REVIEW: Urokinase as a Multidomain Protein and Polyfunctional Cell Regulator
V. V. Stepanova1* and V. A. Tkachuk1,2
1Russian Cardiology Research Center, Ministry of Health of Russian Federation, Cherepkovskaya ul. 15, Moscow, 121552 Russia; fax: (095) 414-6719; E-mail: V.Stepanova@cardio.ru2School of Fundamental Medicine, Lomonosov Moscow State University, Moscow, 119899 Russia
* To whom correspondence should be addressed.
Received July 23, 2001; Revision received October 12, 2001
The urokinase type plasminogen activator (urokinase) plays a pivotal role in the regulation of cell adhesion and migration during tissue remodeling. Urokinase not only specifically cleaves plasminogen and converts it into plasmin but also activates intracellular signaling upon binding to certain receptors on the cell surface. The polyfunctional properties of this protein are associated with its three-domain structure as follows: the C-terminal proteolytic domain containing the serine protease active center, the central kringle domain, and the N-terminal domain homologous to epidermal growth factor. This review considers functional properties of urokinase and of its fragments generated on the cell surface as a result of proteolytic processing. This review will discuss the mechanisms of urokinase-mediated regulation of cellular function upon binding to membrane receptors.
KEY WORDS: urokinase, adhesion, migration, tissue remodeling, endocytosis, intracellular signaling
Abbreviations: uPA) urokinase-like plasminogen activator; tPA) tissue-like plasminogen activator; uPAR/CD87) glycosyl phosphatidylinositol-anchored urokinase receptor; PAI-1) first type inhibitor of plasminogen activators; LRP/alpha2-MR) receptor related to the low density lipoprotein receptor, or alpha2-macroglobulin receptor; LDLR) low density lipoprotein receptors; GPI) glycosyl phosphatidylinositol.
Extracellular proteolysis plays a key role in blood coagulation,
fibrinolysis, vascular remodeling, angiogenesis, wound healing, and
growth and metastasizing of malignant tumors. These proteases are
located on the cell surface due to specific interaction with
membrane-bound proteins and are responsible for degradation of the
pericellular matrix components and for destruction of cell-cell
contacts. Thus, these proteases remove fibrin deposits and also provide
the cell invasion/migration within tissues.
The urokinase-type and tissue-type plasminogen activators (uPA, or urokinase, and tPA, respectively) are important components of the extracellular protease system because they specifically convert plasminogen into plasmin. Plasmin is a serine protease with a wide substrate specificity. Upon the generation of intravascular thrombi mainly constituted of polymeric fibrin, the system of plasminogen activators is triggered to recover the blood flow, and the plasmin directly degrades fibrin that leads to the thrombus dissolving. The plasminogen activators are believed to be key components of the fibrinolysis system. On the cell surface plasmin activates a number of metalloproteinases that degrade the extracellular matrix proteins and the components of basal membrane, such as collagen, fibronectin, and laminin. The combined effects of the plasminogen activators, plasmin, and metalloproteinases on the plasma membrane promote a vector cell movement due to destruction of the cell-cell contacts and the matrix and also due to the activation or releasing of latent or matrix-bound growth factors possessing chemotactic properties.
The tissue-type plasminogen activator is mainly involved in fibrinolysis, but the urokinase-type plasminogen activator, in addition to the fibrinolytic function, plays the most important role in cell migration and tissue remodeling.
On the cell surface urokinase binds to the high affinity receptor (uPAR/CD87) which is located on the leading edge of the migrating cells. The binding of urokinase to the receptor provides a strictly local proteolysis of the extracellular matrix proteins in the direction of the cell movement. Moreover, the urokinase-receptor complex activates the intracellular signaling and thus regulates the cell adhesion, migration, and proliferation. Furthermore, the urokinase receptor and its specific inhibitor (PAI-1) can interact with adhesion receptors, extracellular matrix proteins, and also with proteins which mediate the activation of intracellular signaling. These findings allowed to consider the system of plasminogen activators as a separate group of regulators of cell movement and communication which provide both proteolytic and “signaling” functions.
The present review considers mechanisms of the urokinase involvement in cell adhesion and migration.
THE STRUCTURE AND PROCESSING OF UROKINASE
Urokinase is synthesized by vascular endothelial and smooth muscle cells (SMC), by epithelial cells, fibroblasts, monocytes/macrophages, and also by cells of malignant tumors of different origin [1-3]. This protein is secreted by cells as a single-chain polypeptide with molecular weight of 54 kD that consists of 411 amino acid residues [4]. The urokinase structure is subdivided into three domains: the N-terminal domain homologous to the epidermal growth factor (amino acids 9-45), the kringle domain (amino acids 46-143), and the C-terminal catalytic domain (amino acids 144-411) (Fig. 1). The “growth factor-like domain” (GFD) is responsible for the interaction of urokinase with the uPAR/CD87 receptor [5]. The protease domain of urokinase includes the active site of the enzyme represented by a specific for serine proteases amino acid triad His204, Asp255, and Ser356. The kringle domain of urokinase contains a sequence that interacts with the specific inhibitor PAI-1 [6]. It has also been found that urokinase binds to heparin through the kringle domain, but the functional significance of this interaction is yet unclear [7]. We have found that the kringle domain binds on the cell surface to a specific receptor that is distinct from uPAR/CD87 [8]. The interaction of urokinase with this receptor results in the migration of SMC, fibroblasts, and of a number of other cells [8, 9]. According to data of nuclear magnetic resonance, the urokinase has extensive, but not unrestricted, motion between the different domains [10, 11]. Each domain of the protein has a rigid structure supported by internal disulfide bonds, three of which are located in the “growth” factor-like domain, three in the kringle domain, and six bonds are located in the proteolytic domain. Intramolecular disulfide bonds maintain the due orientation of amino acid residues in the active center of urokinase proteolytic domain, since the loss of the enzymatic activity was observed after the destruction or irregular formation of S-S-bonds [12].
During the post-translational modification, carbohydrate residues can be incorporated in two regions of the urokinase molecule. N-Glycosylation of Asn302 in the proteolytic domain and O-glycosylation of Thr18 in the growth factor-like domain increase by 10-15% the molecular weight of the urokinase. The role of urokinase glycosylation has not been determined conclusively, but some data suggest that the Asn302-glycosylated urokinase is more susceptible to activation by plasmin and is more resistant to inhibitors [13]. The deglycosylation of the Thr18 residue within the growth factor-like domain of uPA can cause the loss of the mitogenic properties, despite its unaltered affinity for uPAR/CD87 [14].Fig. 1. Structure of single-chain and two-chain urokinase forms. G, growth factor-like domain; K, kringle domain; P, protease domain.
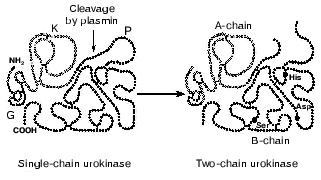
The single-chain urokinase has no peptidase activity to synthetic substrates. Therefore, this urokinase form was for a long time considered as the enzyme precursor, or prourokinase [15, 16]. However, prourokinase can convert plasminogen into plasmin. Plasmin, in its turn, activates urokinase because it cleaves the Lys158-Ile159 peptide bond and converts the single-chain urokinase into the two-chain form (Fig. 1). The two-chain urokinase reveals protease activity with respect to both synthetic substrates and plasminogen, and the rate of plasminogen cleavage by the two-chain urokinase is more than 200-fold higher than the rate of cleavage by the single-chain form [17]. Other extracellular proteases, such as kallikrein, blood coagulation factor XIIa, and cathepsin B, also cleave the Lys158-Ile159 bond of urokinase. In the two-chain urokinase the polypeptide chains A and B (light and heavy chains, respectively) are connected by the Cys148-Cys279 disulfide bond. The A-chain includes the “growth” and the kringle domains, whereas the proteolytic domain is a part of the B-chain. Upon cleavage of the Lys158-Ile159 bond a newly generated amino terminal tail of the B-chain with Ile159 on the end is relocated into the region of the substrate-binding pocket of the protease. This is associated with the formation of an ion pair between the NH2-group of the Ile159 and the side chain of Asp255. These conformational changes result in the opening of the substrate-binding pocket and, possibly, of the active site of the enzyme [18].
A 32-kD urokinase was isolated from adenoma cell culture [19]. This low-molecular-weight form of the protein is a single-chain fragment of urokinase containing amino acids 144-411. The enzymatic properties of this fragment are virtually the same as the properties of the full-length urokinase. The low-molecular-weight two-chain urokinase is generated after the cleavage of the Lys135-Lys136 bond by plasmin in the region between the kringle and protease domains, and its proteolytic activity is comparable to the activity of the full-length two-chain urokinase [20]. Because the urokinase cleavage in this region can separate the A- and B-chains, it is obvious that in addition to the low-molecular-weight urokinase an amino terminal fragment of the protein is generated [21]. Nevertheless, up to now the amino terminal fragment is prepared by the cleavage of the full-length urokinase by protease V8 [22]. Moreover, in studies on urokinase processing by plasmin we have shown that the amino terminal fragment is a minor product among the polypeptides produced. The urokinase cleavage products within varied time intervals were analyzed by SDS-electrophoresis with subsequent N-terminal sequencing. The formation of the two-chain urokinase by the cleavage of the Lys158-Ile159 bond was followed by the cleavage of the Lys46-Ser47 bond in the region between the growth factor-like and the kringle domains. This resulted in the elimination of the growth factor-like domain and in the generation of a two-chain urokinase form with molecular weight of 36-40-kD lacking the growth factor-like domain. More prolonged exposure to plasmin resulted in the elimination of the kringle domain and in the generation of a 32-kD urokinase [23]. These findings suggest that in addition to the proteolytic activation, plasmin can sequentially split off the N-terminal domains of urokinase with production of several proteolytically active forms (Fig. 2). The urokinase form lacking the growth factor-like domain is also generated on the cell surface under plasmin action [23].
Proteolysis of the full-length urokinase by thrombin proceeds quite differently. Thrombin hydrolyzes the Arg156-Phe157 peptide bond that results in formation of a two-chain urokinase variant that is proteolytically inactive and fails to be activated by other proteases (Fig. 2). Such processing is a mechanism for inactivation of the enzyme [24, 25]. However, the resulting inactive form probably has other functions on the cell surface that are not associated with proteolysis.Fig. 2. Urokinase fragments generated upon proteolytic processing of urokinase on the cell surface. G, growth factor-like domain; K, kringle domain; P, protease domain.
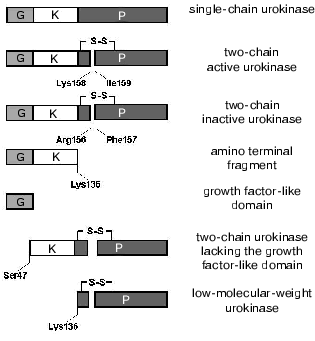
Thus, a number of bonds in urokinase undergo proteolytic cleavage with formation of several variants of the protein on the cell surface (Fig. 2). The full-length, single-, and two-chain forms of urokinase and its amino terminal fragment can bind to the uPAR/CD87 receptor on the cell surface. We have shown that urokinase lacking the growth factor-like domain is unable to interact with the uPAR/CD87 and, consequently, can bind to the surface of SMC and of other cells either through the kringle domain or through the protease domain (see below). This suggests the presence of additional urokinase receptors on the plasma membrane. It seems that the urokinase fragments generated upon its proteolytic processing by extracellular proteases can affect cellular function independently or in cross-talk with uPAR/CD87.
LOCALIZATION OF UROKINASE ON THE CELL SURFACE
Many cell types contain the specific receptor uPAR/CD87, which binds urokinase with high affinity (Kd ~ 10-10-10-9 M). The urokinase receptor was first identified on human monocytes and on U937 line cells [26]. The mature receptor consists of 283 amino acids and has molecular weight 55-60 kD [27, 28]. The receptor molecule contains several N-glycosylation sites, and the weight of sugar residues comprises about half of the molecular weight of the mature protein [29]. The N-terminal growth factor-like domain of urokinase is responsible for its binding to uPAR/CD87 [5]. Chemical modification and site-specific mutagenesis showed that the Omega-loop (amino acid residues 22-30) within the growth factor-like domain of urokinase contains the key amino acids important for the interaction with the receptor, namely, Asn22, Asn27, His29, and Trp30 [29-32]. Other components of the blood coagulation system and fibrinolysis that contain the epidermal growth factor-like domains/repeats fail to interact with uPAR/CD87.
uPAR/CD87 contains three homologous sequence repeats/domains (D1, D2 and D3), which are not very similar in primary structure but possess a conserved pattern of cysteine residues. An internal repetition of the spacing of cysteines in polypeptide chain is present also in some other single-domain proteins of the Ly-6 receptor family [33-35].
The first, N-terminal domain of the receptor is shown to play the key role in the binding of urokinase [36]. However, to provide a high affinity receptor-ligand interaction, all three domains of the receptor are required because the apparent urokinase binding affinity of isolated first receptor domain is 2000-fold lower than that of the intact receptor [37]. Photoaffinity labeling has also shown that the sites of the urokinase contact with the receptor are located not only in the first but also in the two other domains of uPAR/CD87 [29, 31]. N-Glycosylation of the Asn52 residue also affected the affinity of the receptor-ligand interaction, since the recombinant receptor lacking the sugar residue at the Asn52 had lower affinity for urokinase than the native glycosylated protein [38].
The urokinase receptor lacks a transmembrane sequence and is anchored in the plasma membrane by a glycosyl phosphatidylinositol (GPI) moiety. During the post-translational processing, 30 amino acids are removed from the C-end of the receptor precursor, and the GPI-anchor covalently binds to the Gly283 residue [38, 39]. Thus, the urokinase receptor was long considered to be a “trap”, or a protein which localizes urokinase in discrete areas of the cell surface. Due to the GPI-anchorage, the urokinase receptor has high mobility in the plasma membrane, and its location depends on the functional state of the cell. In fact, if in the resting cell uPAR is uniformly distributed on the surface, the migrating cell forms clusters of the urokinase receptors on the leading edge [40]. The concentration of the proteolytic potential provides the vector movement of the cell along the chemoattractant gradient.
Similarly to other GPI-anchored proteins, the urokinase receptor is concentrated in special intrusions of the plasma membrane, caveolae, which contain great amounts of glycosphingolipids, sphingomyelin, polyphosphoinositols, and cholesterol. The structure of these formations is thought to be maintained by the membrane protein caveolin [41]. This protein is thought to form a hairpin-like structure within the membrane in such a way that the C-terminal and the N-terminal domains face the cytoplasm. Cells which do not express caveolin form on their plasma membrane the cholesterol-enriched flat clusters, so-called rafts, which contain the GPI-anchored proteins.
The function of uPAR is not limited by the locating of urokinase on the cell surface. The receptor-bound single-chain urokinase is activated by plasmin more efficiently than the free urokinase [42].
We have shown that uPAR can protect bound urokinase from the further degradation by plasmin. Thus, plasmin-mediated elimination of the growth factor-like domain in urokinase takes place more slowly if it is bound to uPAR. Unlike the full-length protein, urokinase lacking the growth factor-like domain is unable to interact with uPAR and undergoes rapid endocytosis and intracellular degradation [23]. Thus, uPAR increases the “half-life” of the functionally active urokinase on the cell surface.
An abundance of experimental data suggests that urokinase binding to the uPAR/CD87 receptor on the plasma membrane activates intracellular signaling systems that regulate cell migration, adhesion, proliferation, and differentiation (see below). Since, unlike most of classic receptors, uPAR lacks the transmembrane and cytoplasmic domains, alone it is not capable to initiate intracellular signaling. An obligatory partner(s) is probably required which associate(s) with uPAR or urokinase and transduce(s) the signal across the plasma membrane.
The single- and two-chain urokinase forms also bind to receptors of the low density lipoprotein receptors (LDLR) family: to the LDLR-relative protein/alpha2-macroglobulin receptor (LRP/alpha2-MR) [43-45], and to the very low density lipoprotein receptor (VLDLR) [46]. These receptors provide the clearance of the various protease-inhibitor complexes from the cell surface by endocytosis through clathrin-coated pits (see below). Despite the significant difference in the structure, these receptors consist of the same structural motifs. They are transmembrane proteins which pierce the membrane once, with the N-terminus faced into the extracellular space and the C-terminal end directed into the cytoplasm. These receptors are built from four main blocks: the cysteine-enriched repeats in the ligand-binding domain, the repeats homologous to the epidermal growth factor, the transmembrane domain, and one or several copies of the signal sequence N-P-X-Y located in the cytoplasmic domain and responsible for the receptor internalization through coated pits. The affinity of these receptors for urokinase (Kd ~ (1-2)*10-8 M) is an order of magnitude lower than affinity of uPAR/CD87. Since LRP/alpha2-MR interacts with urokinase via sites located in its A-chain, it is suggested that uPAR and LRP/alpha2-MR should compete for the binding of urokinase. It is suggested that high affinity uPAR-urokinase interaction could prevent binding of uPA to LRP/alpha2-MR and subsequent intracellular degradation (see below) [47].
Since not only full-length single- or two-chain urokinase forms capable of binding to uPAR (see above) could associate with the cell surface, it seems that other receptors could interact with truncated urokinase forms via their kringle or protease domains. In the search for such receptors, we have isolated a membrane protein with molecular weight 200 kD (p200) that binds urokinase via its proteolytic domain. Consequently, unlike uPAR/CD87 or LRP/alpha2-MR, this receptor can bind the truncated urokinase fragments lacking the growth factor-like and kringle domains. The structure of this protein is not yet established; however, we have found that it participates in the process of urokinase internalization (see below).
We have also shown the presence of a receptor on the surface of SMC and of some other cells which binds urokinase through its kringle domain. This receptor is also not studied in detail, but we have shown its key role in the urokinase-induced activation of cell migration [8].
Thus, through the different domains urokinase can specifically interact with a number of target receptors that can regulate the functional activity of cells.
MECHANISMS OF REGULATION OF CELL FUNCTION BY UROKINASE
Cell adhesion, migration, and proliferation underlie tissue remodeling. These processes in a large extent depend on urokinase, which generates plasmin on the cell surface. Wide substrate specificity allows plasmin to digest some matrix proteins or activate a number of metalloproteases, which, in their turn, further degrade the components of the extracellular matrix. Matrix proteins are the ligands of the integrin receptors, which are being associated with the intracellular signaling systems regulate cytoskeleton rearrangements, adhesive contacts, and chemotaxis. Since urokinase modifies extracellular matrix environment, it affects the integrin-regulated cell function. Apart the plasminogen urokinase either directly or via plasmin can activate or release from extracellular matrix a number of growth factors: hepatocyte growth factor (HGF/SF), vascular endothelial growth factor (VEGF165), transforming growth beta-factor (TGF-beta), fibroblast growth factor (FGF-2) [1, 48, 49]. These growth factors bind to their receptors on the cell surface and activate intracellular signaling pathways that regulate the cell behavior.
There is much evidence suggesting that in addition to providing extracellular proteolysis, urokinase upon binding to the membrane receptors activates intracellular signaling. Urokinase-induced cytoskeleton rearrangements and redistribution of adhesive contacts affect cell adhesion and migration that may be unrelated to proteolysis.
The signaling effects of urokinase are suggested to be mediated by uPAR/CD87, LRP.alpha2-MR, or other membrane uPA-binding proteins. It was shown that urokinase-induced cell migration is associated with the activation of Src- and Janus-kinases [50-52]. It has been also demonstrated that uPAR/CD87 can be coprecipitated with the following tyrosine protein kinases: Hck, Fyn, Lyn, Frg, Jak1, and Tyk2 [50, 52-54]. uPAR-dependent chemotaxis could be observed on the wild-type cells, but not on the cells with the Src gene knockout [55]. It has been also shown that the heterotrimeric GTP-binding proteins (G-proteins) mediate the urokinase-induced cell chemotaxis [52].
Urokinase activates a number of signaling pathways that regulate cytoskeleton rearrangements. Occupancy of uPAR by urokinase was also shown to result in activation of Hsk kinase, focal adhesion kinase (FAK), paxillin, the mitogen-activated protein kinase (MAP-kinase), and to promote phosphorylation of p130CAS protein and of DNA-binding activators of transcription STAT-1 and STAT-2 [51, 52, 56-58]. It was observed that occupancy of uPAR/CD87 by urokinase was followed by activation of protein kinase Cepsilon (PKCepsilon) and serine phosphorylation of cytokeratins 8 and 18 [59]. Since PKCepsilon regulates Raf-kinase activity [60], it is reasonable to suggest that urokinase binding to uPAR could result in activation of this kinase family.
At present, it is suggested that urokinase binding to and endocytosis via LRP/alpha2-MR or VLDLR can induce intracellular signaling. The binding of urokinase and of some other ligands to LRP/alpha2-MR results in an increase in the intracellular concentration of cAMP with subsequent activation of protein kinase A. Inhibition of this response by cholera toxin suggested the involvement of Gs-protein. The alpha-subunit of Gs-protein was shown to coprecipitate with LRP/alpha2-MR [61]. It was demonstrated that interaction of LRP/alpha2-MR with the cytoplasmic adaptor protein Disabled-1 (Dab-1) through the sequence N-P-X-Y results in association with and activation of Src and Abl nonreceptor tyrosine kinases [62, 63] and of the microtubule stabilizing protein tau [64]. LDL receptor family members are also involved in the activation of MAP-kinases and in the regulation of cell adhesion [65].
The role of uPAR/CD87 in urokinase-induced intracellular signaling is usually studied using the following forms: the active full-length urokinase, the enzyme irreversibly inactivated with diisopropyl fluorophosphate, the low-molecular-weight form consisting only of the protease domain, and also the amino terminal fragment which consists of the growth factor-like and the kringle domains. In most cases the ability to activate the intracellular signaling and cell migration was inherent to the urokinase fragments possessing the growth factor-like domain, which were able to bind the uPAR/CD87. Since only one high affinity urokinase receptor was known, the ability to transduce a signal across membrane was attributed toward the uPAR/CD87, even despite the fact that it lacks the transmembrane and the cytoplasmic domains. The coupling of the uPAR/CD87 with the intracellular signaling machinery was thought to occur by the way of interaction of the urokinase-occupied receptor with certain transmembrane proteins.
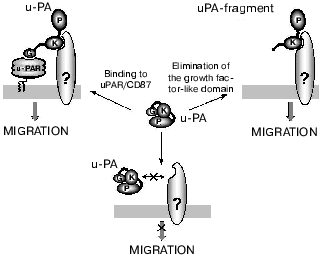
We have shown that the kringle domain of urokinase mediates its chemotactic properties, and the kringle domain, the kringle-containing urokinase fragments, and the full-length uPA elicited similar chemotactic effects. The deletion of the growth factor-like domain from uPA, and, as a consequence, abolishing its ability to bind uPAR/CD87, did not abolish its chemotactic properties. The low-molecular-weight form of urokinase lacking both the growth factor-like and kringle domains failed to activate cell migration. Thus, it was shown that the kringle domain of urokinase can be involved in the induction of cell chemotaxis by uPA even in the absence of uPAR/CD87. Our data indicated the presence of two different classes of binding sites for urokinase. The high affinity binding sites represent binding of the uPA growth factor-like domain to the uPAR/CD87, whereas the binding to the low affinity site(s) is mediated by the urokinase kringle domain [8]. It seems that the major mechanism responsible for uPA-induced cell motility involves signaling dependent on kringle interaction with definite membrane target. We found that the specific activation of the p38 MAP kinase cascade was required for the uPA's kringle-induced migration, and inhibition of this signaling pathway blocked both the uPA and uPA's kringle-stimulated cell migration [66]. In fact, the chemotactic effects of the uPA forms lacking the growth factor-like domain are independent of uPAR/CD87, and the kringle domain-containing uPA variants are chemotactic even for uPAR-deficient cells. However, ability of the full-length urokinase to induce chemotaxis in uPAR-expressing cells requires both the kringle domain binding sites (kringle-associating receptor) and the sites which bind the growth factor-like domain (uPAR). Based on our findings, we suggest that the growth factor-like domain in urokinase could shield the effector site on the kringle domain which is responsible for binding/activation of the kringle-associating receptor. It is feasible to suggest that the effector site within the kringle domain could be unmasked either when the growth factor-like domain in the full-length urokinase binds to uPAR/CD87 (activation of cell migration by full-length uPA), or upon proteolytic elimination of the growth factor-like domain in urokinase (cell migration activated by the growth factor-like domain-deficient urokinase form) (Fig. 3). Recall that a urokinase fragment lacking the growth factor domain could be generated on the cell surface when acted upon by a plasmin (see above).
Despite a steadily increasing flow of reports describing signaling effects of urokinase, precise mechanisms of the signal transduction from the plasma membrane to the “executive” cell systems are not clear since adaptor transmembrane proteins that are capable of interaction with urokinase or with its known receptors on one hand, and with the cellular interior on the other hand have not been identified. A possible candidate for such a partner is the integrin family of adhesion receptors. It was reported that uPAR/CD87 co-precipitates [53] and is co-localized [67] on the cell surface with the integrin Mac-1 (complement receptor 3 also denoted as CR3 or CD11b/CD18) of the beta2-integrin family. A physical association between Mac-1 and uPAR/CD87 on leukocytes has been confirmed by the methods of immunolocalization and resonance energy transfer [68, 69]. It was shown that uPAR/CD87-Mac-1 interaction is inhibited by addition of N-acetyl-D-glucosamine, this suggesting a lectin-like interaction of these receptors [70]. A region in the alpha-subunit of the Mac-1 which interacts with the uPAR/CD87 molecule has been recently localized [71]. This region (amino acid residues 400-424) is located close to the fibrinogen-binding I-domain of Mac-1. And the inhibitory effect of uPAR/CD87 on fibrinogen binding to Mac-1 could be explained by the closeness of the contact sites. Functional analysis has shown that Mac-1 can mediate the signaling effects of the urokinase receptor [72]. The interaction of uPAR/CD87 with Mac-1 is not permanent and depends on the functional state of the cell. Thus, in resting neutrophils these receptors are co-localized and randomly distributed throughout the cell surface, but dissociate as cells polarize to migrate and uPAR/CD87 is moving to the lamelipodia of the cells and Mac-1 to the uropodia [69]. On other cell types, uPAR/CD87 appears to be stably associated with beta1- and beta3-integrins [70, 73-75]. Together these data account for presence of uPAR at focal adhesion sites [76, 77] at which integrins accumulate and interact with the cellular cytoskeleton [40]. However, the interaction of uPAR/CD87 with integrins explains only partially the mechanisms which underlie the urokinase-induced cell adhesion and migration.Fig. 3. Putative mechanism that describes the involvement of uPAR/CD87 and of the kringle-associating receptor in urokinase-induced cell migration. G, growth factor-like domain; K, kringle domain; P, protease domain; uPAR, urokinase receptor uPAR/CD87; ?, kringle-associating receptor; uPA, urokinase.
uPAR is also a high-affinity receptor for the extracellular matrix protein vitronectin. Vitronectin-binding integrins alphanubeta3-, alphanubeta5 and uPAR provide adhesion of cells on vitronectin. In addition vitronectin binds to urokinase inhibitor PAI-1 and stabilizes its inhibitory activity serving as an extracellular depot of the functionally active uPA inhibitor. It was shown that PAI-1 and uPAR compete for binding to vitronectin [78-80]. The vironectin-binding site on uPAR is distinct from the urokinase-binding site. Further, binding of urokinase to uPAR strongly promotes vitronectin binding by uPAR [79-81]. We recently demonstrated that in vascular SMC uPA/uPAR are functionally associated with the ecto-protein kinase CK2 (casein kinase 2) [82]. Cell surface-located CK2 is capable of phosphorylating the vitronectin. We found that vitronectin is selectively phosphorylated by CK2 in a uPA/uPAR-dependent manner. It seems that the phosphorylated vitronectin is a better ligand for integrins and uPAR when compared to unphosphorylated form. Our results indicate that the uPA-dependent cell adhesion is a function of selective vitronectin phosphorylation by the ecto-kinase CK2, which activity appears to be regulated by uPA. We also reported that uPAR/CD87 and CK2 form a functional complex with the shuttle protein nucleolin. Nucleolin is an abundant nuclear phosphoprotein that shuttles between the nucleus and cytoplasm and that also can locate on the cell surface. We demonstrated that uPA can induce cell proliferation through the activation of the complex, that includes uPAR/CD87, CK2 and nucleolin [82].
Thus, the polyfunctional feature of urokinase could be attributed to its multidomain structure and hence this protein possesses several recognizing sites both for the substrates and for different receptors on the cell surface.
INACTIVATION AND REMOVAL OF UROKINASE FROM THE CELL
SURFACE
Several specific inhibitors inactivate two-chain urokinase on the cell surface: the PAI-1, the PAI-2, the protease nexin-1 (PN-1), and the protein C inhibitor [83-86]. They belong to the superfamily of serpins (serine proteinase inhibitors) that function by acting as suicide substrates or pseudosubstrates and form an irreversible covalent complex with their target protease. PAI-1 is one of the main inhibitors of urokinase. Inhibition of urokinase by PAI-1 is followed by the endocytosis and intracellular degradation of the protease-inhibitor complex. Kinetic analyses of the interaction between uPA and PAI-1 suggest that the interaction between the two proteins is a two-step mechanism. The first reaction is a rapid and reversible association of urokinase and inhibitor. The urokinase molecule possesses several reversible PAI-1 binding sites. One of these sites is located on the kringle domain (amino acids 91-94) and two others on the C-terminal region (amino acids 373-380 and 386-390) [6]. During a second step the inhibitor forms the irreversible covalent complex with the urokinase. The uPAR-bound urokinase-inhibitor complex is internalized through clathrin-coated pits with the aid of receptors of the LDLR family. The LDLR-relative protein, or alpha2-macroglobulin receptor (LRP/alpha2-MR) [43-45], glycoprotein 330, or megalin (gp300/megalin) [87], and the receptor of very low density lipoproteins (VLDLR) are members of the LDLR family [46]. When uPAR-bound uPA is complexed to the inhibitor, the complex is immediately internalized and degraded in the lysosomes.
The LRP/alpha2-MR, gp330/megalin, and VLDLR are multiligand receptors and can bind the lactoferrin, lipoprotein lipase, and exotoxin A of Pseudomonas aeruginosa, and the complexes of the proteases with alpha2-macroglobulin, the complexes of the plasminogen activators with their inhibitors [62, 87-91]. The single-chain urokinase binds to uPAR/CD87, then is turned into the proteolytically active two-chain form, and finally complexes with inhibitors such as PAI-1. Then the resulting tripartite complex uPAR/uPA/PAI-1 binds with high affinity (~0.5-1 nM) to the LDLR family receptors and is internalized [44, 92]. In the endosomes, urokinase-inhibitor complex dissociates from the uPAR/CD87 and is degraded in lysosomes [92, 93], whereas uPAR/CD87 is recycled back to the cell surface [47]. The sites of a contact between the uPAR/uPA/PAI-1 complex and the LRP/alpha2-MR are located on the PAI-1 molecule and on the urokinase within its catalytic domain and the amino terminal fragment [94, 95]. It was demonstrated that the single- and two-chain urokinase forms bound to the LRP/alpha2-MR can be internalized and degraded even in the absence of PAI-1 [88], but urokinase inactivation by PAI-1 significantly facilitates the endocytosis [93, 96, 97].
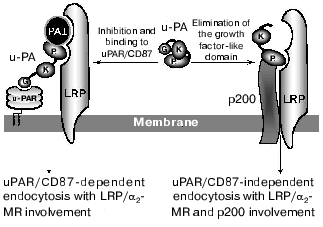
Considering that the urokinase degradation products, among them those unable to bind to uPAR/CD87, could occur on the cell surface, it is feasible to suggest the additional mechanism that mediates the clearance of the truncated urokinase forms without contribution of the uPAR. We have demonstrated that urokinase via its PD interacts with the surface protein with mass of 200 kD (p200). Using a competitive binding approach we found that the p200 along with the LRP/alpha2-MR is involved in binding and endocytosis of the intact uPA and uPA forms lacking the growth factor-like domain regardless of uPAR/CD87. When we compared the rates of internalization of the full-length urokinase and the growth factor-like domain-lacking form we found that the latter undergoes more efficient intracellular degradation [23]. The “classic” endocytosis of the urokinase, mediated by uPAR/CD87 and LRP/alpha2-MR, seems to proceed at lower rate if compared with the rate of urokinase internalization and degradation with the participation of the p200. Thus, we demonstrate the existence of an additional clearance mechanism for urokinase that could proceed without contribution of the uPAR (Fig. 4). This mechanism can be triggered either when uPAR/CD87 receptors are saturated and the excess of uPA still persists or when the urokinase fragments failing to bind to uPAR should be removed from the cell surface. It should also be reminded that the urokinase fragments lacking the growth factor-like domain are chemotactic for the cells through the kringle-dependent mechanism (see above). We suggest that the newly discovered pathway of urokinase endocytosis might be utilized by cell either for protection themselves from excessive proteolytic attack of the active urokinase fragments or for modulation of urokinase-mediated regulatory signals.
The complexity and diversity of urokinase-activated signaling pathways suggests the existence of several effector proteins that mediate its cellular effects. Due to multidomain structure urokinase seems to be able to bring several receptors into a united functional complex providing the cross-talk of regulatory signals. On the other hand, it might be suggested that urokinase fragments generated upon its proteolytic processing on the cell surface can bind to their own target receptors and provide the physiological responses. Different expression levels of receptors capable of binding urokinase or its fragments, the presence or absence of uPAR/CD87, and also the combination of urokinase-activated signaling pathways seem to play important roles in regulation of cellular function.Fig. 4. Mechanisms of endocytosis of full-length urokinase and of its fragments (see text). G, growth factor-like domain; K, kringle domain; P, protease domain; uPAR, urokinase receptor uPAR/CD87; uPA, urokinase; PAI-1, type 1 plasminogen activator inhibitor; LRP/alpha2-MR, LDLR-related protein/alpha2-macroglobulin receptor.
REFERENCES
1.Tkachuk, V., Stepanova, V., Little, P. J., and
Bobik, A. (1996) Clin. Exp. Pharmacol. Physiol., 23,
759-765.
2.Clowes, A. W., Clowes, M. M., Au, Y. P. T., Reidy,
M. A., and Belin, D. (1990) Circ. Res., 67, 61-67.
3.Eaton, D. L., Scott, R. W., and Baker, J. B. (1984)
J. Biol. Chem., 259, 6241-6247.
4.Holmes, W. E., Pennica, D., Blaber, M., Rey, M. W.,
Guenzler, W. A., Steffens, G. L., and Heyneker, H. L. (1985)
Biotechnology, 3, 923-929.
5.Appella, E., Robinson, E. A., Ullrich, S. J.,
Stoppelli, M. P., Corti, A., Cassani, G., and Blasi, F. (1987) J.
Biol. Chem., 262, 4437-4440.
6.Mimuro, J., Kaneko, M., Murakami, T., Matsuda, M.,
and Sakata, Y. (1992) Biochim. Biophys. Acta, 1160,
325-334.
7.Stephens, R. W., Bokman, A. M., Myohanen, H. T.,
Reisberg, T., Tapiovaara, H., Pedersen, N., Gröndahl-Hansen, J.,
Llinas, M., and Vaheri, A. (1992) Biochemistry, 31,
7572-7579.
8.Mukhina, S., Stepanova, V., Traktouev, D.,
Poliakov, A., Beabealashvilly, R., Gursky, Ya., Minashkin. M.,
Shevelev, A., and Tkachuk, V. (2000) J. Biol. Chem.,275,
16450-16458.
9.Poliakov, A. A., Mukhina, S. A., Traktouev, D. O.,
Bibilashvily, R. S., Gursky, Y. G., Minashkin, M. M., Stepanova, V. V.,
and Tkachuk, V. A. (1999) J. Recept. Signal Transduct. Res.,
19, 939-951.
10.Nowak, U. K., Li, X., Teuten, A. J., Smith, R.
A., and Dobson, C. M. (1993) Biochemistry,
32,298-309.
11.Bogusky, M. J., Dobson, C. M., and Smith, R. A.
(1989) Biochemistry, 28, 6728-6735.
12.Kobayashi, H., Gotoh, J., Hirashima, Y., Fujie,
M., Sugino, D., and Terao, T. (1995) J. Biol. Chem., 270,
8361-8366.
13.Lenich, C., Pannell, R., Henkin, J., and
Gurewich, V. (1992) Thromb. Haemost., 68, 539-544.
14.Rabbani, S. A., Mazar, A. P., Bernier, S. M.,
Haq, M., Bolivar, I., Henkin, J., and Goltzman, D. (1992) J. Biol.
Chem., 267, 14151-14156.
15.Nolan, C., Hall, L. S., Barlow, G. H., and
Tribby, I. I. E. (1977) Biochim. Biophys. Acta, 496,
384-400.
16.Wun, T. C., Ossowski, L., and Reich, E. (1982)
J. Biol. Chem., 257, 7262-7268.
17.Lijnen, H. R., Zamarron, C., Blaber, M., Winkler,
M. E., and Collen, D. (1990) J. Biol. Chem., 265,
5232-5236.
18.Goldberg, G. I., Frisch, S. M., He, C., Wilhelm,
S. M., Reich, R., and Collier, I. E. (1990) Ann. N. Y. Acad.
Sci., 580, 375-384.
19.Stump, D. C., Lijnen, H. R., and Collen, D.
(1986) J. Biol. Chem., 261, 17120-17126.
20.Stump, D. C., Thienpont, M., and Collen, D.
(1986) J. Biol. Chem., 261, 1267-1273.
21.Kobayashi, O., Matsui, K., Minamiura, N., and
Yamamoto, T. (1985) J. Biochem. (Tokyo), 97, 37-44.
22.Mazar, A. P., Buko, A., Petros, A., Barnathan, E.
S., and Henkin, J. (1992) Fibrinolysis, 6 (Suppl. 1),
49-55.
23.Poliakov, A., Tkachuk, V., Ovchinnikova, T.,
Potapenko, N., Bagryantsev, S., and Stepanova, V. (2001) Biochem.
J.,355,639-645.
24.Ichinose, A., Fujikawa, K., and Suyama, T. (1985)
J. Biol. Chem., 261, 3486-3489.
25.Braat, E. A., Levi, M., Bos, R., Haverkate, F.,
Lassen, M. R., de Maat, M. P., and Rijken, D. C. (1999) J. Lab.
Clin. Med., 134,161-167.
26.Vassali, J.-D., Baccino, D., and Belin, D. (1985)
J. Cell Biol., 100, 86-92.
27.Behrendt, N., Rønne, E., Ploug, M., Petri,
T., Løber, D., Nielsen, L. S., Schleuning, W.-D., Blasi, F.,
Appella, E., and Danø, K. (1990) J. Biol. Chem.,
265, 6453-6460.
28.Ploug, M., Rønne, E., Behrendt, N., Jensen,
A. L., Blasi, F., and Danø, K. (1991) J. Biol. Chem.,
266, 1926-1933.
29.Ploug, M. (1998) Biochemistry, 37,
16494-16505.
30.Quax, P. H., Grimbergen, J. M., Lansink, M.,
Bakker, A. H., Blatter, M. C., Belin, D., van Hinsbergh, V. W., and
Verheijen, J. H. (1998) Arterioscler. Thromb. Vasc. Biol.,
18, 693-701.
31.Ploug, M., Rahbek-Nielsen, H., Ellis, V.,
Roepstorff, P., and Dano, K. (1995) Biochemistry, 34,
12524-12534.
32.Magdolen, V., Rettenberger, P., Koppitz, M.,
Goretzki, L., Kessler, H., Weidle, U. H., Konig, B., Graeff, H.,
Schmitt, M., and Wilhelm, O. (1996) Eur. J. Biochem.,
237,743-751.
33.Shevach, E. M., and Korty, P. E. (1989)
Immunol. Today, 10, 195-200.
34.Ploug, M., Kjalke, M., Ronne, E., Weidle, U.,
Hoyer-Hansen, G., and Dano, K. (1993) J. Biol. Chem.,
268, 17539-17546.
35.Sugita, Y., Nakano, Y., Oda, E., Noda, K., Tobe,
T., Miura, N. H., and Tomita, M. (1993) J. Biochem. (Tokyo),
114,473-477.
36.Behrendt, N., Ploug, M., Patthy, L., Houen, G.,
Blasi, F., and Dano, K. (1991) J. Biol. Chem., 266,
7842-7847.
37.Ploug, M., Ellis, V., and Dano, K. (1994)
Biochemistry, 33,8991-8997.
38.Moller, L. B., Pollanen, J., Ronne, E., Pedersen,
N., and Blasi, F. (1993) J. Biol. Chem., 268,
11152-11159.
39.Moller, L. B., Ploug, M., and Blasi, F. (1992)
Eur. J. Biochem., 208,493-500.
40.Andreasen, P. A., Kjoller, L., Christensen, L.,
and Duffy, M. J. (1997) Int. J. Cancer, 72, 1-22.
41.Monier, S., Parton, R. G., Vogel, F., Behlke, J.,
Henske, A., and Kurzchalia, T. V. (1995) Mol. Biol. Cell,
6, 911-927.
42.Ellis, V., Scully, M. F., and Kakkar, V. V.
(1989) J. Biol. Chem., 264, 2185-2188.
43.Herz, J., Clouthier, D. E., and Hammer, R. E.
(1992) Cell, 71,411-421.
44.Conese, M., Olson, D., and Blasi, F. (1994) J.
Biol. Chem., 269, 17886-17892.
45.Conese, M., Nykjaer, A., Petersen, C. M.,
Cremona, O., Pardi, R., Andreasen, P. A., Gliemann, J., Christensen, E.
I., and Blasi, F. (1995) J. Cell Biol., 131,
1609-1622.
46.Argraves, K. M., Battey, F. D., MacCalman, C. D.,
McCrae, K. R., Gåfvels, M., Kozarsky, K. F., Chappell, D. A.,
Strauss, J. F., III, and Strickland, D. K. (1995) J. Biol.
Chem., 270, 26550-26557.
47.Nykjaer, A., Conese, M., Christensen, E. I.,
Olson, D., Cremona, O., Gliemann, J., and Blasi, F. (1997) EMBO
J., 16,2610-2620.
48.Plouet, J., Moro, F., Bertagnolli, S.,
Coldeboeuf, N., Mazarguil, H., Clamens, S., and Bayard, F. (1997) J.
Biol. Chem., 272, 13390-13396.
49.Saksela, O., and Rifkin, D. B. (1990) J. Cell
Biol., 110, 767-775.
50.Dumler, I., Weis, A., Mayboroda, O. A., Maasch,
C., Jerke, U., Haller, H., and Gulba, D. C. (1998) J. Biol.
Chem., 273, 315-321.
51.Dumler, I., Kopmann, A., Weis, A., Mayboroda, O.
A., Wagner, K., Gulba, D. C., and Haller, H. (1999) Arterioscler.
Thromb. Vasc. Biol., 19, 290-297.
52.Resnati, M., Guttinger, M., Valcamonica, S.,
Sidenius, N., Blasi, F., and Fazioli, F. (1996) EMBO J.,
15, 1572-1582.
53.Bohuslav, J., Horejsí, V., Hansmann, C.,
Stöckl, J., Weidle, U. H., Majdic, O., Bartke, I., Knapp, W., and
Stockinger, H. J. (1995) Exp. Med., 181, 1381-1390.
54.Konakova, M., Hucho, F., and Schleuning, W. D.
(1998) Eur. J. Biochem., 253, 421-429.
55.Fazioli, F., Resnati, M., Sidenius, N.,
Higashimoto, Y., Appella, E., and Blasi, F. (1997) EMBO J.,
16, 7279-7286.
56.Nguyen, D. H., Hussaini, I. M., and Gonias, S. L.
(1998) J. Biol. Chem., 273, 8502-8507.
57.Tang, H., Kerins, D. M., Hao, Q., Inagami, T.,
and Vaughan, D. E. (1998) J. Biol. Chem., 273,
18268-18272.
58.Dumler, I., Petri, T., and Schleuning, W.-D.
(1993) FEBS Lett., 322, 37-40.
59.Busso, N., Masur, S. K., Lazega, D., Waxman, S.,
and Ossowski, L. (1994) J. Cell Biol.,126, 259-270.
60.Cai, H., Erhardt, P., Troppmair, J., Diaz-Meco,
M. T., Sithanandam, G., Rapp, U. R., Moscat, J., and Cooper, G. M.
(1993) Mol. Cell. Biol., 13, 7645-7651.
61.Goretzki, L., and Mueller, B. M. (1998)
Biochem. J., 336 (Pt. 2), 381-386.
62.Willnow, T. E., Nykjaer, A., and Herz, J. (1999)
Nat. Cell Biol., 1, E157-E162.
63.Howell, B. W., Gertler, F. B., and Cooper, J. A.
(1997) EMBO J., 16, 121-132.
64.Hiesberger, T., Trommsdorff, M., Howell, B. W.,
Goffinet, A., Mumby, M. C., Cooper, J. A., and Herz, J. (1999)
Neuron, 24,481-489.
65.Gotthardt, M., Trommsdorff, M., Nevitt, M. F.,
Shelton, J., Richardson, J. A., Stockinger, W., Nimpf, J., and Herz, J.
(2000) J. Biol. Chem., 275, 25616-25624.
66.Goncharova, E. A., Tkachuk, V. A., Ratner, E. I.,
Parfyonova, Ye. V., and Vorotnikov, A. B. (2001) Zh. Evol. Biokhim.
Fiziol.,36, 569-575.
67.Xue, W., Kindzelskii, A. L., Todd, R. F., III,
and Petty, H. R. (1994) J. Immunol., 152, 4630-4640.
68.Sitrin, R. G., Pan, P. M., Harper, H. A., Todd,
R. F., III, Harsh, D. M., and Blackwood, R. A. (2000) J.
Immunol., 165, 3341-3349.
69.Kindzelskii, A. L., Laska, Z. O., Todd, R. F.,
III, and Petty, H. R. (1996) J. Immunol., 156,
297-309.
70.Xue, W., Mizukami, I., Todd, R. F., III, and
Petty, H. R. (1997) Cancer Res., 57, 1682-1689.
71.Simon, D. I., Wei, Y., Zhang, L., Rao, N. K., Xu,
H., Chen, Z., Liu, Q., Rosenberg, S., and Chapman, H. A. (2000) J.
Biol. Chem., 275, 10228-10234.
72.Todd, R. F., and Petty, H. R. (1997) J. Lab.
Clin. Med., 129, 492-498.
73.May, A. E., Kanse, S. M., Lund, L. R., Gisler, R.
H., Imhof, B. A., and Preissner, K. T. (1998) J. Exp. Med.,
188,1029-1037.
74.Ghosh, S., Brown, R., Jones, J. C., Ellerbroek,
S. M., and Stack, M. S. (2000) J. Biol. Chem., 275,
23869-23876.
75.Wei, Y., Yang, X., Liu, Q., Wilkins, J. A., and
Chapman, H. A. (1999) J. Cell Biol., 144,1285-1294.
76.Pollanen, J., Hedman, K., Nielsen, L. S., Dano,
K., and Vaheri, A. (1988) J. Cell Biol., 106, 87-95.
77.Hebert, C. A., and Baker, J. B. (1988) J. Cell
Biol., 106, 1241-1247.
78.Deng, G., Curriden, S. A., Wang, S., Rosenberg,
S., and Loskutoff, D. J. (1996) J. Cell. Biol.,
134,1563-1571.
79.Hoyer-Hansen, G., Behrendt, N., Ploug, M., Dano,
K., and Preissner, K. T. (1997) FEBS Lett.,
420,79-85.
80.Kanse, S. M., Kost, C., Wilhelm, O. G.,
Andreasen, P. A., and Preissner, K. T. (1996) Exp. Cell. Res.,
224,344-353.
81.Wei, Y., Waltz, D. A., Rao, N., Drummond, R. J.,
Rosenberg, S., and Chapman, H. A. (1994) J. Biol. Chem.,
269,32380-32388.
82.Dumler, I., Stepanova, V., Jerke, U., Mayboroda,
O. A., Vogel, F., Bouvet, P., Tkachuk, V., Haller, H., and Gulba, D. C.
(1999) Curr. Biol., 9, 1468-1476.
83.Manchanda, N., and Schwartz, B. S. (1995) J.
Biol. Chem., 270, 20032-20035.
84.Reinartz, J., Schaefer, B., Bechtel, M. J., and
Kramer, M. D. (1996) Exp. Cell Res., 223,91-101.
85.Baker, J. B., Low, D. A., Simmer, R. L., and
Cunningham, D. D. (1980) Cell, 21,37-45.
86.Geiger, M., Huber, K., Wojta, J., Stingl, L.,
Espana, F., Griffin, J. H., and Binder, B. R. (1989) Blood,
74,722-728.
87.Stefansson, S., Kounnas, M. Z., Henkin, J.,
Mallampalli, R. K., Chappell, D. A., Strickland, D. K., and Argraves,
W. S. (1995) J. Cell Sci., 108, 2361-2368.
88.Kounnas, M. Z., Henkin, J., Argraves, W. S., and
Strikland, D. K. (1993) J. Biol. Chem., 268,
21862-21867.
89.Willnow, T., Goldshtein, J. L., Orth, K., Brown,
M., and Herz, J. (1992) J. Biol. Chem., 267,
26172-26180.
90.Orth, K., Willnow, T., Herz, J., Gething, M. J.,
and Sambrook, J. (1994) J. Biol. Chem.,269,
21117-21122.
91.Filippova, M. P., Bochkov, V. N., and Tkachuk, V.
A. (1998) Usp. Biol. Khim., 38, 115-141.
92.Olson, D., Pollanen, J., Høyer-Hansen, G.,
Rønne, E., Sakaguchi, K., Wun, T.-Ch., Appella, E., Danø, K.,
and Blasi, F. (1992) J. Biol. Chem., 267, 9129-9133.
93.Cubellis, M. V., Wun, T.-C., and Blasi, F. (1990)
EMBO J., 9, 1079-1085.
94.Nykjaer, A., Kjoller, L., Cohen, R. L., Lawrence,
D. A., Garni-Wagner, B. A., Todd, R. F., van Zonneveld, A. J.,
Gliemann, J., and Andreasen, P. A. (1994) J. Biol. Chem.,
269, 25668-25676.
95.Rodenburg, K. W., Kjoller, L., Petersen, H. H.,
and Andreasen, H. H. (1998) Biochem. J., 329 (Pt. 1),
55-63.
96.Stoppelli, M. P., Corti, A., Soffientini, A.,
Cassani, G., Blasi, F., and Assolian, R. K. (1985) Proc. Natl. Acad.
Sci. USA, 82, 4939-4943.
97.Nykjaer, A., Petersen, C. M., Møller, B.,
Jensen, P. H., Moestrup, S. K., Holtet, T. L., Etzerodt, M.,
Thøgersen, H. C., Munch, M., Andreasen, P. A., and Gliemann, J.
(1992) J. Biol. Chem., 267, 14543-14546.