin. In a sequential, multicomponent model of Meta II formation (see Fig. 1), Meta II is clearly stabilized through proton uptake. That is, the proton exchange between the Schiff base and Glu113 is biased towards the latter upon proton uptake by E134, suggesting that the pKa of Glu113 is higher when Glu134 is protonated than when it is deprotonated.
The basic observation that the protonation state of Glu134 determines the amount of Meta I or Meta II is prima facie evidence for coupling in the Meta state since coupling of pKas means that the pKa of the counter-ion, Glu113, depends on the protonation state of a second residue, Glu134. Further evidence that the pKa of the counter-ion depends on the protonation state of Glu134 is that mutants of 134 that mimic protonated Glu134 by substitution with a neutral, polar species all shift the equilibrium between Meta I and Meta II towards Meta II [86].
Evidence for the converse interaction of these two groups, that the protonation state of Glu113 affects the pKa of Glu134 is more tenuous, but plausible. The order of events with respect to proton uptake by Glu134 is that first light-induced Schiff base deprotonation/Glu134 protonation occurs, and then a proton is taken up by Glu113 [21]. This suggests that protonation of the counter-ion can raise the pKa of Glu134.
Models for the coupling of the pKa of the Schiff base counter-ion, Glu113, with a protonatable group on the surface, probably Glu134. Case 1. Unphotolyzed rhodopsin. For rhodopsin, we propose a coupling between the Schiff base counter-ion, Glu113, and a protonatable surface group, Z. Z is probably Glu134, but Glu134 may only constitute part of Z; in any case, Z does not have to be identified for the following analysis. The protonation of Z controls the pKa of Glu113, and vice versa. The best evidence for such coupling is the pH dependence of Meta I<-->Meta II equilibrium. The interaction of Glu113 and Z can be described by a 4-corner diagram, as was done for Asp85 and Proton Release Complex in BR [60, 69], which reflects the protonation states of the groups involved:
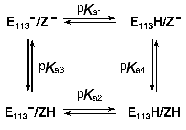
The pKas refer to Glu113 (E113 in the scheme) and Z when the Schiff base is protonated. The sum of the pKas to go from the upper left to the lower right is pKa1 + pKa4 = pKa2 + pKa3. In bacteriorhodopsin, the change in the pKa of one of the coupled residues upon a change in the protonation state of the other is similar whether the protonation change occurs during the photocycle, or is induced in the unphotolyzed pigment by experimental titration of the bulk pH. This is the case even though the absolute pKas of the residues differ in the two situations. The same is probably also true in rhodopsin. If so, we expect that protonation of either Glu113 or Glu134 in the dark, under acidic conditions, would lead to an increase of several units in the pKa of the other residue, and the concomitant partial protonation of that residue (see below, the model for Meta I<-->Meta II transition).
As mentioned above, we were not able to titrate either residue at acidic pH without denaturing the pigment, so the pKa values are unknown at present. However, there is another way to determine whether there is coupling between the two residues, namely by titrating Glu113 in a neutral mutant of Glu134 (which approximates a protonated Glu134). According to the model above, the pKa of Glu113 in such a pigment should be about seven units higher than in wild type rhodopsin. Thus, Glu113 may be titratable in the dark at high enough pH values above the denaturation pH of rhodopsin. Another option may be to examine neutral mutant of Arg135, which normally forms a salt bridge with Glu134 in an otherwise rather hydrophobic environment between helices III and VI in the dark state of rhodopsin [48]. Mutation of Arg135 may result in the destabilization of the anionic state of Glu134, and raise its pKa to within the titratable range. We are proceeding to test both of these options.
Case 2. Metarhodopsin. Coupling of internal proton transfer in Meta I<-->Meta II with the protonation of Z. A simple version of the coupling model for Metarhodopsin (Fig. 6) that probably can account for the main features of the pH dependence of Meta I<-->Meta II transition would be to assume that the proton transfer between the Schiff base and Glu113 is an internal transfer, meaning that the Schiff base and Glu113 exchange a proton only with each other, a possible scenario for the buried ion pair which has been discussed earlier [64, 87]. These internal reactions are shown in the scheme (Fig. 6) with block arrows. The second assumption would be that the thermodynamics of this exchange might depend on the protonation state of another residue Z. Z would be the only group that equilibrates with the bulk.
The fraction of Meta II as a function of pH can be calculated from the following formula:Fig. 6. A scheme describing effect of protonation of a group Z (presumably Glu134) on the proton transfer from the Schiff base (SB) to Glu113 (E113) and simulation of the pH dependence of the equilibrium amount of Meta II using Eq. (1) for the case of pKa4 = 7. Solid lines are for K2 = 0.1 (pK2 = 1), dashed lines are for K2 = 0.01 (pK2 = 2). Note that the apparent pKa of the curves differ from pKa = 7. Thus, for K1 = 10 and K2 = 0.1, the pKa = 6.
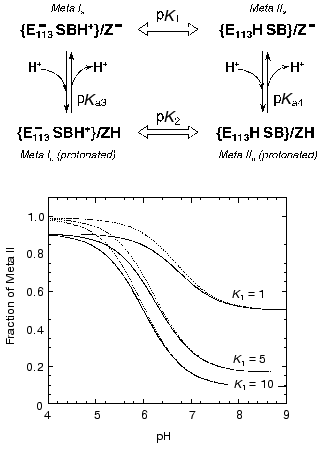
fMII = 1 + 10(pH - pKa4)/(1 + K2 + (1 + K1) 10(pH - pKa4)), (1)
where K1 = [Meta Ia]/[Meta IIa], K2 = [Meta Ib]/[Meta IIb].
The effect of the protonation of group Z on Meta I<-->Meta II equilibrium depends on the strength of the linkage (coupling strength) between the two reactions. We define coupling strength of two protonatable groups as the size of the pK shift (or equilibrium constant shift) of one group due to the protonation of the other [64, 88]. If the coupling strength is 1, then protonation of Z could change the ratio of Meta I/Meta II 10-fold (which is equivalent to change of the equilibrium constant by one pK unit). If the coupling strength is 2, then the ratio can be changed 100-fold. Correspondingly the pKa of Z should also change by one or two pKa units upon the Meta I to Meta II transition. Thus if the pKa of Z in Meta II is 7 and coupling strength is 2, the pKa of Z in Meta I would be 5. Figure 7 depicts three curves generated with Eq. (1) for different K1 and K2 assuming that the pKa of Z in the Meta II state (pKa4) is 7. The amount of Meta II formed when group Z is deprotonated depends on the equilibrium constant K1. When K1 = 1, equilibrium mixture would contain 50% Meta I and Meta II at high pH, K1 = 10 only about 10% of Meta II will be present. An increase in the equilibrium amount of Meta II at lower pH depends on the coupling strength (and correspondingly on the value of K2). The solid curves correspond to K2 equal to 0.1 (pK2 = 1), whereas dashed lines correspond to K2 = 0.01 (pK2 = 2). Comparison of the curves with the experimental data shown in Fig. 3 indicates that the coupling strength between Meta I<-->Meta II transition and protonation of group Z for bovine rhodopsin in digitonin is at least 1. More exact determination of an actual coupling strength and equilibrium constants would require measurements of the amount of Meta I present.
Analysis of the complex Meta II formation kinetics.The formation of Meta II in digitonin-solubilized rhodopsin can be readily decomposed into two observable components, fast and slow (Figs. 2 and 3). Both the apparent lifetimes and amplitudes of these components are dependent on pH. We analyzed the kinetics of Meta II formation in light of several plausible reaction schemes that include proton uptake (Fig. 7). The fast component of Meta II formation (Fig. 2) directly represents the (millisecond) equilibration of the Schiff base proton between the Schiff base and Glu113. Further formation of Meta II represents a shift in that equilibrium due to depletion of an initial Meta II subspecies into isospectral, later subspecies of Meta II, in analogy with the formation of M in bacteriorhodopsin [52]. One of the simplest implementations of such a Meta II formation scheme is shown as Scheme 1 (Fig. 7). In this case, the depletion of the initial Meta II subspecies (Meta IIa) is due to proton uptake from the bulk. This model was presented initially by Arnis and Hofmann and used by others since then to analyze Meta II formation [20].Fig. 7. Schemes to explain the pH dependence of Meta II formation.
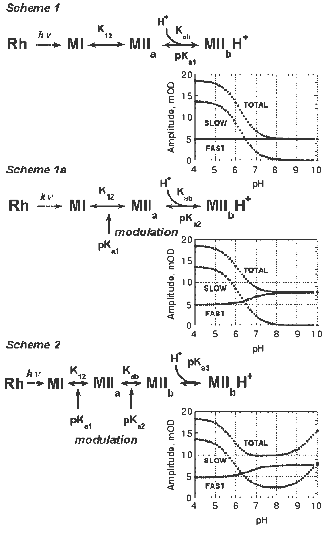
The scheme can be analyzed in the following manner. If it is assumed that the formation of the fast Meta II component reflects a pseudoequilibration between Meta I and Meta IIa, the amplitude of the fast component can be used to estimate the pseudoequilibrium constant K12. Since no Meta I is present when Meta I and Meta IIa have fully equilibrated,

The maximal Meta II amplitude is the maximum total Meta II amplitude observed (in the entire pH range); Scheme 1 suggests that this is approached at low pH. This analysis assumes that the equilibrium constants between the photointermediates and subspecies are such that an experimental situation can be reached at which the entire photoexcited sample accumulates in Meta II; that is, Meta I decays completely. This should occur at low pH, since the equilibrium between Meta IIa and Meta IIb will be biased completely forward due to the abundance of bulk protons. The value of pKab, the pKa of the putative proton uptake group, can be estimated using similar reasoning. At a given pH, when Meta II formation has saturated, a pseudoequilibrium exists between Meta I, Meta IIa, and Meta IIb.
Hence,
[Meta IItotal] = [Meta IIa] + [Meta IIb]
or:
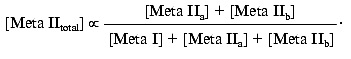
Since: 1) K12 = [Meta IIa]/[Meta I] and 2) Kab = ([Meta IIa][H+])/[Meta IIb] = K12[Meta I][H+]/[Meta IIb], the above expression becomes:
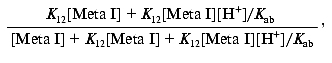
which reduces to:
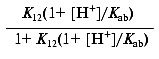
or:
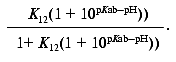
The proportionality constant is calculated during curve fitting of the above expression to the data and should be equal to the maximum experimentally observed Meta II amplitude. Note that Scheme 1 does not provide for any pH dependent change in the amplitude of the fast component, since K12 is pH independent. It can be posited that the protonation state of a nearby residue modulates the equilibrium of the Schiff base proton between the Schiff base and the counter-ion, as is the case in bacteriorhodopsin. In this case, K12 has two values, depending on the protonation state of this “modulator” residue. This results in a simple modification of Scheme 1 (Scheme 1a). K12 is substituted by an expression of the form:
K12lo/(1 + 10pH-pK1) + K12hi/(1 + 10pK1-pH).
Since we observe a small pH dependent increase in the amplitude of the fast component, we suggest that a residue near the Schiff base/counter-ion charge pair is being titrated and influences the proton affinities of these two residues (see proposed Scheme 2, Fig. 7). Several residues in the binding pocket of bacteriorhodopsin, notably Arg82 and Tyr57, have been shown to have a strong effect on kinetics of M formation [56, 89]. Note that this residue is not Glu134. The coupling model presented above would suggest that titration (protonation) of Glu134 would result in an increased pKa for Glu113 (or, equivalently, a larger value for K12 in Scheme 1) at lower pH. Thus, one would expect to observe an increase in the amplitude of the fast component with decreasing, rather than increasing pH.
Schemes 1/1a are similar to schemes proposed by Hofmann and coworkers [5, 21, 26], who have suggested formation of an early Meta II intermediate, followed by proton uptake inferred from pH-sensitive dye measurements. As seen from model data generated using Schemes 1/1a (Fig. 7), the slower component of Meta II formation should decrease in amplitude to zero with increasing pH, as the bulk proton concentration becomes extremely small. According to our observations, however, the amplitude of the slower component, though decreasing between pH 5 and 7, saturates at a definite value; subsequently, the amplitude increases with pH up to 10. This suggests that a simple model consisting of only two Meta II subspecies (related to a proton uptake) is inadequate to describe the entire Meta II formation process and that the sequence is more complicated. The fact that the amplitude of the slow component saturates above zero suggests that what appears as a single component probably consists of several additional subspecies. The kinetics of the transitions between these subspecies may be similar to each other, or the later transitions may be rate-limited by the preceding transitions.
A representative model is shown as Scheme 2, along with model data generated using the pKa and pseudoequilibrium constant values shown, using an analysis similar to that detailed above. The generated data is quite similar to the experimental data we obtained (Fig. 7). The inclusion of the extra subspecies Meta II´a allows the amplitude of the slow component to saturate instead of decreasing to zero with increasing pH. In addition, the presence of this subspecies provides a rationale for why the kinetics of the slow component are less sensitive to pH than would be predicted by, say, Scheme 1/1a, since the Meta ..a-->Meta II´a transition could be rate-limiting for the subsequent proton uptake step. Inclusion of this subspecies provides another “point of modulation”, to explain the increase in the amplitude of the slow component at high pH. Just as the protonation state of a residue could influence the proton transfer between the Schiff base and the counter-ion, a second residue could influence the equilibrium between Meta IIa and Meta II´a depending on its protonation state. Obviously, Scheme 2 is far from unique in rationalizing the experimental amplitude data presented in Fig. 3. The increase in the amplitude of the slow component at high pH could also be due to, for example, a proton release process, although there is a controversy if one occurs late in Meta II (see introductory section and [34]). Identification of putative “modulating” amino acids through mutagenesis is necessary to further verify or disprove Scheme 2 or similar models.
How can Schemes 1/1a or 2 be integrated with the idea of residue coupling, presented in the previous section? Coupling explains why there is no substantial back reaction between Meta IIb and Meta I or a Meta I-like intermediate. As the pKa of Glu113 is high after the protonation of Glu134, Glu113 will be much less prone to release its proton back to the Schiff base. The coupling model also suggests that, in neutral mutants of Glu134, the amplitude of the fast component should be larger than that observed in the wild type pigment. That is, K12 should have a higher value in such mutants, since K12 is related to the difference in the pKas of the Schiff base and Glu113, and since the pKa of Glu113 should be relatively higher in the mutants than in the WT. We will be testing this in the near future as well.
Deuterium and azide effects on kinetics of Meta II formation. The use of deuterium in the study of the bacteriorhodopsin photocycle has helped to identify those transitions that include proton translocation steps (see, for example, [90]). Deuterium effects occur either because: 1) a translocated proton is substituted by a deuteron; 2) internal water molecules or exchangeable protons whose reorientation is required for a transition are substituted. In light of this, we expect to observe deuterium effects for both components of Meta II formation, as both involve proton translocation steps. We observe a ~3.5-fold increase in the apparent time constants of both the fast and slow components of Meta II formation, after incubating our samples for several days in deuterium solution (Fig. 4). Interestingly, the magnitude of the deuterium isotope effect is larger than that expected for proton transitions between freely rotating groups or disordered proton transfer chains, in which reorientations of the participating groups limit the rate of proton transfer [90, 91]. The magnitudes of the deuterium isotope effects are more consistent with protonation changes involving fixed oriented groups, or with complex multiproton transfer events [90]. These results seem appropriate for the fast component of Meta II, which likely reflects direct proton transfer between the Schiff base and Glu113. Both groups participate in a complex hydrogen- and charge-bonded network in the retinal binding pocket [48], which may limit their rotational freedom but keeps them in orientations favorable for proton transfer. The results are more surprising for the slow component of Meta II formation, which ostensibly includes the uptake of a proton by Glu134 from the bulk. One would expect the deuterium isotope effect to be closer to 1.5 [91], if this reprotonation comes from transient water chains at the membrane surface. The magnitude of the isotope effect suggests instead that there are oriented donor groups (possibly, strongly bound water molecules) accessible to Glu134 in Meta IIa/Meta II´a.
Azide has similarly been used in the analysis of the bacteriorhodopsin photocycle to influence the kinetics of specific photocycle intermediates [92-94]. Azide and other weak acids appear to act as “proton shuttles” in some contexts, directly participating in proton exchange between the intramembrane portions of bacteriorhodopsin and the bulk aqueous phase [94]. In other contexts, azide may bind within bacteriorhodopsin and alter the internal hydrogen-bonded water structures within the pigment [93]. We noted that azide increases the rate of the slow component of Meta II formation in a concentration-dependent manner (Fig. 5), suggesting that azide can aid in the protonation of Glu134. This implies a shuttle-type mechanism for azide in this step, but somewhat contradicts our suggestion, above, that Glu134 may be reprotonated from a strongly bound water molecule. We were surprised to note an azide effect on the fast component of Meta II as well; in this case, azide slows down rather than speeding up the kinetics. The effect on the absorbance at 380 nm is relatively independent on the azide concentration. This suggests that an azide molecule may bind in the retinal pocket of the pigment and alter the local structure in such a manner as to increase the barrier for proton translocation between the Schiff base and Glu113. Because the concentration of rhodopsin in our samples is much less than 50 mM, the lowest concentration of azide tested, the putative azide binding sites may be saturated at this concentration, eliminating any effect of azide concentration. Thus, azide affects the kinetics of both the fast and slow components of Meta II formation in ways that suggest two different mechanisms of action.
Azide also appears to accelerate the decay of Meta II (Fig. 5). With increasing azide concentration, the peak amount of accumulated Meta II decreases and the slope of the absorbance change at 380 nm becomes negative, while the absorbance at 480 nm recovers. This may be due to the decay of Meta II into Meta III, which absorbs at ~460 nm [95]. In the absence of azide, the decay of Meta II to Meta III is very slow, taking ~100 sec at 20°C in washed ROS membranes [95]. In our experiments, we do not detect any Meta III at subsecond timescales in the absence of azide. However, in the presence of azide, Meta II decays with apparent kinetics of seconds to a photointermediate with a protonated Schiff base. Whether this intermediate is a “genuine” Meta III intermediate, similar to that observed in more physiological settings at long timescales, is open to question. To our knowledge, this is one of relatively few lines of evidence suggesting that proton transfers are involved in the formation of Meta III from Meta II (see, for example, [96]). The effect of azide is most easily explained as the direct reprotonation of the Schiff base through a proton-shuttling mechanism. However, other explanations, such as an azide-mediated change in the pKa of the Schiff base through a change in the retinal binding pocket are certainly plausible.
This work was supported by NIH grant EYO1323. We thank Sergei Balashov for many illuminating conversations.
REFERENCES
1.Kaulen, A. D. (2000) Biochim. Biophys. Acta,
1460, 204-219.
2.Drachev, L. A., Kalamkarov, G. R., Kaulen, A. D.,
Ostrovsky, M. A., and Skulachev, V. P. (1980) FEBS Lett.,
119, 125-131.
3.Drachev, L. A., Kalamkarov, G. R., Kaulen, A. D.,
Ostrovsky, M. A., and Skulachev, V. P. (1981) Eur. J. Biochem.,
117, 471-481.
4.Hofmann, K. P. (1986) Photobiochem.
Photobiophys., 13, 309-327.
5.Hofmann, K. P. (1999) in Rhodopsins and
Phototransduction (Wiley, C., ed.) Vol. 224, Novartis Foundation
Symposium, pp. 158-180.
6.Kliger, D. S., and Lewis, J. W. (1995) Isr. J.
Chem., 35, 289-307.
7.Litman, B. J., and Mitchell, D. C. (1996) in
Biomembranes (Lee, A., ed.) Vol. 2A, JAI Press, Inc., Greenwich,
CT, pp. 1-32.
8.Thorgeirsson, T. E., Lewis, J. W.,
Wallace-Williams, S. E., and Kliger, D. S. (1992) Photochem.
Photobiol., 56, 1135-1144.
9.Thorgeirsson, T. E., Lewis, J. W.,
Wallace-Williams, S. E., and Kliger, D. S. (1993) Biochemistry,
32, 13861-13872.
10.Matthews, R. G., Hubbard, R., Brown, P. K., and
Wald, G. (1963) J. Gen. Physiol., 47, 215-240.
11.Doukas, A., Aton, B., Callender, R., and Ebrey,
T. (1978) Biochemistry, 17, 2430-2435.
12.Fong, S., Tsin, A. T. C., Bridges, C. D. B., and
Liou, G. I. (1982) Meth. Enzymol., 81, 133-140.
13.McDowell, J. H., Khan, S. M. A., Bolen, D. W.,
Santaro, M. M., and Hargrave, P. A. (1991) in Signal Transduction in
Photoreceptor Cells (Hargrave, P. A., Hofmann, K. P., and Kaupp, U.
B., eds.) Springer-Verlag, Berlin-New York, pp. 31-41.
14.Jäger, S., Szundi, I., Lewis, J. W., Mah, T.
L., and Kliger, D. S. (1998) Biochemistry, 37,
6998-7005.
15.Jäger, F., Fahmy, K., Sakmar, T. P., and
Siebert, F. (1994) Biochemistry, 33, 10878-10882.
16.Abrahamson, E. W. (1973) in Biochemistry:
Physiology of Visual Pigments (Langer, H., ed.) Springer-Verlag,
New York-Berlin, pp. 47-56.
17.Hoffmann, W., Siebert, F., Hofmann, K. P., and
Kreutz, W. (1978) Biochim. Biophys. Acta, 503,
450-461.
18.Lewis, J. W., Winterle, J. S., Powers, M. A.,
Kliger, D. S., and Dratz, E. A. (1981) Photochem. Photobiol.,
34, 375-384.
19.Straume, M., Mitchell, D. C., Miller, J. L., and
Litman, B. J. (1990) Biochemistry, 29, 9135-9142.
20.Dickopf, S., Mielke, T., and Heyn, M. P. (1998)
Biochemistry, 37, 16888-16897.
21.Arnis, S., and Hofmann, K. P. (1993) Proc.
Natl. Acad. Sci. USA, 90, 7849-7853.
22.Arnis, S., Fahmy, K., Hofmann, K. P., and Sakmar,
T. P. (1994) J. Biol. Chem., 269, 23879-23881.
23.Parkes, J. H., and Liebman, P. A. (1984)
Biochemistry, 23, 5054-5061.
24.Gibson, S. K., Parkes, J. H., and Liebman, P. A.
(1999) Biochemistry, 38, 11103-11114.
25.Miller, A., and Oesterhelt, D. (1990) Biochim.
Biophys. Acta, 1020, 57-64.
26.Hofmann, K. P., Jäger, S., and Ernst, O.
(1995) Isr. J. Chem., 35, 339-355.
27.Radding, C. M., and Wald, G. (1956) J. Gen.
Physiol., 39, 909-922.
28.Falk, G., and Fatt, P. (1966) J. Physiol.,
183, 211-224.
29.Emrich, H. M. (1971) Z. Naturforsch.,
26b, 352-356.
30.Wong, J. K., and Ostroy, S. E. (1973) Arch.
Biochem. Biophys., 154, 1-7.
31.Emrich, H. M., and Reich, R. (1974) Z.
Naturforsch., 29, 577-591.
32.Schleicher, A., and Hofmann, K. P. (1985) Z.
Naturforsch., 40c, 400-405.
33.Bennett, N. (1978) Biochem. Biophys. Res.
Commun., 83, 457-465.
34.Bennett, N. (1980) Biochem. Biophys. Res.
Commun., 96, 1695-1701.
35.Cooper, A., and Converse, C. A. (1976)
Biochemistry, 15, 2970-2978.
36.Bennett, N., Michel-Villaz, M., and Dupont, Y.
(1980) Eur. J. Biochem., 111, 105-110.
37.Arnis, S., and Hofmann, K. P. (1995)
Biochemistry, 34, 9333-9340.
38.Kaupp, U. B., Schnetkamp, P. P. M., and Junge, W.
(1981) Biochemistry, 20, 5500-5510.
39.Szundi, I., Mah, T. L., Lewis, J. W., Jäger,
S., Ernst, O. P., Hofmann, K. P., and Kliger, D. S. (1998)
Biochemistry, 37, 14237-14244.
40.Meyer, C. K., and Hofmann, K. P. (2000) Meth.
Enzymol., 315, 377-387.
41.Cone, R. A., and Pak, W. L. (1971) in Handbook
of Sensory Physiology: Principles of Receptor Physiology
(Lowenstein, W. R., ed.) Vol. 1, Springer-Verlag, Berlin-New York,
pp. 345.
42.Sullivan, J. M., and Shukla, P. (1999)
Biophys. J., 77, 1333-1357.
43.Cone, R. A. (1967) Science, 155,
1128-1131.
44.Cone, R. A. (1969) in Proc. Int. School of
Physics “Enrico Fermi” (Reichardt, W., ed.) Academic
Press, New York-London, pp. 187-200.
45.Spalink, J. D., and Stieve, H. (1980) Biophys.
Struct. Mech., 6, 171-174.
46.Cafiso, D. S., and Hubbel, W. L. (1980)
Biophys. J., 30, 243-264.
47.Sullivan, J. M., Brueggemann, L., and Anumonwo,
J. M. (2001) Biophys. J., 80, 19a.
48.Palczewski, K., Kumasaka, T., Hori, T., Behnke,
C. A., Motoshima, H., Fox, B. A., Le Trong, I., Teller, D. C., Okada,
T., Stenkamp, R. E., Yamamoto, M., and Miyano, M. (2000)
Science, 289, 739-745.
49.Fahmy, K., Sakmar, T. P., and Siebert, F. (2000)
Biochemistry, 39, 10607-10612.
50.Heberle, J., Riesle, J., Thiedemann, G.,
Oesterhelt, D., and Dencher, N. A. (1994) Nature, 370,
379-382.
51.Cao, Y., Brown, L. S., Sasaki, J., Maeda, A.,
Needleman, R., and Lanyi, J. K. (1995) Biophys. J., 68,
1518-1530.
52.Zimányi, L., Váró, G.,
Chang, M., Ni, B., Needleman, R., and Lanyi, J. K. (1992)
Biochemistry, 31, 8535-8543.
53.Misra, S. (1998) Biophys. J., 75,
382-388.
54.Misra, S., Martin, C., Kwon, O., Ebrey, T. G.,
Chen, N., Crouch, R. K., and Menick, D. R. (1997) Photochem.
Photobiol., 66, 774-783.
55.Luecke, H., Schobert, B., Cartailler, J.-P.,
Richter, H.-T., Rosengarth, A., Needleman, R., and Lanyi, J. K. (2000)
J. Mol. Biol., 300, 1237-1255.
56.Balashov, S. P., Govindjee, R., Kono, M.,
Imasheva, E., Lukashev, E., Ebrey, T. G., Crouch, R. K., Menick, D. R.,
and Feng, Y. (1993) Biochemistry, 32, 10331-10343.
57.Scharnagl, C., Hettenkofer, J., and Fischer, S.
F. (1995) J. Phys. Chem., 99, 7787-7800.
58.Dickopf, S., and Heyn, M. P. (1997) Biophys.
J., 73, 3171-3181.
59.Luecke, H., Schobert, B., Richter, H.-T.,
Cartailler, J.-P., and Lanyi, J. K. (1999) Science, 286,
255-260.
60.Balashov, S. P., Imasheva, E. S., Govindjee, R.,
and Ebrey, T. G. (1996) Biophys. J., 71, 1011-1023.
61.Liu, S. Y. (1990) Biophys. J., 57,
943-950.
62.Kono, M., Misra, S., and Ebrey, T. G. (1993)
FEBS Lett., 331, 31-34.
63.Balashov, S. P., Govindjee, R., and Ebrey, T. G.
(1991) Biophys. J., 60, 475-490.
64.Balashov, S. P. (2000) Biochim. Biophys.
Acta, 1460, 75-94.
65.Bridges, C. D. B. (1977) Vision Res.,
23, 331-341.
66.Yuan, C., Kuwata, O., Liang, J., Misra, S.,
Balashov, S., and Ebrey, T. G. (1999) Biochemistry, 38,
4649-4654.
67.Govindjee, R., Dancshazy, Z., Ebrey, T. G.,
Longstaff, C., and Rando, R. R. (1988) Photochem. Photobiol.,
48, 493-496.
68.Liu, S. Y., and Ebrey, T. G. (1988) Biophys.
J., 54, 321-329.
69.Balashov, S. P., Govindjee, R., Imasheva, E. S.,
Misra, S., Ebrey, T. G., Feng, Y., Crouch, R. K., and Menick, D. R.
(1995) Biochemistry, 34, 8820-8834.
70.Nathans, J. (1990) Biochemistry,
29, 9746-9752.
71.Sakmar, T. P., Franke, R. R., and Khorana, H. G.
(1989) Proc. Natl. Acad. Sci. USA, 86, 8309-8313.
72.Zhukovsky, E. A., and Oprian, D. D. (1989)
Science, 246, 928-930.
73.Steinberg, G., Ottolenghi, M., and Sheves, M.
(1993) Biophys. J., 64, 1499-1502.
74.Subramaniam, S., Marti, T., and Khorana, H. G.
(1990) Proc. Natl. Acad. Sci. USA, 87, 1013-1017.
75.Turner, G. J., Miercke, L. J. W., Thorgeirsson,
T. E., Kliger, D. S., Betlach, M. C., and Stroud, R. (1993)
Biochemistry, 32, 1332-1337.
76.Lythgoe, R. J. (1937) J. Physiol.,
89, 331-358.
77.Wald, G. (1938) J. Gen. Physiol.,
47, 215-240.
78.Hanamoto, J. H., Dupuis, P., and El-Sayed, M. A.
(1984) Proc. Natl. Acad. Sci. USA, 81, 7083-7087.
79.DeLange, F., Merkx, M., Bovee-Geurts, P. H. M.,
Pistorius, A. M. A., and DeGrip, W. J. (1997) Eur. J. Biochem.,
243, 174-180.
80.Osborne, H. B., and Nabedryk-Viala, E. (1977)
FEBS Lett., 84, 217-220.
81.Deng, H., Huang, L., Callender, R. H., and Ebrey,
T. G. (1994) Biophys. J., 66, 1129-1136.
82.Ebrey, T. G., and Koutalos, Y. (2001) Prog.
Retin. Eye Res., 20, 49-94.
83.Cohen, G. B., Oprian, D. D., and Robinson, P. R.
(1992) Biochemistry, 31, 12592-12601.
84.Cohen, G. B., Yang, T., Robinson, P. R., and
Oprian, D. D. (1993) Biochemistry, 32, 6111-6113.
85.Ebrey, T. G. (2000) Meth. Enzymol.,
315, 196-207.
86.Weitz, C. J., and Nathans, J. (1993)
Biochemistry, 32, 14176-14182.
87.Honig, B., Ottolenghi, M., and Sheves, M. (1995)
Isr. J. Chem., 35, 429-446.
88.Imasheva, E. S., Balashov, S. P., Ebrey, T. G.,
Chen, N., Crouch, R. K., and Menick, D. R. (1999) Biophys. J.,
77, 2750-2763.
89.Govindjee, R., Misra, S., Balashov, S. P., Ebrey,
T. G., Crouch, R. K., and Menick, D. R. (1996) Biophys. J.,
71, 1011-1023.
90.Brown, L. S., Needleman, R., and Lanyi, J. K.
(2000) Biochemistry, 39, 938-945.
91.Le Coutre, J., and Gervert, K. (1996) FEBS
Lett., 398, 333-336.
92.Tittor, J., Soell, C., Oesterhelt, D., Butt, H.
J., and Bumberg, E. (1989) EMBO J., 8, 3477-3482.
93.Le Coutre, J., Tittor, J., Oesterhelt, D., and
Gerwert, K. (1995) Proc. Natl. Acad. Sci. USA, 92,
4962-4966.
94.Brown, L. S., and Lanyi, J. K. (1996) Proc.
Natl. Acad. Sci. USA, 93, 1731-1734.
95.Lewis, J. W., van Kuijk, F. J. G. M., Carruthers,
J. A., and Kliger, D. S. (1997) Vision Res., 37, 1-8.
96.Klinger, A. L., and Braiman, M. S. (1992)
Biophys. J., 63, 1244-1255.