REVIEW: Genetic Mechanisms of Hereditary Hemostasis Disorders
L. I. Patrushev
Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, Moscow, 117997 Russia; E-mail: patrush@mail.ibch.ru
Received July 3, 2001; Revision received October 1, 2001
This review summarizes known human genes whose mutations are associated with inherited hemostasis defects. These genes are divided into three groups. The genes of the first group are responsible for platelet adhesion, activation, and aggregation. The genes of the second group control the biosynthesis of blood-clotting factors and cofactors. The genes of the third group are required for the functioning of proteins involved in the anticoagulant system and fibrinolysis.
KEY WORDS: hemostasis, platelets, blood-clotting factors, fibrinolysis, mutations, hereditary diseases
Abbreviations: kb) kilobases; ADP) adenosine diphosphate; AP3) adapter complex; APC) activated protein C; ATIII) antithrombin; FG) fibrinogen; FV) factor V; FVa) activated factor V; FVII) factor VII; FVIII) factor VIII; FIX) factor IX; FX) factor X; FXI) factor XI; FXII) factor XII; FXIII) factor XIII; GP) glycoprotein; HCF II) heparin cofactor II; HRG) histidine-rich glycoprotein; IP3) inositol 1,4,5-triphosphate; PAI-1) plasminogen activator inhibitor 1; PC) protein C; PL) plasminogen; PS) protein S; TF) tissue factor; TFPI) tissue factor pathway inhibitor; TI) prostacyclin receptor; tPA) tissue plasminogen activator; TSP-1) thrombospondin 1; TR) thromboxane A2 receptor; TXA2) thromboxane A2; vWD) von Willebrand's disease; TM) thrombomodulin.
Hemostasis is a universal life-supporting system in humans and animals;
it, along with all other biochemical systems and processes, is under
the control of genes. Genetic information may be distorted due to
mutations. All phenotypic diversities of mutations result from one or
more of three general processes: 1) complete termination of gene
expression; 2) quantitative alteration in gene expression level; and 3)
qualitative modulation of gene function [1-3].
Complete termination of gene expression leads to the absence of biological activity of the encoded protein product. It may be either an effect of alteration in the polypeptide chain structure of a protein itself or a result of blocking the various steps of genetic information transmission from gene to the protein, for example, due to regulatory site damage in genes controlling its transcription or translation, or its mRNA splicing failures. Mutations may also lead to quantitative changes in the gene expression level. In this case, the quantity of the protein synthesized is either higher or lower than the norm, or its specific activity is altered (usually diminished). The same result is observed when mutations modify the protein or its mRNA stability. Upon the qualitative modulation of gene function, a new biological activity appears in the protein. As this takes place, either a substrate specificity of the enzyme or a character of its interaction with other cell macromolecules may become changed. All these three types of molecular-genetic consequences occur in inborn hemostasis function lesions.
It should be taken in account that a pathologic phenotype of the organism is not always a result of mutations in corresponding genes, but may develop from other causes. For example, autoantibodies arising against a protein may inhibit its activity and imitate an inborn defect of the protein [4]. Covalent binding of homocysteine to factor Va (FVa) under physiological conditions is one of the causes of FVa resistance to the activated protein C (APC-resistance) in hyperhomocysteinemia [5].
The inborn hemostasis defects are a serious medical problem because they strongly affect life quality, and thrombosis is one of the most prevalent causes of death [6, 7]. Below we consider the genes of the hemostasis system whose mutational damages are associated with its inherited defects.
MUTATIONS AFFECTING THE PLATELET ADHESION, ACTIVATION, AND
AGGREGATION
The initiation of hemostasis occurs within a few seconds after an injury to the blood vessels in response to the interaction of platelets with collagen fibers on the surface of subendothelium, adhesion, and following aggregation. Platelet adhesion is mediated by the von Willebrand factor that plays a role of molecular adapter between a specific glycoprotein complex on the platelet surface (glycoproteins Ib-IX-V) and collagen molecules. The genes supporting the initial steps of hemostasis, whose mutations are associated with defects of platelet adhesion, activation, and aggregation, are summarized in Table 1.
Table 1. Human genes responsible for
platelet activity and mutations affecting hemostasis
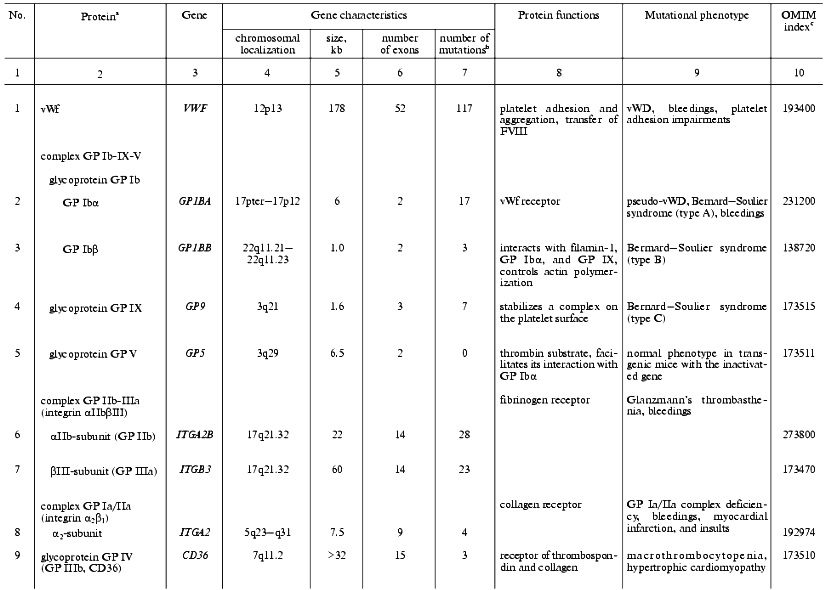
aThe genes are listed whose known mutations are associated
with hereditary hemostasis disorders.
bTotal of known mutations in HGMD (Human Gene Mutation
Database, http://www.uwcm.ac.uk/uwcm/mg/hgmd0.html).
cNumber of protein and associated disorders in the OMIM
(Online Mendelian Inheritance in Man) database [3].
dIn parentheses, total number of found mutations is given.
Von Willebrand factor (vWf). In hemostasis, vWf executes three main functions: provides platelet aggregation on the subendothelium matrix of damaged vessels, activates cells, and transfers FVIII in plasma and protects it against degradation [8-12]. A complete loss of von Willebrand factor activity is a rare cause of the homonymous disease (vWD). This phenotype, called vWD type 3, is distributed in the World's populations with a frequency of (1-5)/1,000,000. A total of fifteen different missense and nonsense mutations, three microdeletions and insertions, and five large gene rearrangements were described which led to a complete loss or drastic decrease in biological activity and/or biosynthesis of the factor [13-16].
In vWD type 2, which is divided into four subtypes, certain biochemical properties of vWf rather than its quantity are affected by missense mutations. The type 2A (10-15% of all clinical cases) is caused by mutations clustering in exon 28 that modify the A2 repeat of the vWf gene resulting in intracellular transfer defects and accelerated proteolytic degradation of vWf after its secretion [17-21]. Type 2B (<5% of clinical cases) is a consequence of changes in the A1 repeat of the vWf gene (21 missense mutations and one small deletion), which is accompanied by an increased affinity of the factor to GP Ib of platelets with the enhancement of their interaction (including a spontaneous one) leading to the fast excretion of the complexes from the circulating blood and to thrombocytopenia [22]. vWD type 2M is caused by rare (sporadic cases) missense mutations or small deletions in the A1 repeat encoding part of exon 28 that do not alter the reading frame. In these cases the biological activity of the factor decreases, but its multimerization remains normal or increased [23]. In vWD type 2N, mutations alter the N-terminal part of the factor and thus affect its interaction with factor VIII. Lastly, vWD type 1 is the most frequent form of this disease, its frequency being (1-30)/1,000 persons [11, 24]. Patients with vWD type 1 are characterized by moderate bleedings developing against the backdrop of decrease in both the protein itself and its activity levels to 20-50% of the norm. The ability of vWf for multimerization remains unaffected. Symptoms of the vWD type 1 are caused by many various molecular-genetic mechanisms. In particular, many patients with vWD type 1 symptoms are heterozygous carriers of mutations that lead to vWD type 3 in the homozygous state [25].
Glycoprotein complex GP Ib-IX-V. Glycoprotein transmembrane complex GP Ib-IX-V of platelets is one of the main components participating in their adhesion to the subendothelial matrix and signal transduction from collagen via vWf inside the cells [26, 27]. An important role of this complex in platelet functioning is evident from the fact that mutations inactivating either GP Ib or IX lead to the development of rare hereditary disease, Bernard-Soulier syndrome, which is characterized by uncommonly large platelet size and prolonged bleedings that appear due to the broken adhesion of platelets on the subendothelial matrix of damaged vessels [28]. GP Ib-IX-V complex consists of three components: integrin GP Ib composed of two subunits, alpha and beta, connected with a disulfide bond, and glycoproteins GP IX and GP V. All of the protein components of the complex are encoded by separate genes.
An extracellular domain of GP Ibalpha contains a sequence with three Tyr residues that are sulfated by tyrosylsulfate sulfotransferase of the Golgi apparatus and form the binding site for thrombin. Here in the intracellular part of GP Ibalpha, two binding sites for vWf are located (residues Tyr276-Glu282 and His1-Leu275). The mutational substitutions of these residues are associated with a pseudo-vWD because they do not affect the vWf itself [29].
A point mutation in the GP Ibbeta gene, which causes the premature termination of its mRNA translation at amino acid residue 21 and the termination of biosynthesis of the protein, leads to the lack of the entire glycoprotein complex on the platelet surface and to the progression of Bernard-Soulier syndrome [30].
The conservative sequence Leu-Arg-Met in GP IX is responsible for its interaction with GP Ibbeta and the stability of entire complex on the cell surface. Mutations modifying this region of the polypeptide chain lead to a deficiency of the entire GP Ibalpha and, as a result, to platelet adhesion impairment [31, 32].
When in a complex, GP V facilitates the interaction of thrombin with the GP Ibalpha subunit and at the same time acts as its substrate [33]. In transgenic mice with completely inactivated GP V encoding gene this subunit is not necessary for the expression of GP Ib-IX complex on the platelet surface [33]. However, its absence leads to the enhancement of the action of thrombin on the platelets followed by their earlier and higher aggregation. GP V is apparently an inhibitor of platelet activation by thrombin. There are no known mutations in the GP5 gene.
Integrin alphaIIbbeta3 is a fibrinogen receptor on the platelet surface. This glycoprotein complex is composed of two noncovalently interacting subunits. The subunits are encoded by separate genes located closely on the chromosome 17 [34]. In response to platelet activation by collagen, thrombin, ADP, or other ligands, the complex undergoes a conformational change (inside-out signaling) and, as a result, gains the ability to interact with fibrinogen and cause platelet aggregation [35]. In turn, the interaction of integrin with fibrinogen is accompanied by a clustering of integrin receptors and signal transduction inside the cell (outside-in signaling), which is one of the mechanisms controlling the level of irreversible platelet aggregation and clot consolidation.
A key role of integrin alphaIIbbeta3 in clot formation is indicated by severe clinical consequences of its mutational damage. Such mutations, of which no less than 50 have been counted in both subunit genes, lead to the progression of Glanzmann's thrombasthenia, a rare hereditary autosomal recessive disease characterized by sporadic mucosal bleedings [36].
Glycoprotein GP IV (CD36, GP IIIb) is a thrombospondin receptor and one of the primary collagen receptors; it offers an additional mechanism of platelet adhesion on the surface of collagen matrix [37]. Mutations in the CD36 gene have been found in 2-3% of the Japanese population, with 40% of patients with hypertrophic cardiomyopathy exhibiting its dysfunction [38]. Using biochemical tests and transgenic mice deficient in GP IV, it was found that GP IV provides for fatty acid transfer into muscle and fat tissue cells [39].
Glycoprotein complex GP Ia/IIa (integrin alpha2beta1) expressed on the surface of megakaryocytes and platelets interacts with collagen, although being expressed on other cell types it plays a role of laminin receptor [40]. This receptor offers the primary platelet adhesion to subendothelial surfaces. Allele variants of the alpha2-subunit gene have been found, these being associated with either low or high expression levels of the complex on the platelet surface. The high expression level is a risk factor for myocardial infarction or brain insult in young people [41, 42], whereas the low level is associated with bleeding and inclination to von Willebrand's disease [43]. Mutations were found recently in promoter nucleotide sequence of the alpha2-subunit gene of this complex, these determining 10-fold differences in its expression level. Transcription factors SP1 and SP3 interact with promoter sites in which these mutations are located [44].
Glycoprotein GP VI acts as one of the collagen receptors on the surface of platelets that control their activation and aggregation [45]. Patients with GP VI deficiency display symptoms of hemorrhagic diathesis. Autoantibodies to GP VI, which block collagen-induced aggregation, were found in a single case [46]. These hereditary platelet anomalies have not been characterized on the gene level.
Thromboxane A2 receptors (TR) belong to the family of receptors associated with G-proteins. Being synthesized preferentially by platelets, TXA2 interacting with TR induces changes in platelet shape and aggregation and also a contraction of blood vessel walls. In response to agonist actions, signal transduction occurs accompanied by activation of phospholipase C beta-isoform, which leads in turn to the elevation of intracellular concentrations of diacylglycerol and IP3 accompanied by intracellular calcium mobilization. In contrast, prostacyclin, another prostanoid, acting via a specific receptor TI functionally bounded to TR, is a vasodilator and inhibits platelet aggregation. The inhibition, at least in part, results from a desensitization of TR alpha-isoform (TRalpha) [47]. In patients with disturbed platelet aggregation accompanied by moderate bleedings, a point mutation was found in the TRalpha gene that blocks adenylate cyclase activation due to the Arg60Leu substitution in the polypeptide chain of the receptor [48].
ADP receptors. There are three ADP receptors expressed on the platelet surface, two of them being involved in signal transduction via G-proteins [49]. P2Y1 receptor associated with Gq-protein activates phospholipase Cbeta, which is necessary for intracellular calcium mobilization, and induces some cytoskeleton rearrangements resulting in changes in platelet shape [50]. The second receptor, P2YAC, is associated with Gi-protein. Its stimulation accompanied by adenylate cyclase inhibition is necessary for the successful completion of the aggregation process. Small deletions in gene (P2Y12) of this receptor were associated with platelet aggregation defects [51]. The functions of the third receptor, P2X1, which is an ADP-induced calcium channel, are unknown [49].
Gelsolin in submicromolar concentrations reversibly depolymerizes the intracellular actin filaments and simultaneously participates in the clearance of actin circulating in the bloodstream. A point mutation G654T (Asp187Asn) in the gelsolin gene is accompanied by platelet shape changes and bleedings in patients with hereditary gelsolin-associated amyloidoses [52]. The importance of gelsolin for hemostasis was confirmed using transgenic mice with the gene inactivated [53].
Defects of Platelet Storage Granules and Secretion
A heterogeneous group of hereditary diseases associated with defects in the delta- or/and alpha-granules themselves and also in their contents is accompanied by hemorrhagic symptoms of various severity [54].
Hermansky-Pudlak syndrome. This syndrome is a rare hereditary autosomal recessive disorder characterized by albinism, bleedings, and defects of cell organelles: melanosomes, lysosomes, and dense granules of platelets [55]. Two genes were identified whose mutations are associated with this disease: HPS1, encoding a novel cytosolic and transmembrane protein entering in the composition of cytoplasmic organelles, and HPS2 (AP3B1). The latter encodes the beta3A subunit of the AP3 adapter complex, whose role in this disease was confirmed with mutant mice [56]. In the European population, deletions and insertions in the codons 321-322 of the HPS1 gene leading to a reading frame shift are most frequent [55].
Gray platelet syndrome is caused by alpha-granule deficiency. The same symptoms were described in the deficiency of P-selectin, a platelet glycoprotein responsible for the interaction of activated platelets and blood cells with endothelium [57, 58]. This adhesion receptor stimulates TF synthesis in monocytes and provides for leukocyte gathering around the vessel injury.
Thrombospondin 1 (TSP-1) is a homotrimer glycoprotein, a component of alpha-granules, which is secreted by activated platelets [59]. In the presence of bivalent cations, it interacts with platelet membranes promoting platelet aggregation. TSP-1 also interacts specifically with TFPI, immobilizing it on vessel endothelial cells in the place the clot is formed [60]. Mutations in the human TSP-1 gene are not known, but acute pneumonia and pulmonary bleedings accompany complete inactivation of this gene in mice [61], indicating its possible involvement in corresponding human pathologies.
MUTATIONS AFFECTING THE ACTIVITY OF BLOOD-CLOTTING FACTORS
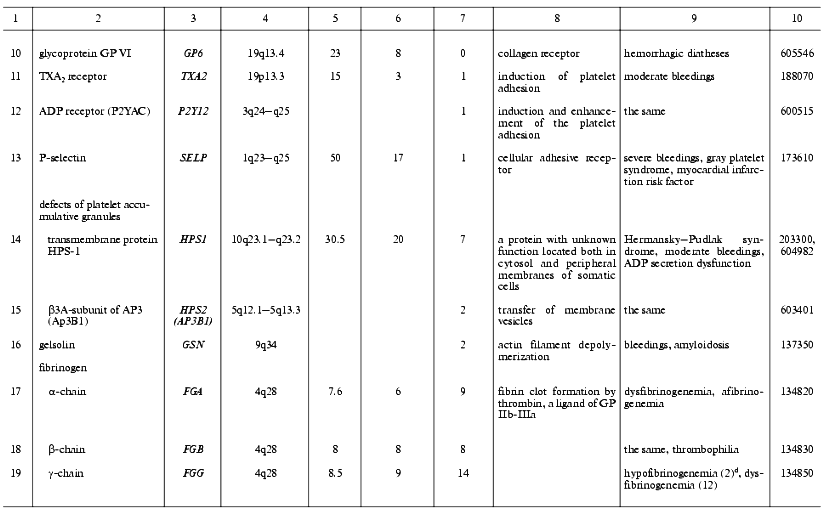
Thrombin is a multifunctional proteinase and one of the main hemostasis elements that acts as a powerful procoagulant: it activates platelets via PAR1 and PAR4 receptors and cleaves fibrinogen into fibrin. Also, thrombin plays a role of specific anticoagulant activating the protein C system [62]. It plays an important role in inflammatory processes and embryogenesis, particularly in atherogenesis, and development of the nervous system and some of its pathologies [63-66]. Known mutations in the thrombin gene are often associated with hereditary lesions of the blood-clotting system. Two main types of phenotypic traits were described for these mutations. In prothrombin deficiency, moderate bleedings occur due to the clot formation defects [67]. The elevated level of prothrombin in plasma, for example as a result of the mutation G20210A, is associated with thrombophilia [68]. The latter mutation is located in the 3´-terminal non-coding region of the prothrombin gene and does not affect its polypeptide chain, but apparently stabilizes its mRNA. This mutation is widely distributed in the United States and in European populations and is one of the most serious risk factors for venous and arterial thromboses [69, 70] (Table 2). Our studies suggest that this mutation, as well as the Leiden mutation to be discussed below, is associated with thromboses in Russia too [71, 72].
Table 2. Human genes encoding the main
blood-clotting factors and cofactors and mutations associated with
hemostasis disorders
aSee footnotes to the Table 1.
Factor V (FV) is a single-chain glycoprotein. As a result of proteolytic activation with thrombin, it converts to the activated FV (FVa), a cofactor of the factor X (FXa) [73]. Two types of mutational damage have been found in the FV gene. The first type of mutations lead to the FV deficiency, the second lead to FVa resistance to inactivation with APC (APC-resistance) resulting phenotypically in bleedings or thrombophilia, respectively [74, 75]. Unlike experimental animals, a complete loss of FV due to homozygous mutations is not lethal but rarely distributed (about 1/1,000,000) [75, 76]. APC-resistance due to the factor V Leiden (a point mutation that leads to the substitution Arg506Gln in the FV polypeptide chain) is one of the most frequent causes of hereditary thrombophilia in both European and Russian populations [74, 77]. Other, less frequent mutations in the FV gene have been found (Cambridge, Arg306Thr and Hong Kong, Arg306Gly) that also lead to APC-resistance [78, 79]. Thrombophilia is also characteristic of compound homozygotes with one allele of the FV gene carrying the factor V Leiden and the other allele inactivated [80].
Factor VII (FVII) is a precursor of a vitamin K-dependent serine proteinase. In a complex with TF, VIIa activates factors FIX and FX, thus being involved in the initiation of blood clotting. Inherited FVII deficiency is a rare autosomal recessive disease characterized by bleedings of various severities [81, 82]. Patients suffering from the most severe form of FVII deficiency (complete loss of its activity) are nonviable and die of bleedings shortly after the birth.
Factor VIII (FVIII) circulating in plasma is present in giant glycoprotein complexes with vWf that protect it against degradation. The activated form, FVIIIa, acts as a cofactor of FX activation with factor IXa on a phospholipid surface, many times accelerating this process [83, 84]. Upon the activation, FVIII undergoes a proteolysis, releases from the complex with vWf, and after the loss of a short peptide FVIII processed becomes able to combine into active heterotrimeric form. In this form, FVIIIa is able to interact with phospholipid surfaces and with the FX-FIXa complexes residing on them. In these complexes, FVIIIa can undergo inactivation by APC, thrombin, FIXa, and/or FXa. This abundance of specific protein-to-protein and other interactions of FVIII is a basis for their impairments by mutations accompanied by hemophilia A that is distributed (in the USA) with a frequency of about 1/10,000 men [84]. Indeed, more than 600 various mutations affecting FVIII functioning have been described in all of the 26 exons of its large gene located on the sex chromosome X [85].
Factor IX (FIX) circulates in the blood flow as a zymogen of a serine proteinase. It can be transformed into the active form (FIXa) in two proteolytic steps by the complexes FXIa-Ca2+ or FVIIa-TF-Ca2+ and therewith becomes able to activate FX in the presence of FVIIIa as a cofactor [86]. Multiple mutations affecting protein-to-protein interactions, processing, and/or inactivating FIX are accompanied by bleedings (hemophilia B). More than 650 mutations are known in all exons of its gene [87]. Hemophilia B is inherited as a chromosome X-coherent disease and is diagnosed in 1/30,000 men [88].
Factor X (FX) circulates in the blood as a zymogen of a vitamin K-dependent proteinase that plays a key role in the conversion of prothrombin to thrombin. The activation of FX with formation of FXa occurs under the action of the complexes FVIIa-TF (outer tenase) or FIXa-FVIIIa (inner tenase). FXa, when in a membrane-associated prothrombinase complex with FVa-Ca2+, executes a proteolytic processing of prothrombin [62]. Also, FXa interconnects the blood-clotting and inflammatory processes: it activates leucocytes, endothelial, and smooth muscle cells via interaction with their surface receptors resulting in the release of multiple growth factors and cytokines [89]. A mutational FX deficiency is accompanied by explicit clinical consequences, such as bleedings of various severities [90].
Factor XI (FXI) is a zymogen of a serine proteinase; unlike all of the other factors, it circulates in the blood flow in a complex of its homodimer bound with disulfide bonds and high-molecular-weight kininogen [91, 92]. As soon as the FXI-kininogen-FXIIa complex is formed on the platelet surface, the activated FXIa that is necessary for the activation of FIX is formed as a result of restricted proteolysis. Genetic defects of FXI are rare and are primarily specific for Ashkenazi Jews [93, 94]. The peculiarity of bleedings accompanying these genetic defects is the absence of correlation between disease severity and the level of FXI activity in patients. For instance, no symptoms can be present in a total deficiency of FXI [93, 95]. This kind of phenotypic trait heterogeneity may be partly explained from the FXI isoform present in platelets and expressed on the cell surface, whose amino acid sequence encoded by exon 5 is missed due to the alternative splicing of its pre-mRNA [96]. Data on the existence of this isoform in humans are contradictory [97].
Factor XII (Hageman factor, FXII) circulating in blood is a glycoprotein precursor of a multifunctional serine proteinase. Activated FXII (FXIIa) that is formed due to the single proteolytic split of the precursor polypeptide chain plays an important role in the contact phase of blood clotting by the proteolytic activation of FXI. FXIIa is also involved in the fibrinolytic process as an activator of prekallikrein, because the latter in turn activates FXII itself, the kinin system, and a urokinase-type plasminogen activator u-PA. FXIIa involvement in the activation of complement was also reported [98]. Anticoagulant functions of FXIIa are also expressed via its interaction with GP Ib-IX-V on the platelet surface followed by the inhibition of platelet activation by thrombin and their subsequent aggregation [99]. Inborn defects of FXII affecting its biosynthesis, activation, and protein-to-protein interactions are, as a rule, asymptomatic or associated with thrombophilia [100].
Factor XIII (fibrin-stabilizing factor, FXIII) circulating in the blood flow is a thrombin-activated heterodimeric (A2B2) transglutaminase precursor composed of two subunits: catalytic (A) and non-catalytic (B) [101]. There are tissue homodimeric (A2) forms of this factor in megakaryocytes/platelets, monocytes, macrophages, and placenta cells, which lack B-subunits [102]. Activated FXIII (FXIIIa) cross-links the polypeptide chains of fibrin to form amide bonds between gamma-carboxyl groups of glutamate residues and epsilon-amino groups of lysine residues. Collagen, fibronectin, and alpha2-plasmin inhibitor are also substrates of FXIIIa. These reactions are expressed physiologically in increased thrombus strength and elasticity and also in its resistance to plasmin. Inborn FXIII deficiencies are accompanied by the lasting bleedings associated with hampered anapleroses and spontaneous abortions [103]. Most FXIII deficiencies are associated with mutations in the gene F13A1 encoding the A-subunit, and a few of them are associated with the gene F13B encoding the B-subunit [104]. Great interest is given to the recently found widespread polymorphism of the F13A1 gene resulting in the Val34Leu mutation in the A-subunit, which is localized close to the site of proteolytic activation of FXIII [105]. Homozygous mutations were found to lead to a significant increase in FXIIIa activity, which inscrutably protects the organism against thromboses and cardiac infarctions [106, 108].
Tissue factor (TF) is a membrane glycoprotein, receptor, and FVII/FVIIa cofactor involved in the outer blood-clotting pathway [109, 110]. TF initiates a blood-clotting cascade, which partially explains the low frequency of its inborn defects. Actually, in mice with inactivated tissue factor gene most of progeny die in early embryogenesis [111]. Now the only mutation known affecting the factor functions is a small insertion in the 5´-terminal non-coding region of the gene [107].
Fibrinogen (FG) is a plasma glycoprotein composed of two sets of three structurally different polypeptides: alpha (FGA), beta (FGB), and gamma (FGG); each is encoded by a separate gene [112]. FG is a platelet adhesion protein. Thrombin converts FG into fibrin. Due to alternative splicing and intense posttranslational modifications, a set of highly heterogeneous FG molecules is present in plasma. Mutations in all three FG genes have been described, which lead to dysfibrinogenemia or afibrinogenemia with lowered or absent FG contents in plasma [113-117]. However, even afibrinogenemia is accompanied by only moderate bleedings.
MUTATIONS AFFECTING THE ANTICOAGULANT FACTORS AND FIBRINOLYTIC
SYSTEM
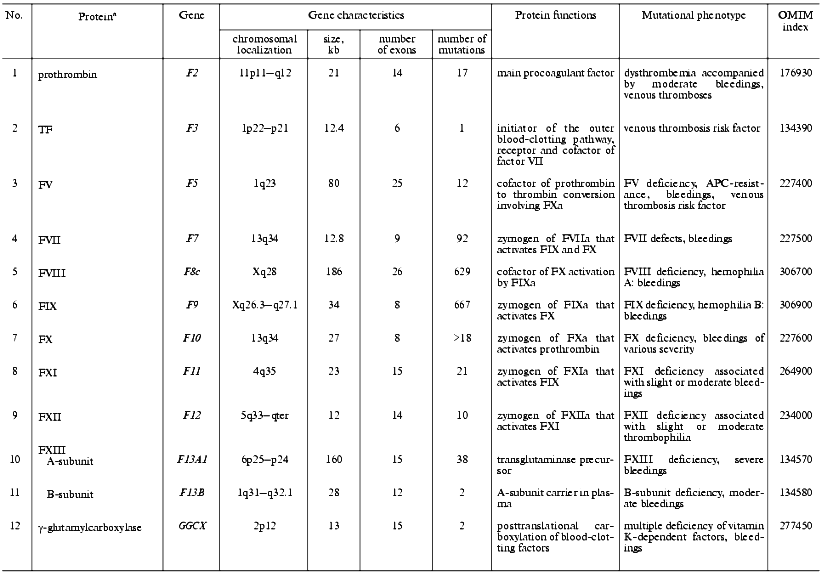
Thrombomodulin (TM) is a glycoprotein that is expressed on the endothelial cell surface and forms a complex with thrombin (1 : 1) [118]. In the complex with TM, thrombin alters its specificity for some proteins, for example, 1000-fold increases its affinity to protein C and activates it to form the natural anticoagulant. Mutations in the TM gene are the cause of thrombophilia accompanied by thrombembolia and cardiac infarction [119, 120] (Table 3).
Table 3. Characteristics of genes of
anticoagulant system factors and fibrinolysis in humans and mutations
associated with disturbances in hemostasis
aSee footnotes to the Table 1.
Antithrombin (ATIII) belongs to a superfamily of protein serine proteinase inhibitors (serpins) and is one of the significant inhibitors of thrombin [121]. Alike TFPI, ATIII is one of the most important negative regulators for the outer blood-clotting pathway. ATIII interacts with the TF-FVIIa complex and causes its dissociation [122]. The polypeptide chain of ATIII contains two main functional domains: the C-terminal domain interacts with proteinases, and the N-terminal domain carries two binding sites for heparin and heparin sulfate proteoglycans of endothelial cell surface. Inborn ATIII deficiency caused by mutations in its gene is distributed with a frequency of 1/3,000 persons. These mutations have been found over the entire length of the gene and are often associated with thrombophilia [123]. Two types of inborn ATIII defects are distinguished: the classic deficiency type I is connected with nearly complete loss of the protein or its functional activity; type II deficiency is associated with genetically changed protein variants with damaged active center (subtype RS, reactive site), heparin binding domain (subtype HBS, heparin binding site), and multiple damages (subtype PE, pleiotropic effect).
Protein C (PC)is a vitamin K-dependent glycoprotein, precursor of activated PC (APC), and natural anticoagulant. APC acts as an anticoagulant in a complex with PS in the presence of phospholipids by means of proteolytic cleavage of activated FVa and FVIIIa [124]. PC deficiency due to various mutations is a serious risk factor of venous thromboses and is inherited as an autosomal dominant trait. The frequency of mutations resulting in PC deficit in heterozygous state is 1/16,000-30,000 persons [125, 126]. Severe forms of PC deficiency due to either homozygous or compound heterozygous mutations are accompanied by rapid lethal outcome resulting from thromboses in the early postnatal period [127].
Protein S (PS) is a vitamin K-dependent cofactor of activated PC [128]. When in a complex with APC, PS increases ~10-fold the APC affinity to the phospholipid surface of endothelial cells and platelets, wherein the interaction with the surface increases ~20-fold the ability of the PC-PS complex to inactivate FVa. About 40% of PS is free, and the remainder exists in a complex with C4b-BP--the protein regulator of the classic complement pathway. The free PS only acts as a cofactor [129]. Autosomal dominant inheritance type is characteristic for inborn PS deficiency that is found in 1-5% of patients with venous thromboses [130-132]. Corresponding heterozygous mutations in the human genome increase 5-10-fold the risk of venous and arterial thromboses. There are three distinctive types of hereditary PS deficiency: in type I, II, or III a decrease in the total protein, its specific activity, or free PS level in plasma, respectively, is observed.
Tissue factor pathway inhibitor (TFPI) is a glycoprotein that belongs to the Kunitz inhibitor superfamily. Most of TFPI is associated with the vessel endothelium cells via complex formation with TSP-1 [60]. TFPI inhibits FXa activity and, as a result, the activity of the VIIa-TF complex [133]. A heterozygous mutation in exon 7 of the TFPI gene is possibly associated with deep vein thromboses [134], and a polymorphism in a promoter region of the gene is accompanied by alterations in the level of TFPI synthesis [135]. An important role of TFPI in hemostasis is indicated by severe consequences of total inactivation of the gene in transgenic mice, which die of bleedings in the prenatal period [136].
Heparin cofactor II (HCF II) is an inhibitor of thrombin [137]. A HCF II deficiency was described in a patient with a coronary disorder and recidivating stenoses after coronary angioplasties [138]. A homozygous mutation in HCF2 gene in combination with a heterozygous mutation in ATIII gene was described in a woman with venous thromboses. Thromboembolic epiphenomena did not occur in her sister also carrying homozygous mutation in the HCF2 gene, but not in the ATIII gene[139].
Plasminogen is the precursor of plasmin (PL), whose broad substrate specificity provides potential danger of its uncontrolled activation [140]. Fibrinogen and fibrin are the main PL substrates. The blood-clotting factors, such as V/Va, VII, VIII, IX, X/Xa, and XII, are also substrates of PL [141-143]. PL also influences cell adhesion and migration via its activity towards the extracellular matrix proteins [140]. Inborn defects of PL are often associated with thrombophilia [144, 145].
Tissue plasminogen activator (tPA) is synthesized by the vessel endothelium cells in the form of a PL-activated precursor [140]. tPA is mainly located and expressed in the vicinity of a thrombus due to its high affinity to fibrin, which is an activator of tPA [146]. Inborn hemostasis disorders associated with elevated tPA level in plasma and bleedings and also with its impaired secretion by endothelial cells were accompanied by familial thromboses [147, 148].
Plasminogen activator inhibitor 1 (PAI-1) is an important component of the fibrinolytic system. Its inborn deficiency is accompanied by bleedings, and overexpression by the development of thrombophilia [149-151]. Insertion/deletion polymorphism of a single G-base in the promoter region of the PAI-1 gene (4G/5G) is currently under study; a homozygous deletion (4G/4G-genotype) leads to elevated PAI-1 level due to the activation of its gene transcription [149, 152]. Patients with homozygous deletion much more often display hereditary coronary disorders due to elevated PAI-1 level and corresponding inhibition of PA activity.
Histidine-rich glycoprotein (HRG) that is abundantly present in plasma forms complexes with fibrinogen, FXIIIa, plasminogen, and heparin [153]. The point substitution Gly85Glu identified in the polypeptide chain of HRG results in a dysfunction of its secretion from the platelet alpha-granules with a decrease in its level to 21% of control [154, 155]. HRG deficiency was associated with inherited thromboses, indicating an anticoagulant function of this glycoprotein.
Recent studies show the important role of genetic factors in many human diseases. Hemostasis system disorders are no exception in this respect. In this review we have considered the genes whose known mutations modify the biochemical properties of the encoded proteins involved in the hemostasis system. Numerous syndromes of blood-clotting disturbances with familial inheritance but unknown genes involved in pathological process are out of the scope of consideration. Many such examples are given in the OMIM database [3]. Taken together, these facts suggest the possibility of a rapid increase in the number of known genes and mutations associated with inherited hemostasis defects in the offing.
Nevertheless, it would be a mistake to assume that simple accumulation of such information rapidly leads to qualitative changes in diagnostics and prevention of blood-clotting associated diseases. Since any human gene acts within the integrated genetic system formed by a unique set of allele variants of other genes, evaluation of its actual contribution to the development of a complex phenotypic (pathologic) trait is possible in a very restricted number of cases, for instance, in mutant genes F8 and F9. Population investigations on the abundant statistic material reveal associations between certain mutations and diseases providing the possibility to subsume a mutant individual to a specific risk group for disease development, but do not allow exact prediction of the development of the pathologic process. In connection with this, illustrative results have been obtained with inbred transgenic mice in which both alleles of one distinct gene are inactivated by means of gene knock-out. Phenotypic manifestation of such null-mutations are very diverse on the background of “identical” gene pool.
The solution of this problem of ambiguous correspondence between certain genotype and forming phenotype of a multicellular organism may be the development of new effective methods for determination of actual genotype of a particular organism, and also of the number of phenotypic traits on the molecular level. A real step on this way will be the determination of the complete primary human genome structure, which is already largely unscrambled [156]. A complete human genetic portrait composed on this basis and supported by the data on simultaneous expression levels of numerous genes, for example, using advanced microchip technologies enabling at now to follow the expression of thousands of genes simultaneously [157], offers great opportunities to solve the problem in the near future.
I am very pleased to express my gratitude to I. N. Bokarew, who guided me through the world of hemostasis, and also to S. M. Strukova for support and valuable remarks during the preparation of the manuscript.
REFERENCES
1.Patrushev, L. I. (2000) Gene Expression [in
Russian], Nauka, Moscow.
2.Auerbakh, Sh. (1978) Problems of Mutagenesis
[Russian translation], Mir, Moscow.
3.A Database “Online Mendelian Inheritance in
Man” - http://www.ncbi.nlm.nih.gov/omim/
4.Sohngen, D., Specker, C., Bach, D., Kuntz, B. M.
E., Burk, M., Aul, C., Kobbe, G., Heyll A., Hollmig, K. A., and
Schneider, W. (1997) Ann. Hematol., 74, 89-93.
5.Undas, A., Williams, E. B., Butenas, S., Orfeo, T.,
and Mann, K. G. (2001) J. Biol. Chem.,276, 4389-4397.
6.Bertina, R. M. (1999) Thromb. Haemost.,
82, 601-609.
7.Rosendaal, F. R. (1999) Thromb. Haemost.,
82, 610-619.
8.Ruggeri, Z. M. (1999) Thromb Haemost.,
82, 576-584.
9.Mohlke, K. L., Nichols, W. S., and Ginsburg, D.
(1999) Int. J. Clin. Lab. Res., 29, 1-7.
10.Ewenstein, B. M. (1997) Annu. Rev. Med.,
48, 525-542.
11.Veyradier, A., Fressinaud, E., and Meyer, D.
(1998) Int. J. Clin. Lab. Res., 28, 201-210.
12.Goodeve, A. C., Eikenboom, J. C. J., Ginsburg,
D., Hilbert, L., Mazurier, C., Peake, I. R., Sadler, J. E., and
Rodeghiero, F. (2001) Thromb. Haemost., 85, 929-931.
13.Eikenboom, J. C. J., Castman, G., Vos, H. L.,
Bertina, R. M., and Rodeghiero, F. (1998) Thromb. Haemost.,
79, 709-717.
14.Eikenboom, J. C., Ploos van Amstel, H. K.,
Reitsma, P. H., and Briet, E. (1992) Thromb. Haemost.,
68, 448-454.
15.Schneppenheim, R., Krey, S., Gergmann, F., Bock,
D., Budde, U., Lange, M., Linde, R., Mittler, U., Meili, E., Mertes,
G., Olek, K., Plendl, H., and Simeon, W. (1994) Hum. Genet.,
94, 640-652.
16.Zhang, Z. P., Blomback, M., Egberg, N., Falk, G.,
and Anvret, M. (1994) Genomics, 21, 188-193.
17.Bernardi, F., Casonato, A., Marchetti, G.,
Gemmati, D., Bizzaro, N., Pontara, E., and Girolami, A. (1998) Br.
J. Haematol., 103, 885-887.
18.Iannuzzi, M. C., Hidaka, N., Boehnke, M. L.,
Bruck, M. E., Hanna, W. T., Collins, F. S., and Ginsburg, D. (1991)
Am. J. Hum. Genet., 48, 757-763.
19.Ribba, A. N., Hilbert, L., Lavergne, J. M.,
Fressinaud, E., Boyer-Neumann, C., Ternisien, C., Juhan-Vague, I.,
Goudemand, J., Girma, J., Mazurier, C., and Meyer, D. (2001)
Blood,97,952-959.
20.Lavergne, J.-M., de Paillette, L., Bahnak, B. R.,
Ribba, A.-S., Fressinaud, E., Meyer, D., and Pietu, G. (1992) Brit.
J. Haematol., 82, 66-72.
21.Meyer, D., Fressinaud, E., Gaucher, C., Lavergne,
J. M., Hilbert, L., Ribba, A. S., Jorieux, S., and Mazurier, C. (1997)
Thromb. Haemost., 78, 451-456.
22.Hilbert, L., Gaucher, C., de Romeuf, C.,
Horellou, M. H., Vink, T., and Mazurier, C. (1994) Blood,
83, 1542-1550.
23.Lopez-Fernandez, M. F., Gonzalez-Boullosa, R.,
Blanco-Lopez, M. J., Perez, M., and Batlle, J. (1991) Am. J.
Hematol., 36, 163-170.
24.Weiss, H. J., Pietu, G., Rabinowitz, R., Girma,
J.-P., Rogers, J., and Meyer, D. (1983) J. Lab. Clin. Med.,
101, 411-425.
25.Castaman, G., Eikenboom, J. C., Contri, A., and
Rodeghiero, F. (2000) Thromb. Haemost., 84, 351-352.
26.Andrews, R. K., Lopez, J. A., and Berndt, M. C.
(1997) Int. J. Biochem. Cell. Biol., 29, 91-105.
27.Andrews, R. K., Shen, Y., Gardner, E. E., Dong,
J., Lopez, J. A., and Berndt, M. C. (1999) Thromb.
Haemost., 82, 357-364.
28.Lopez, J. A., Andrews, R. K., Afshar-Kharghan,
V., and Berndt, M. C. (1998) Blood, 91, 4397-4418.
29.Kaski, S., Kekomaki, R., and Partanen, J. (1996)
Immunogenetics, 44, 170-176.
30.Moran, N., Morateck, P. A., Deering, A., Ryan,
M., Montgomery, R. R., Fitzgerald, D. J., and Kenny, D. (2000)
Blood, 96, 532-539.
31.Kenny, D., Morateck, P. A., Gill, J. C., and
Montgomery, R. R. (1999) Blood, 93, 2968-2975.
32.Rivera, C. E., Villagra, J., Riordan, M.,
Williams, S., Lindstrom, K. J., and Rick, M. E. (2001) Br. J.
Haemat., 112, 105-108.
33.Ramakrishnan, V., Reeves, P. S., DeGuzman, F.,
Deshpande, U., Ministri-Madrid, K., DuBridge, R. B., and Phillips, D.
R. (1999) Proc. Natl. Acad. Sci. USA, 96,
13336-13341.
34.Thornton, M. A., Poncz, M., Korostishevsky, M.,
Yakobson, E., Usher, S., Seligsohn, U., and Peretz, H. (1999)
Blood, 94, 2039-2047.
35.Payrastre, B., Missy, K., Trumel, C., Bodin, S.,
Plantavid, M., and Chap, H. (2000) Biochem. Pharmacol.,
60, 1069-1074.
36.Patil, S., Newman, D. K., and Newman, P. J.
(2001) Blood, 97, 1727-1732.
37.Tandon, N. N., Kralisz, U., and Jamieson, G. A.
(1989) J. Biol. Chem., 264, 7576-7583.
38.Tanaka, T., Sohmiya, K., and Kawamura, K. J.
(1997) Molec. Cell. Cardiol., 29, 121-127.
39.Coburn, C. T., Knapp, F. F., Febbraio, M., Beets,
A. L., Silverstein, R. L., and Abumrad, N. A. (2000) J. Biol.
Chem., 275, 32523-32529.
40.Zutter, M. M., and Santoro, S. A. (1990) Am.
J. Pathol., 137, 113-120.
41.Carlsson, L. E., Santoso, S., Spitzer, C.,
Kessler, C., and Greinacher, A. (1999) Blood, 93,
3583-3586.
42.Santoso, S., Kunicki, T. J., Kroll, H.,
Haberbosch, W., and Gardemann, A. (1999) Blood, 93,
2449-2453.
43.Di Paola, J., Federici, A. B., Mannucci, P. M.,
Canciani, M. T., Kritzik, M., Kunicki, T. J., and Nugent, D. (1999)
Blood, 93, 3578-3582.
44.Jacquelin, B., Tarantino, M. D., Kritzik, M.,
Rozenshteyn, D., Koziol, J. A., Nurden, A. T., and Kunicki, T. J.
(2001) Blood, 97, 1721-1726.
45.Watson, S. P., and Gibbins, J. (1998) Immunol.
Today, 19, 260-264.
46.Arai, M., Yamamoto, N., Moroi, M., Akamatsu, N.,
Fukutake, K., and Tanoue, K. (1995) Br. J. Haematol., 89,
124-130.
47.Walsh, M.-T., Foley, J. F., and Kinsella, B. T.
(2000) J. Biol. Chem., 275, 20412-20423.
48.Hirata, T., Ushikubi, F., Kakizuka, A., Okuma,
M., and Narumiya, S. (1996) J. Clin. Invest., 97,
949-956.
49.Gachet, C. (2001) Thromb.
Haemost.,86, 222-232.
50.Jin, J., Daniel, J. L., and Kunapuli, S. P.
(1998) J. Biol. Chem., 273, 2030-2034.
51.Hollopeter, G., Jantzen, H.-M., Vincent, D., Li,
G., England, L., Ramakrishnan, V., Yang, R.-B., Nurden, P., Nurden, A.,
Julius, D., and Conley, P. B. (2001) Nature, 409,
202-206.
52.Kiuru, S., Javela, K., Somer, H., and Kekomaki,
R. (2000) Thromb. Haemost., 83, 491-495.
53.Witke, W., Sharpe, A. H., Hartwig, J. H., Azuma,
T., Stossel, T. P., and Kwiatkowski, D. J. (1995) Cell,
81, 41-51.
54.Weiss, H. J., Witte, L. D., Kaplan, K. L., Lages,
B. A., Chernoff, A., Nossel, H. L., Goodman, D. S., and Baumgartner, H.
R. (1979) Blood, 54, 1296-1319.
55.Oh, J., Ho, L., Ala-Mello, S., Amato, D.,
Armstrong, L., Bellucci, S., Carakushansky, G., Ellis, J. P., Fong,
C.-T., Green, J. S., Heon, E., Legius, E., Levin, A. V., Nieuwenhuis,
H. K., Pinckers, A., Tamura, N., Whiteford, M. L., Yamasaki, H., and
Spritz, R. A. (1998) Am. J. Hum. Genet., 62, 593-598.
56.Zhen, L., Jiang, S., Feng, L., Bright, N. A.,
Peden, A. A., Seymour, A. B., Novak, E. K., Elliott, R., Gorin, M. B.,
Robinson, M. S., and Swank, R. T. (1999) Blood, 94,
146-155.
57.Mazurov, A. V., Vinogradov, D. V., Khaspekova, S.
G., Krushinsky, A. V., Gerdeva, L. V., and Vasiliev, S. A. (1996)
Eur. J. Haematol., 57, 38-41.
58.Furie, B., Furie, B. C., and Flaumenhaft, R.
(2001) Thromb. Haemost., 86, 214-221.
59.Frazier, W. A. (1987) J. Cell. Biol.,
105, 625-632.
60.Mast, A. E., Stadanlick, J. E., Lockett, J. M.,
Dietzen, D. J., Hasty, K. A., and Hall, C. L. (2000) J. Biol.
Chem., 275, 31715-31721.
61.Lawler, J., Sunday, M., Thibert, V., Duquette,
M., George, E. L., Rayburn, H., and Hynes, R. O. (1998) J. Clin.
Invest., 101, 982-992.
62.Becker, R. C., and Spencer, F. A. (1998) J.
Thromb. Thrombol., 5, 215-229.
63.Strukova, S. M. (2001) Biochemistry
(Moscow), 66, 8-18.
64.Di Cera, E., Dang, Q. D., and Ayala, Y. M. (1997)
Cell. Mol. Life Sci., 53, 701-730.
65.Gingrich, M. B., and Traynelis, S. F. (2000)
Trends Neurosci., 23, 399-407.
66.Turgeon, V. L., Salman, N., and Houenou, L. J.
(2000) Thrombosis Res., 99, 417-427.
67.Morishita, E., Saito, M., Kumabashiri, I.,
Asakura, H., Matsuda, T., and Yamaguchi, K. (1992) Blood,
80, 2275-2280.
68.Rosendaal, F. R., Siscovick, D. S., Schwartz, S.
M., Psaty, B. M., Raghunathan, T. E., and Vos, H. L. (1997)
Blood, 90, 1747-1750.
69.Rosendaal, F. R., Doggen, C. J. M., Zivelin, A.,
Arruda, V. R., Aiach, M., Siscovick, D. S., Hillarp, A., Watzke, H. H.,
Bernardi, F., Cumming, A. M., Preston, F. E., and Reitsma, P. H. (1998)
Thromb. Haemost., 79, 706-708.
70.Patrushev, L. I. (1998) Rus. Med. Zh., No.
5, 181-185.
71.Reshetnyak, T. M., Patrushev, L. I., Stukacheva,
E. A., Miroshnikov, A. I., Tikhonova, T. L., Nasonov, E. L., and
Alekberova, Z. S. (2000) Terapevt. Arkh.,No. 5,34-38.
72.Ozolinya, L. A., Patrushev, L. I., Shpolyanskaya,
N. Yu., Makarov, O. V., Stukacheva, E. A., Strukova, S. M., and
Miroshnikov, A. I. (2001) Tromboz, Gemostaz,
Reologiya, No. 5, 47-53.
73.Rosing, J., and Tans, G. (1997) Int. J.
Biochem. Cell Biol.,29, 1123-1126.
74.Bertina, R. M., Koeleman, B. P. C., Koster, T.,
Rosendaal, F. R., Dirven, R. J., de Ronde, H., van der Velden, P. A.,
and Reitsma, P. H. (1994) Nature, 369, 64-67.
75.Guasch, J. F., Cannegieter, S., Reitsma, P. H.,
van't Veer-Korthof, E. T., and Bertina, R. M. (1998) Br. J.
Haematol., 101, 32-39.
76.Zehnder, J. L., Hirak, D. D., Jones, C. D.,
Gross, N., and Grumet, F. C. (1999) Thromb. Haemost., 82,
1097-1099.
77.Patrushev, L. I., Zykova, E. S., Kayushin, A. L.,
Korosteleva, M. D., Miroshnikov, A. I., Bokarew, I. N., Leont'ev, S.
G., Koshkin, V. M., and Severin, E. S. (1998) Thrombosis Res.,
92, 251-259.
78.Chan, W. P., Lee, C. K., Kwong, Y. L., Lam, C.
K., and Liang, R. (1998) Blood, 91, 1135-1139.
79.Williamson, D., Brown, K., Luddington, R.,
Baglin, C., and Baglin, T. (1998) Blood, 91,
1140-1144.
80.Zehnder, J. L., and Jain, M. (1996) Blood
Coagul. Fibrin., 7, 361-362.
81.McVey, J. H., Boswell, E., Mumford, A. D.,
Kemball-Cook, G., and Tuddenham, E. G. D. (2001) Hum. Mutat.,
17, 3-17.
82.Wulff, K., and Herrmann, F. H. (2000) Hum.
Mutat., 15, 489-496.
83.Mann, K. G., Nesheim, M. E., Church, W. R.,
Haley, P., and Krishnaswamy, S. (1990) Blood, 76,
1-16.
84.Soucie, J. M., Evatt, B., and Jackson, D. (1998)
Am. J. Hematol., 59, 288-294.
85.Tuddenham, E. G., Schwaab, R., Seehafer, J.,
Millar, D. S., Gitschier, J., Higuchi, M., Bidichandani, S., Connor, J.
M., Hoyer, L. W., Yoshioka, A., Peake, I. R., Olek, K., Kazazian, H.
H., Lavergne, J.-M., Giannelli, F., Antonarakis, S. E., and Cooper, D.
N. (1994) Nucleic Acids Res., 22, 4851-4868.
86.Diuguid, D. L., Rabiet, M. J., Furie, B. C.,
Liebman, H. A., and Furie, B. (1986) Proc. Natl. Acad. Sci.
USA,83,5803-5807.
87.Giannelli, F., Green, P. M., Sommer, S. S., Poon,
M.-C., Ludwig, M., Schwaab, R., Reitsma, P. H., Goossens, M., Yoshioka,
A., Figueiredo, M. S., and Brownlee, G. G. (1997) Nucleic Acids
Res., 25, 133-135.
88.Green, P. M., Bentley, D. R., Mibashan, R. S.,
Nilsson, I. M., and Giannelli, F. (1989) EMBO J.,
8, 1067-1072.
89.Cirino, G., Cicala, C., Bucci, M., Sorrentino,
L., Ambrosini, G., DeDominicis, G., and Altieri, D. C. (1997) J.
Clin. Invest., 99, 2446-2451.
90.Millar, D. S., Elliston, L., Deex, P., Krawczak,
M., Wacey, A. I., Reynaud, J., Nieuwenhuis, H. K., Bolton-Maggs, P.,
Mannucci, P. M., Reverter, J. C., Cachia, P., Pasi, K. J., Layton, D.
M., and Cooper, D. N. (2000) Hum. Genet., 106,
249-257.
91.Bouma, B. N., and Griffin, J. H. (1977) J.
Biol. Chem.,252,6432-6437.
92.Thompson, R. E., Mandle, R., and Kaplan, A. P.
(1977) J. Clin. Invest., 60, 1376-1380.
93.Asakai, R., Chung, D. W., Davie, E. W., and
Seligsohn, U. (1991) New Engl. J. Med., 325,
153-158.
94.Bolton-Maggs, P. H. (2000) Haemophilia,
6 (Suppl. 1), 100-109.
95.Bolton-Maggs, P. H., Young Wan-Yin, B., McCraw,
A. H., Slack, J., and Kernoff, P. B. (1988) Br. J. Haematol.,
69, 521-528.
96.Hsu, T., Shore, S., Seshsmma, T., Bagasra, O.,
and Walsh, P. (1998) J. Biol. Chem., 273,
13787-13793.
97.Martincic, D., Kravtsov, V., and Gailani, D.
(1999) Blood, 94, 3397-3404.
98.Pixley, R. A., and Colman, R. W. (1993) Meth.
Enzymol., 222,51-65.
99.Bradford, H. N., Pixley, R. A., and Colman, R. W.
(2000) J. Biol. Chem., 275, 22756-22763.
100.Schloesser, M., Zeerleder, S., Lutze, G.,
Halbmayer, W.-M., Hofferbert, S., Hinney, B., Koestering, H., Lammle,
B., Pindur, G., Thies, K., Kohler, M., and Engel, W. (1997)
Blood, 90, 3967-3977.
101.Schwartz, M. L., Pizzo, S. V., Hill, R. L., and
McKee, P. A. (1971) J. Biol. Chem., 246, 5851-5854.
102.Poon, M. C., Russell, J. A., Low, S., Sinclair,
G. D., Jones, A. R., Blahey, W., Ruether, B. A., and Hoar, D. I. (1989)
J. Clin. Invest.,84,787-792.
103.Lorand, L., Losowsky, M. S., and Miloszewski,
K. J. M. (1980) Progr. Hemost. Thromb., 5, 245-290.
104.Kera, Y., Nishimukai, H., and Yamasawa, K.
(1981) Hum. Genet., 59, 360-364.
105.Mikkola, H., Syrjala, M., Rasi, V., Vahtera,
E., Hamalainen, E., Peltonen, L., and Palotie, A. (1994) Blood,
84, 517-525.
106.Kohler, H. P., Stickland, M. H., Ossei-Gerning,
N., Carter, A., Mikkola, H., and Grant, P. J. (1998) Thromb.
Haemost., 79, 8-13.
107.Arnaud, E., Barbalat, V., Nicaud, V., Cambien,
F., Evans, A., Morrison, C., Arveiler, D., Luc, G., Ruidavets, J.-B.,
Emmerich, J., Fiessinger, J.-N., and Aiach, M. (2000) Arterioscler.
Thromb. Vasc. Biol., 20, 892-898.
108.Wartiovaara, U., Mikkola, H., Szoke, G.,
Haramura, G., Karpati, L., Balogh, I., Lassila, R., Muszbek, L., and
Palotie, A. (2000) Thromb. Haemost., 84, 595-600.
109.McGilvary, J. D., and Rottstein, O. D. (1999)
Sepsis, 3, 93-101.
110.Ruf, W., and Edgington, T. S. (1994) FASEB
J., 8, 385-390.
111.Erlich, J., Parry, G. C. N., Fearns, C.,
Muller, M., Carmeliet, P., Luther, T., and Mackman, N. (1999) Proc.
Natl. Acad. Sci. USA, 96, 8138-8143.
112.Herrick, S., Blanc-Brude, O., Gray, A., and
Laurent, G. (1999) Int. J. Biochem. Cell Biol., 31,
741-746.
113.Neerman-Arbez, M., Honsberger, A., Antonarakis,
S. E., and Morris, M. A. (1999) J. Clin. Invest., 103,
215-218.
114.Neerman-Arbez, M., de Moerloose, P., Bridel,
C., Honsberger, A., Schonborner, A., Rossier, C., Peerlinck, K.,
Claeyssens, S., Di Michele, D., d'Oiron, R., Dreyfus, M.,
Laubriat-Bianchin, M., Dieval, J., Antonarakis, S. E., and Morris, M.
A. (2000) Blood, 96, 149-152.
115.Neerman-Arbez, M., de Moerloose, P.,
Honsberger, A., Parlier, G., Arnuti, B., Biron, C., Borg, J.-Y., Eber,
S., Meili, E., Peter-Salonen, K., Ripoll, L., Vervel, C., d'Oiron, R.,
Staeger, P., Antonarakis, S. E., and Morris, M. A. (2001) Hum.
Genet., 108, 237-240.
116.Koopman, J., Haverkate, F., Grimbergen, J.,
Engesser, L., Novakova, I., Kerst, A. F. J. A., and Lord, S. T. (1992)
Proc. Natl. Acad. Sci. USA,89, 3478-3482.
117.Cote, H. C. F., Lord, S. T., and Pratt, K. P.
(1998) J. Am. Soc. Hematol., 92, 2195-2212.
118.Esmon, N. L. (1987) Sem. Thromb.
Hemost., 13, 454-463.
119.Doggen, C. J. M., Kunz, G., Rosendaal, F. R.,
Lane, D. A., Vos, H. L., Stubbs, P. J., Cats, V. M., and Ireland, H.
(1998) Thromb. Haemost., 80, 743-748.
120.Ohlin, A.-K., and Marlar, R. A. (1995)
Blood, 85, 330-336.
121.Lane, D. A., Kunz, G., Olds, R. J., and Thein,
S. L. (1996) Blood Rev., 10, 59-74.
122.Rao, L. V., Nordfang, O., Hoang, A., and
Pendurthi, U. (1995) Blood, 85, 121-129.
123.Lane, D. A., Bayston, T., Olds, R. J., Fitches,
A. C., Cooper, D. N., Millar, D. S., Jochmans, K., Perry, D. J.,
Okajima, K., Thein, S. L., and Emmerich, J. (1997) Thromb.
Haemost., 77, 197-211.
124.Esmon, C. T. (2000) Biochim. Biophys.
Acta, 1477, 349-360.
125.Millar, D. S., Johansen, B., Berntorp, E.,
Minford, A., Bolton-Maggs, P., Wensley, R., Kakkar, V., Schulman, S.,
Torres, A., Bosch, N., and Cooper, D. N. (2000) Hum. Genet.,
106, 646-653.
126.Reitsma, P. H. (1996) Nucleic Acids
Res., 24, 157-159.
127.Marlar, R. A., Montgomery, R. R., and
Broekmans, A. W. (1989) J. Paediat., 114, 528-534.
128.Dahlback, B. (1995) Thromb. Res.,
77, 1-43.
129.Dahlback, B. (1991) Thromb. Haemost.,
66, 49-61.
130.Aiach, M., Borgel, D., Gaussem, P., Emmerich,
J., Alhenc-Gelas, M., and Gandrille, S. (1997) Sem. Hematol.,
34, 205-216.
131.Borgel, D., Gandrille, S., and Aiach, M. (1997)
Thromb. Haemost., 78, 351-356.
132.Gandrille, S., Borgel, D., Ireland, H., Lane,
D. A., Simmonds, R., Reitsma, P. H., Mannhalter, C., Pabinger, I.,
Saito, H., Suzuki, K., Formstone, C., Cooper, D. N., Espinosa, Y.,
Sala, N., Bernardi, N., and Alach, M. (1997) Thromb. Haemost.,
77, 1201-1214.
133.Davie, E. W., Fujikawa, K., and Kiesiel, W.
(1991) Biochemistry, 30, 10363-10370.
134.Kleesiek, K., Schmidt, M., Gotting, C.,
Brinkmann, T. S., and Prohaska, W. (1998) Blood, 92,
3976-3977.
135.Moatti, D., Haidar, B., Fumeron, F., Gauci, L.,
Boudvillain, O., Seknadji, P., Olliver, V., Aumont, M. C., and de
Prost, D. (2000) Thromb. Haemost., 84, 244-249.
136.Huang, Z.-F., Higuchi, D., Lasky, N., and
Broze, G. J. (1997) Blood, 90, 944-951.
137.Herzog, R., Lutz, S., Blin, N., Marasa, J. C.,
Blinder, M. A., and Tollefsen, D. M. (1991) Biochemistry,
30, 1350-1357.
138.Matsuo, T., Kario, K., Sakamoto, S., Yamada,
T., Miki, T., Hirase, T., and Kobayashi, H. (1992) Thromb. Res.,
65, 495-505.
139.Villa, P., Aznar, J., Vaya, A., Espana, F.,
Ferrando, F., Mira, Y., and Estelles, A. (1999) Thromb.
Haemost., 82, 1011-1014.
140.Irigoyena, J. P., Munoz-Canoves, P., Monteroa,
L., Koziczaka, M., and Nagaminea, Y. (1999) Cell. Mol. Life
Sci., 56, 104-132.
141.Ewald, G. A., and Eisenberg, P. R. (1995)
Circulation,91, 28-36.
142.Pryzdial, E. L. G., and Kessler, G. E. (1996)
J. Biol. Chem.,271, 16614-16620.
143.Zeibdawi, A. R., and Pryzdial, E. L. G. (2001)
J. Biol. Chem.,276, 19929-19936.
144.Hach-Wunderle, V., Scharrer, I., and
Lottenberg, R. (1988) Thromb. Haemost., 59, 277-280.
145.Carmeliet, P., and Coolen, D. (1996)
Fibrinolysis, 10, 195-213.
146.Lijnen, H. R., and Collen, D. (1982) Sem.
Thromb. Hemost., 8,2-10.
147.Aznar, J., Estelles, A., Vila, V., Reganon, E.,
Espana, F., and Villa, P. (1984) Thromb. Haemost., 52,
196-200.
148.Petaja, J., Rasi, V., Vahtera, E., and Myllyla,
G. (1991) Brit. J. Haematol., 79, 291-295.
149.Dawson, S. J., Wiman, B., Hamsten, A., Green,
F., Humphries, S., and Henney, A. M. (1993) J. Biol. Chem.,
268, 10739-10745.
150.Fay, W. P., Parker, A. C., Condrey, L. R., and
Shapiro, A. D. (1997) Blood, 90, 204-208.
151.Schleef, R. R., Higgins, D. L., Pillemer, E.,
and Levitt, L. J. (1989) J. Clin. Invest., 83,
1747-1752.
152.Margaglione, M., Cappucci, G., Colaizzo, D.,
Giuliani, N., Vecchione, G., Grandone, E., Pennelli, O., and di Minno,
G. (1998) Arterioscler. Thromb. Vasc. Biol., 18,
152-156.
153.Leung, L. (1993) J. Lab. Clin. Med.,
121, 630-631.
154.Shigekiyo, T., Ohshima, T., Oka, H., Tomonari,
A., Azuma, H., and Saito, S. (1993) Thromb. Haemost., 70,
263-265.
155.Shigekiyo, T., Yoshida, H., Matsumoto, K.,
Azuma, H., Wakabayashi, S., Saito, S., Fujikawa, K., and Koide, T.
(1998) Blood, 91, 128-133.
156.Lander, E. S., Linton, L. M., Birren, B.,
Nusbaum, C., Zody, M. C., Baldwin, J., Devon, K., Dewar, K., Doyle, M.,
FitzHugh, W., Funke, R., Gage, D., Harris, K., Heaford, A.,et al.
(2001) Nature, 409, 860-921.
157.Roth, M., and Yarmush, M. L. (1999) Annu.
Rev. Biomed. Eng., 1, 265-297.