REVIEW: Matrix Metalloproteinases and Cellular Fibrinolytic Activity
H. R. Lijnen
Center for Molecular and Vascular Biology, University of Leuven, Campus Gasthuisberg, O & N, Herestraat 49, B-3000 Leuven, Belgium; fax: +32-16-345990; E-mail: roger.lijnen@med.kuleuven.ac.be
Received February 28, 2001; Revision received April 16, 2001
Several molecular interactions between the matrix metalloproteinase (MMP) and the plasminogen/plasmin (fibrinolytic) system may affect cellular fibrinolysis. MMP-3 (stromelysin-1) specifically hydrolyzes urokinase (u-PA), yielding a 17 kD NH2-terminal fragment containing the functionally intact receptor (u-PAR)-binding sequence and a 32 kD COOH-terminal fragment containing the intact serine proteinase domain. MMP-3 generates an angiostatin-like fragment (containing kringles 1-4 with the cellular binding domains) from plasminogen. Treatment with MMP-3 of monocytoid THP-1 cells saturated with bound plasminogen, resulted in a dose-dependent reduction of the amount of u-PA-activatible plasminogen. Treatment with MMP-3 of cell-bound u-PA, in contrast, did not alter cell-associated u-PA activity. These data thus indicate that MMP-3 may downregulate cell-associated plasmin activity by decreasing the amount of activatible plasminogen, without affecting cell-bound u-PA activity. MMP-3 also specifically interacts with the main inhibitors of the fibrinolytic system. Thus, MMP-3 specifically hydrolyzes human alpha2-antiplasmin (alpha2-AP), the main physiological plasmin inhibitor. alpha2-AP cleaved by MMP-3 no longer forms a stable complex with plasmin and no longer interacts with plasminogen. Cleavage and inactivation of alpha2-AP by MMP-3 may constitute a mechanism favoring local plasmin-mediated proteolysis. Furthermore, MMP-3 specifically hydrolyzes and inactivates human plasminogen activator inhibitor-1 (PAI-1). Stable PAI-1 bound to vitronectin is cleaved and inactivated by MMP-3 in a comparable manner as free PAI-1; the cleaved protein, however, does not bind to vitronectin. Cleavage and inactivation of PAI-1 by MMP-3 may thus constitute a mechanism decreasing the antiproteolytic activity of PAI-1 and impairing the potential inhibitory effect of vitronectin-bound PAI-1 on cell adhesion and/or migration. These molecular interactions of MMP-3 with enzymes, substrates and inhibitors of the fibrinolytic system may thus play a role in the regulation of (cellular) fibrinolysis. Furthermore, the temporal and topographic expression pattern of MMP components, as well as studies in gene-deficient mice, suggest a functional role in neointima formation after vascular injury.
KEY WORDS: matrix metalloproteinases, cellular fibrinolysis, restenosis, stromelysin-1
Abbreviations:alpha2-AP) alpha2-antiplasmin; MMP) matrix metalloproteinase; PAI-1) plasminogen activator inhibitor-1; PAI-2) plasminogen activator inhibitor-2; ProMMP) proenzyme form of MMP; SMC) smooth muscle cells; TIMP) tissue inhibitor of MMP; t-PA) tissue-type plasminogen activator; u-PA) urokinase-type plasminogen activator; u-PAR) u-PA receptor; WT) wild-type; X-/-) gene deficient.
The fibrinolytic (plasminogen/plasmin) system contains a proenzyme,
plasminogen, which is converted to the active enzyme, plasmin, by
tissue-type (t-PA) or urokinase-type (u-PA) plasminogen activator.
Plasmin degrades fibrin into soluble degradation products.
t-PA-mediated plasminogen activation is mainly involved in the
dissolution of fibrin at the site of vascular injury. u-PA binds to a
specific cellular receptor (u-PAR) resulting in enhanced activation of
cell-bound plasminogen. The main role of u-PA appears to be in the
induction of pericellular proteolysis via the degradation of matrix
components or via activation of latent proteinases or growth factors.
Inhibition of the fibrinolytic system may occur either at the level of
the PA, by specific plasminogen activator inhibitors (PAI-1 and PAI-2),
or at the level of plasmin, mainly by
alpha2-antiplasmin [1].
Physiological fibrinolysis is regulated by specific molecular
interactions between its main components as well as by controlled
synthesis and release, primarily from endothelial cells, of PAs and
PAIs. The fibrinolytic system is not only responsible for the removal
of fibrin from the circulation, but is believed to play a role in
several other biological processes, including ovulation, embryogenesis,
intima proliferation, angiogenesis, tumorigenesis, and atherosclerosis.
Recent observations in mice with inactivation of the main components of
the fibrinolytic system have confirmed the physiological relevance of
the system in these processes, but also suggested involvement of other
proteolytic systems, mainly the matrix metalloproteinase (MMP) system
[2-6].
Matrix metalloproteinases (MMPs) are classified on the basis of substrate specificity. Collagenases (MMP-1, interstitial collagenase; MMP-8, neutrophil collagenase; MMP-13, collagenase-3) degrade connective tissue collagens; gelatinases (MMP-2, gelatinase A; MMP-9, gelatinase B) degrade collagen types IV, V, VII, and X, elastin and denatured collagens; stromelysins (MMP-3, stromelysin-1; MMP-10, stromelysin-2) and MMP-7, matrilysin, degrade the proteoglycan core proteins, laminin, fibronectin, elastin, gelatin, and nonhelical collagen, whereas stromelysin-3 (MMP-11) is unusual and does not degrade any of the major extracellular matrix components; macrophage metalloelastase (MMP-12) degrades insoluble elastin, collagen IV, fibronectin, laminin, entactin, and proteoglycans. Twenty-four members of the MMP family have been identified, among which six are membrane-type (MT) MMPs [7].
MMPs are generally secreted as zymogens that are extracellularly activated by several proteinases. In vitro, plasmin directly activates proMMP-1, proMMP-3, proMMP-9, proMMP-10, and proMMP-13 [8-11]. Activation of proMMP-2 involves hydrolysis by MT1-MMP, yielding an intermediate that is activated by plasmin [12]. ProMMP-2 is also hydrolyzed by u-PA [13]. Activation of proMMP-9 may occur via plasmin-dependent mechanisms, or via plasmin-independent mechanisms [14]. Thus, plasmin may play an important role in the in vivo activation of proMMPs. Several active MMPs can further activate other proMMPs, thus constituting positive feedback mechanisms [9, 11, 15]. MMPs are inhibited by specific tissue inhibitors of MMPs (TIMPs). Four members of the TIMP family have been identified, of which TIMP-1, synthesized by most types of connective tissue cells as well as by macrophages, acts against all members of the collagenase, stromelysin, and gelatinase classes [16].
MMPs, alone or in concert with the plasminogen/plasmin system, are involved in the degradation of extracellular matrix components (figure), a requirement for cell migration and tissue remodeling, which play an essential role in many (patho)physiological processes.
Schematic representation of potential interactions between the fibrinolytic (plasminogen/plasmin) and matrix metalloproteinase systems. The fibrinolytic system contains a proenzyme, plasminogen, that is converted to the active enzyme plasmin by tissue-type (t-PA) or urokinase-type (u-PA) plasminogen activator. Plasmin degrades fibrin and converts latent matrix metalloproteinases (pro-MMP's) into active MMPs, which in turn degrade extracellular matrix (ECM). Activation is also regulated by positive feedback mechanisms in which several active MMPs can activate other pro-MMP's. Plasmin-mediated effects are inhibited by alpha2-antiplasmin and MMP-mediated effects by tissue inhibitors (TIMPs). Inhibition may also occur at the level of the plasminogen activators by plasminogen activator inhibitors (mainly PAI-1 and PAI-2) (solid line, activation/conversion; dotted line, inhibition)
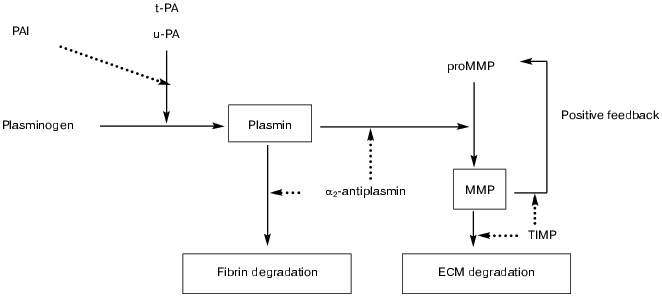
INTERACTIONS BETWEEN THE PLASMINOGEN/PLASMIN AND MMP SYSTEMS
The physiological relevance of plasmin in the in vivo activation of proMMPs has not been clearly established, nor was the role of the physiological plasminogen activators t-PA and u-PA. The availability of mice with targeted inactivation of the main components of the plasminogen/plasmin system has allowed this to be studied in more detail.
The distribution of latent (pro-) or active MMP-2 and MMP-9 was monitored in aorta extracts and in serum-free conditioned cell culture medium obtained from wild-type (WT) mice and from mice with deficiency of t-PA (t-PA-/-), u-PA (u-PA-/-), PAI-1 (PAI-1-/-) or plasminogen (Pg-/-). This was achieved by zymography on gelatin-containing gels, which detects molecular forms of latent MMP-2 (70 and 65 kD) and of active MMP-2 (61 kD and mainly 58 kD), in addition to 94 kD proMMP-9 and 83 kD active MMP-9. In vivo activation of proMMP-2 was observed independently of plasmin(ogen) [17]. This was substantiated by the findings that: 1) total levels of MMP-2 (active plus latent) were comparable in fibroblasts, smooth muscle cells (SMC), or aorta extracts of the different gene-deficient mice; 2) the contribution of active 58 kD MMP-2 to the total MMP-2 level was comparable in WT, t-PA-/-, u-PA-/-, PAI-1-/- and also in Pg-/- mice; and 3) addition of plasmin(ogen) to the cell culture medium did not significantly affect activation of proMMP-2. These data also indicate that direct activation of proMMP-2 by u-PA or t-PA does not play a major physiological role. Active MMP-9 was detected in samples from WT, t-PA-/-, u-PA-/-, and PAI-1-/- mice, indicating that its activation does not require both physiological plasminogen activators. In contrast, no active MMP-9 was detected in macrophages or SMC derived from Pg-/- mice, indicating that efficient proMMP-9 activation in these cells is plasmin(ogen)-dependent. However, active MMP-9 was detected in Pg-/- fibroblasts indicating that proMMP-9 activation may also occur independently of plasminogen. These data obtained with gene-deficient mice thus indicate that in vivo activation of proMMP-2 occurs independently of plasmin(ogen), whereas activation of proMMP-9 may occur via plasmin-dependent or plasmin-independent (MMP-mediated) mechanisms [17].
Because MMP-3 can activate proMMP-9, we hypothesized that plasmin may be involved in proMMP-9 activation indirectly through activation of proMMP-3. Therefore, a potential physiological role of MMP-3 in the expression and activation of MMP-2 or MMP-9in the wall of injured arteries was studied with the use of homozygous MMP-3-deficient (MMP-3-/-) mice [14]. One week after perivascular electric injury of the carotid or femoral artery in wild-type (MMP-3+/+) or MMP-3-/- mice, 70 and 65 kD proMMP-2 levels were enhanced by 2- to 4-fold, with corresponding increases of 20- to 40-fold for active 61 and 58 kD MMP-2. Active MMP-2 species represented approximately one third of the total MMP-2 concentration for both MMP-3+/+ and MMP-3-/- mice. ProMMP-9 levels were enhanced 10- to 80-fold; MMP-9 was not detected in non-injured carotid or femoral arteries, whereas one week after injury its contribution to the total MMP-9 level was 11 to 18% for MMP-3+/+ and MMP-3-/- mice. Cell culture experiments confirmed comparable ratios of active versus latent MMP-2 in skin fibroblasts and SMC derived from MMP-3+/+ and MMP-3-/- mice. In MMP-3+/+ and MMP-3-/- macrophages, comparable levels of 94 kD proMMP-9 were detected, and plasmin(ogen)-mediated conversion to 83 kD MMP-9 was obtained in both genotypes. ProMMP-9 activation thus appears to be possible via plasmin-dependent and MMP-3 -independent mechanisms. MMP-3 or other MMPs that critically depend on MMP-3 for their activation thus do not play a critical role in physiological activation of proMMP-2 or proMMP-9 [14].
CELL-ASSOCIATED PLASMINOGEN ACTIVATION
A striking analogy between the role of cell surfaces and that of fibrin in plasminogen activation has been recognized. Many cell types bind plasminogen activators and plasminogen, resulting in enhanced plasminogen activation [18-21] and protection of bound plasmin from inhibition by alpha2-antiplasmin [22, 23].
Binding of plasminogen to cultured human umbilical vein endothelial cells (HUVEC) was reported with a dissociation constant of 310 nM, and approximately 106 binding sites per cell [19]. Other studies report that most cells bind plasminogen via its lysine binding sites with a high capacity (>107 sites per cell) but a relatively low affinity (dissociation constant of approximately 1 µM). Gangliosides [24], as well as a class of membrane proteins with COOH-terminal lysine residues such as alpha-enolase [25], play an important role in binding of plasminogen to cells. The catalytic efficiency of t-PA for activation of cell-bound plasminogen is about 10-fold higher than in solution, possibly as a result of conversion of the plasminogen conformation to the more readily activatable “Lys-plasminogen-like” structure [26]. Alternatively, it was shown that vascular cells have the capacity to regulate pericellular fibrinolysis by modulating the expression of plasminogen receptors; enhanced receptor occupancy results in enhanced plasminogen activation by t-PA [27].
Specific, saturable and reversible binding of t-PA to HUVEC has been demonstrated [18, 28]. A high affinity binding site with a low number of binding sites and a lower affinity binding site with a high number of binding sites have been identified. With ligand blot techniques a Mr 40,000 membrane protein was identified which was suggested to represent the functional t-PA receptor; this receptor is related to annexin II [29]. Cell surface-bound t-PA retains it enzymatic activity and is protected from inhibition by PAI-1.
The binding of u-PA to its receptor (u-PAR) at the cell surface is believed to be crucial for its activity under physiological conditions. Binding results in a strongly enhanced plasmin generation, due to effects on both the activation of plasminogen [30] and on the feedback activation of scu-PA to tcu-PA by generated plasmin [31]. Both of these effects are also critically dependent on the cellular binding of plasminogen. Cell associated plasmin is protected from rapid inhibition by alpha2-antiplasmin, which further favors the activation of receptor-bound scu-PA. This system can however be efficiently inhibited by both PAI-1 and PAI-2 [32]. The observation that direct anchorage of u-PA to the cell surface (using a GPI-anchored u-PA mutant) leads to a potentiation of plasmin generation equivalent to that observed in the presence of u-PAR, suggests that u-PAR mainly functions to localize u-PA at the cell-surface [33]. Assembly of u-PAR-mediated plasminogen activation complexes also requires direct interactions between u-PA and plasminogen not involving the active site [34]. Concentration of proteolytic activity at the cell surface may also occur via vitronectin, trapping soluble u-PAR-u-PA complexes [35]. Both u-PA and u-PAR have been found in a complex with beta1-, beta2-, and beta3-integrins, thereby allowing mutual interactions and regulatory processes between cell adhesion and proteolysis (reviewed in [36]).
Assembly of plasminogen and plasminogen activators at the endothelial cell surface thus provides a focal point for plasmin generation and may play an important role in maintaining blood fluidity and non-thrombogenicity.
EFFECT OF MMPs ON CELL-ASSOCIATED PROTEOLYTIC ACTIVITY
We have investigated whether interactionsbetween the MMP and plasminogen/plasmin systems may affect cellular fibrinolysis. Our studies revealed that MMP-3 specifically hydrolyzes the Glu143-Leu144 peptide bond in u-PA, yielding a 17 kD NH2-terminal fragment containing the receptor (u-PAR)-binding sequence and a 32 kD COOH-terminal fragment containing the serine proteinase domain [37]. The 17 kD u-PA moiety specifically binds to the u-PAR on THP-1 cells, indicating that the receptor-binding site remained intact. The 32 kD moiety derived from single chain u-PA (scu-PA) has intact functional properties with respect to its activation by plasmin, and the specific amidolytic activity and plasminogen activating potential of its two-chain derivative. Thus, on one hand, u-PA may play a role in the activation of proMMP-3 via generation of plasmin, and on the other hand, active MMP-3 may prevent cellular receptor-binding of u-PA. This interaction may play a role in regulation of cell-associated u-PA activity, and may represent a mechanism whereby u-PAR-dependent and u-PAR-independent functions of u-PA are controlled.
Subsequent studies revealed that MMP-3 generates an angiostatin-like fragment (containing kringles 1-4 with the cellular binding domains) from plasminogen. MMP-3 indeed hydrolyzes the Glu59-Asn60, Pro447-Val448 and Pro544-Ser545 peptide bonds in plasminogen, yielding a 55 kD NH2-terminal fragment comprising kringles 1-4, a 14 kD domain corresponding to kringle 5 and a 30 kD domain comprising the serine proteinase domain [38].
Because of these specific molecular interactions between MMP-3 and plasminogen or u-PA, we investigated a potential role of MMP-3 in the regulation of cellular fibrinolytic activity by affecting binding and/or activation of plasminogen and/or scu-PA [39]. Plasminogen bound specifically to human monocytoid THP-1 cells, to murine MMP-3-/- SMC and fibroblasts. Treatment with MMP-3 of cells saturated with bound plasminogen, resulted in a dose-dependent reduction of the amount of u-PA activatible plasminogen (reduction to 25-40% of the value in the absence of MMP-3). Immunoblotting with specific monoclonal antibodies and autoradiography of eluates of the cells treated with MMP-3 revealed cleavage of plasminogen into the 55 kD fragment and mini-plasminogen (kringle 5 plus the proteinase domain).
Treatment with MMP-3 of cell-bound u-PA (THP-1 or HT1080 cells) did not alter cell-associated u-PA activity, measured in a direct chromogenic substrate assay or in a plasminogen-coupled chromogenic substrate assay (residual u-PA activity always >=85% of that without MMP-3 treatment). Autoradiography of 125I-labeled u-PA moieties, removed from the cells by treatment with acid or with phosphatidylinositol phospholipase C, confirmed that u-PA remained essentially intact after MMP-3 treatment.
These data thus indicate that MMP-3 may downregulate cell-associated plasmin activity by decreasing the amount of activatible plasminogen, without affecting cell-bound u-PA activity [39]. In in vitro migration assays on collagen-coated surfaces, it was furthermore observed that SMC migration was significantly (p < 0.005) reduced for MMP-3-/- SMC as compared to WT [40]. Plasmin-mediated MMP-3 activity may thus play a role in SMC migration, an important contributor to neointima formation after vascular injury.
MMP-3 also specifically interacts with the main inhibitors of the fibrinolytic system. Thus MMP-3 specifically hydrolyzes the Met374-Ser375 (P3-P2) peptide bond in human alpha2-antiplasmin (alpha2-AP), the main physiological plasmin inhibitor. Cleavage is completely abolished in the presence of the MMP inhibitors EDTA or 1,10-phenanthroline. At enzyme/substrate ratio of 1 : 10 at 37°C, alpha2-AP protein cleavage occurs with a half-life of 8 min, and is associated with rapid loss of inhibitory activity towards plasmin with a half-life of 5 min. alpha2-AP cleaved by MMP-3 does no longer form a stable complex with plasmin, as shown by SDS-PAGE, and does no longer interact with plasminogen, as shown by crossed immunoelectrophoresis with plasminogen added to the gel. These data are compatible with the removal of a COOH-terminal fragment containing the reactive site peptide bond and the plasmin(ogen)-binding site. In addition, MMP-3 cleaves the Pro19-Leu20 peptide bond in alpha2-AP thereby removing the fibrin-binding site from the inhibitor. A dysfunctional alpha2-AP variant (Ala-alpha2-AP or alpha2-AP Enschede), with an alanine insertion in the reactive site sequence converting it from a plasmin inhibitor into a substrate, was also efficiently cleaved by MMP-3 (half-life of 13 min at 37°C and enzyme/substrate ratio of 1 : 10). Cleavage and inactivation of alpha2-AP by MMP-3 may constitute a mechanism favoring local plasmin-mediated proteolysis [41].
Furthermore, MMP-3 specifically hydrolyzes the Ser337-Ser338 (P10-P9) and Val341-Ile342 (P6-P5) peptide bonds in human plasminogen activator inhibitor-1 (PAI-1) [42]. Cleavage is completely abolished in the presence of the metal chelators EDTA or 1,10-phenanthroline. A stabilized active PAI-1 variant was also cleaved by MMP-3. At enzyme/substrate ratio of 1 : 10 at 37°C, PAI-1 protein cleavage occurred with half-lives of 27 or 14 min for active or stable PAI-1, and was associated with rapid loss of inhibitory activity towards tissue-type plasminogen activator with half-lives of 15 or 13 min, respectively. A substrate-like variant of PAI-1, lacking inhibitory activity but with exposed reactive site loop, was cleaved with a half-life of 23 min, whereas latent PAI-1 in which a major part of the reactive loop is inserted into the molecule, was resistant to cleavage. Biospecific interaction analysis indicated comparable binding of active, stable and substrate PAI-1 to both proMMP-3 and MMP-3 (Ka of 12 to 22*106 M-1), whereas binding of latent PAI-1 occurred with lower affinity (1.7 to 2.3*106 M-1). Stable PAI-1 bound to vitronectin was cleaved and inactivated by MMP-3 in a comparable manner as free PAI-1; the cleaved protein did, however, not bind to vitronectin. Cleavage and inactivation of PAI-1 by MMP-3 may thus constitute a mechanism decreasing the antiproteolytic activity of PAI-1 and impairing the potential inhibitory effect of vitronectin-bound PAI-1 on cell adhesion and/or migration.
The effect of MMP-3 (stromelysin-1), MMP-7 (matrilysin), MMP-9 (gelatinase B), or MMP-12 (metalloelastase) on cellular fibrinolytic activity was also studied with the use of smooth muscle cells (SMC) and fibroblasts derived from mice with specific inactivation of these genes [40]. Activation of cell-bound plasminogen by two-chain urokinase-type plasminogen activator (tcu-PA) was not significantly different with SMC or fibroblasts from the gene-deficient mice (78 to 140% of wild-type). For all cell types very limited conversion of plasminogen to angiostatin-like kringle-containing fragments was observed (<3% of the total cell-bound plasminogen). Activation of plasminogen in solution by cell-associated tcu-PA was also comparable for SMC or fibroblasts of the different genotypes (54 to 160% of wild-type). In vitro SMC migration on scrape wounded collagen-coated surfaces was comparable for wild-type, MMP-7-/-, MMP-9-/-, and MMP-12-/- SMC, but was significantly reduced for MMP-3-/- SMC (p < 0.005 versus wild-type). Serum-free conditioned medium of MMP-3-/- and MMP-7-/- SMC or fibroblasts induced similar lysis of fibrin films as wild-type cells.
These findings indicate that several interactions that have been described above between these MMPs and the plasminogen/plasmin system in a purified system, do not significantly affect plasmin-mediated cellular fibrinolytic activity under cell culture conditions.
CELLULAR PROTEOLYTIC ACTIVITY AND NEOINTIMA FORMATION
Proteinases from the plasminogen/plasmin and the MMP system may participate in the proliferation and migration of SMC, and in matrix remodeling during arterial wound healing [43-45]. To assess the role of the plasminogen/plasmin system in neointima formation, a perivascular electric injury model was applied to mice with targeted inactivation of the main components of the system [46]. These studies revealed that the degree and the rate of arterial neointima formation after injury was significantly reduced in u-PA-/- and Pg-/- mice, as compared to WT and t-PA-/- mice [46, 47] apparently as a result of impaired migration of SMC and leukocytes from the uninjured border into the central injured region. These data thus substantiate a physiological role of u-PA-mediated plasmin proteolysis in SMC migration and neointima formation. However, alpha2-antiplasmin deficiency in mice did not significantly affect SMC migration and neointima formation after vascular injury [48].
The temporal and topographic expression pattern of MMPs (MMP-2, -3, -9, - 12, -13) in this model appeared to be compatible with a role in neointima formation [49]. Indeed, electron microscopic immunogold labeling and immunofluorescence confocal microscopy revealed gradually enhanced expression during vessel remodeling, with the highest levels obtained 1 to 2 weeks after injury, thus coinciding with neointima formation [50]. In non-injured femoral arteries and in injured arteries after 1 week, MMP-2 and MMP-3 were produced by adventitial cells which were not stained for alpha-actin (fibroblasts and/or non-contractile SMC), whereas two weeks after injury, when the SMC have regained their contractile phenotype, MMP-2 and MMP-3 were also detected in some alpha-actin stained cells. MMP-9, MMP-12, and MMP-13 were found in macrophages located mainly in the adventitia and only occasionally in subintimal areas. MMP-2, MMP-3, and MMP-9 levels were always somewhat higher in WT than in u-PA-/- mice, with the intensity of positive immunostaining being proportional to the number of producing cells. Furthermore, the increased labeling of MMP-2 and MMP-3 on the route of their secretory pathway reflects a high level of synthesis. A high amount of MMP-2 remains associated with the cell surface suggesting the presence of a specific binding site, possibly one of the membrane-type metalloproteinases. Interestingly, MMP-2 was found in high concentration extracellularly, associated with the internal elastic lamina and with elastin bundles in the neointima, suggesting that it plays an active role in the degradation of elastin-containing structures. In contrast, virtually no extracellular MMP-9 was detected. Total MMP-9 levels (active plus latent) also markedly increased after injury, reaching a maximum at about one week after injury both in WT and u-PA-/- mice. Active MMP-9 levels were consistently detected in WT arteries after injury, but were very low to undetectable in u-PA-/- arteries. MMP-3 expression after injury gradually increased to reach a maximum after about two weeks. It was produced mainly by fibroblasts (and smooth muscle cells) in the adventitia, where it was found in typical storage granules and was extracellularly associated with the basement membrane and occasionally with collagen fibers. The presence of MMP-3 in storage granules suggests the existence of an intracellular storage pool, which could be released by non-identified triggers. MMP-12 and MMP-13 expression after injury appeared somewhat lower in u-PA-/- than in WT arteries, in agreement with lower amounts of producing macrophages. Highest levels were also observed about two weeks after injury. All three macrophage-secreted MMPs (MMP-9, MMP-12 and MMP-13) were found in intracellular storage granules, suggesting a pathway controlling their acute release when required.
Because of this temporal and topographic expression pattern of MMPs after vascular injury, arterial neointima formation after vascular injury was also studied in mice with deficiency of TIMP-1 [50]. At one to three weeks after injury, the intimal areas were significantly larger in TIMP-1-/- mice as compared to wild-type mice, and contained abundant SMC, whereas the medial areas were comparable, resulting in significantly higher intima/media ratios in the TIMP-1-/- mice. These data thus confirm a physiological role of TIMP-1 in vascular remodeling, most likely via monitoring of MMP activity. These data are compatible with the finding that stromelysin mRNA antisense oligonucleotides inhibited phenotypic modulation of rat arterial SMC and thereby caused a decrease in migration and proliferation and in neointima formation after vessel wall injury [51].
Stromelysin-3 (MMP-11), a unique member of the matrix metalloproteinase (MMP) family, which does not directly cleave the main components of the extracellular matrix, also appears to play a role in neointima formation after vascular injury [6]. Indeed, neointima formation two to three weeks after electric injury of the femoral artery was significantly enhanced in MMP-11-/- as compared to MMP-11+/+ mice, both in a pure 129SV genetic background and in a 50/50 mixed 129SV/BL6 background. The medial areas were comparable, resulting in intima/media ratios that were significantly increased in MMP-11-/- as compared to MMP-11+/+ arteries, both in the 129SV and in the mixed background. Nuclear cell counts in cross-sectional areas of the intima of the injured region were higher in MMP-11-/- as compared to MMP-11+/+ arteries. Immunocytochemical analysis revealed that alpha-actin-positive and CD45-positive cells were more abundant in intimal sections of MMP-11-/- mice. Degradation of the internal elastic lamina was more extensive in MMP-11-/- than in MMP-11+/+ arteries. The mechanisms by which MMP-11 could impair elastin degradation and cellular migration in this model remain, however, enigmatic [6].
Studies with gene-deficient mice have allowed novel interactions between the fibrinolytic and MMP systems to be established. Smooth muscle cell migration and neointima formation after vascular injury appears to be promoted by several MMP system components, which may be activated via plasmin-dependent or plasmin-independent mechanisms.
REFERENCES
1.Lijnen, H. R., and Collen, D. (1995)
Baillière's Clin. Haematol., 8, 277-290.
2.Lijnen, H. R., and Collen, D. (1999) Thromb.
Haemost., 82, 837-845.
3.Lijnen, H. R. (2000) Fibrinolysis
Proteolysis, 14, 175-181.
4.Carmeliet, P., Moons, L., Herbert, J.-M., Crawley,
J., Lupu, F., Lijnen, H. R., and Collen, D. (1997) Circ. Res.,
81, 829-839.
5.Carmeliet, P., Moons, L., Lijnen, H. R., Baes, M.,
Lemaître, V., Tipping, P., Drew, A., Eeckhout, Y., Shapiro, S.,
Lupu, F., and Collen, D. (1997) Nature Genet., 17,
439-444.
6.Lijnen, H. R., van Hoef, B., Vanlinthout, I.,
Verstreken, M., Rio, M. C., and Collen, D. (1999) Arterioscler.
Thromb. Vasc. Biol., 19, 2863-2870.
7.Nagase, H., and Woessner, J. F., Jr.
(1999) J. Biol. Chem., 274, 21491-21494.
8.Okada, Y., Gonoji, Y., Naka, K., Tomita, K.,
Nakanishi, I., Iwata, K., Yamashita, K., and Hayakawa, T. (1992) J.
Biol. Chem., 267, 21712-21719.
9.Suzuki, K., Enghild, J. J., Morodomi, T., Salvesen,
G., and Nagase, H. (1990) Biochemistry, 29,
10261-10270.
10.Eeckhout, Y., and Vaes, G. (1977) Biochem.
J., 166, 21-31.
11.He, C. S., Wilhelm, S. M., Pentland, A. P.,
Marmer, B. L., Grant, G. A., Eisen, A. Z., and Goldberg, G. I. (1989)
Proc. Natl. Acad. Sci. USA, 86, 2632-2636.
12.Baramova, E. N., Bajou, K., Remacle, A., L'Hoir,
C., Krell, H. W., Weidle, U. H., Noel, A., and Foidart, J. M. (1997)
FEBS Lett., 405, 157-162.
13.Keski-Oja, J., Lohi, J., Tuuttila, A.,
Tryggvason, K., and Vartio, T. (1992) Exp. Cell Res.,
202, 471-476.
14.Lijnen, H. R., Silence, J., van Hoef, B., and
Collen, D. (1998) Blood, 91, 2045-2053.
15.Ogata, Y., Enghild, J. J., and Nagase, H. (1992)
J. Biol. Chem., 267, 3581-3584.
16.Brew, K., Dinakarpandian, D., and Nagase, H.
(2000) Biochim. Biophys. Acta, 1477, 267-283.
17.Lijnen, H. R., Silence, J., Lemmens, G.,
Frederix, L., and Collen, D. (1998) Thromb. Haemost., 79,
1171-1176.
18.Hajjar, K. A., Hamel, N. M., Harpel, P. C., and
Nachman, R. L. (1987) J. Clin. Invest., 80,
1712-1719.
19.Hajjar, K. A., Harpel, P. C., Jaffe, E. A., and
Nachman, R. L. (1986) J. Biol. Chem., 261,
11656-11662.
20.Miles, L. A., and Plow, E. F. (1985) J. Biol.
Chem., 260, 4303-4311.
21.Stephens, R. W., Pöllänen, J.,
Tapiovaara, H., Leung, K. C., Sim, P. S., Salonen, E. M., Ronne, E.,
Behrendt, N., Dano, K., and Vaheri, A. (1989) J. Cell Biol.,
108, 1987-1995.
22.Miles, L. A., and Plow, E. F. (1988)
Fibrinolysis, 2, 61-71.
23.Plow, E. F., Freaney, D. E., Plescia, J., and
Miles, L. A. (1986) J. Cell Biol., 103, 2411-2420.
24.Miles, L. A., Dahlberg, C. M., Levin, E. G., and
Plow, E. F. (1989) Biochemistry,28, 9337-9343.
25.Miles, L. A., Dahlberg, C. M., Plescia, J.,
Felez, J., Kato, K., and Plow, E. F. (1991) Biochemistry,
30, 1682-1691.
26.Hajjar, K. A., and Nachman, R. L. (1988) J.
Clin. Invest., 82, 1769-1778.
27.Félez, J., Miles, L. A., Fabregas, P.,
Jardi, M., Plow, E. F., and Lijnen, H. R. (1996) Thromb.
Haemost., 76, 577-584.
28.Barnathan, E. S., Kuo, A., van der Keyl, H.,
McCrae, K. R., Larsen, G. R., and Cines, D. B. (1988) J. Biol.
Chem., 263, 7792-7799.
29.Hajjar, K. A., Jacovina, A. T., and Chacko, J.
(1994) J. Biol. Chem., 269, 21191-21197.
30.Ellis, V., Behrendt, N., and Danø, K. (1991)
J. Biol. Chem., 266, 12752-12758.
31.Ellis, V., Scully, M. F., and Kakkar, V. V.
(1989) J. Biol. Chem., 264, 2185-2188.
32.Ellis, V., Wun, T. C., Behrendt, N., Ronne, E.,
and Dano, K. (1990) J. Biol. Chem., 265, 9904-9908.
33.Lee, S. W., Ellis, V., and Dichek, D. A. (1994)
J. Biol. Chem., 269, 2411-2418.
34.Ellis, V., Whawell, S. A., Werner, F., and
Deadman, J. J. (1999) Biochemistry, 38, 651-659.
35.Chavakis, T., Kanse, S. M., Yutzy, B., Lijnen, H.
R., and Preissner, K. T. (1998) Blood, 91, 2305-2312.
36.May, A. E., Kanse, S. M., Chavakis, T., and
Preissner, K. T. (1998) Fibrinolysis Proteolysis, 12,
205-210.
37.Ugwu, F., van Hoef, B., Bini, A., Collen, D., and
Lijnen, H. R. (1998) Biochemistry, 37, 7231-7236.
38.Lijnen, H. R., Ugwu, F., Bini, A., and Collen, D.
(1998) Biochemistry, 37, 4699-4702.
39.Ugwu, F., Lemmens, G., Collen, D., and Lijnen, H.
R. (1999) Thromb. Haemost., 82, 1127-1131.
40.Ugwu, F., Lemmens, G., Collen, D., and Lijnen, H.
R. (2001) Thromb. Res., 102, 61-69.
41.Lijnen, H. R., van Hoef, B., and Collen, D.
(2001) Biochim. Biophys. Acta, 1547, 206-213.
42.Lijnen, H. R., Arza, B., van Hoef, B., Collen,
D., and Declerck, P. J. (2000) J. Biol. Chem., 275,
37645-37650.
43.Carmeliet, P., and Collen, D. (1988) Thromb.
Res.,91, 255-285.
44.Dollery, C. M., McEwan, J. R., and Henney, A. M.
(1995) Circ. Res., 77, 863-868.
45.Celentano, D. C., and Frishman, W. H. (1997)
J. Clin. Pharmacol., 150, 761-776.
46.Carmeliet, P., Moons, L., Ploplis, V., Plow, E.,
and Collen, D. (1997) J. Clin. Invest., 99, 200-208.
47.Carmeliet, P., Moons, L., Herbert, J.-M.,
Crawley, J., Lupu, F., Lijnen, R., and Collen, D. (1997) Circ.
Res., 81, 829-839.
48.Lijnen, H. R., van Hoef, B., Dewerchin, M., and
Collen, D. (2000) Arterioscler. Thromb. Vasc. Biol., 20,
1488-1492.
49.Lijnen, H. R., Lupu, F., Moons, L., Carmeliet,
P., Goulding, D., and Collen, D. (1999) Thromb. Haemost.,
81, 799-807.
50.Lijnen, H. R., van Hoef, B., Soloway, P., and
Collen, D. (1999) Circ. Res., 85, 1186-1191.
51.Lovdahl, C., Thyberg, J., Cercek, B., Blomgren,
K., Dimayuga, P., Kallin, B., and Hultgardh-Nilsson, A. (1999)
Histol. Histopathol., 14, 1101-1112.