Phosphonates and Their Degradation by Microorganisms
S. V. Kononova and M. A. Nesmeyanova*
Skryabin Institute of Biochemistry and Physiology of Microorganisms, Russian Academy of Sciences, Pushchino, Moscow Region, 142290 Russia; fax: (095) 956-3370; E-mail: aniram@ibpm.serpukhov.su* To whom correspondence should be addressed.
Received July 6, 2001
Phosphonates are a class of organophosphorus compounds characterized by a chemically stable carbon-to-phosphorus (C-P) bond. Wide occurrence of phosphonates among xenobiotics polluting the environment has aroused interest in pathways and mechanisms of their biodegradation. Only procaryotic microorganisms and the lower eucaryotes are capable of phosphonate biodegradation via several pathways. Destruction of the non-activated C-P bond by the C-P lyase pathway is of fundamental importance, and understanding of the process is a basic problem of biochemistry and physiology of microorganisms. This review offers analysis of available data on phosphonate-degrading microorganisms, degradation pathways, and genetic and physiological regulation of this process.
KEY WORDS: phosphonates, C-P bond, biodegradation, C-P lyase
Phosphorus plays a fundamental role in microbial cell physiology and biochemistry, being a part of such important biomolecules as phospholipids, nucleic acids, proteins, polysaccharides, as well as nucleotide cofactors involved in energy transport and catalysis of many cell processes. The vital importance of this nutrient for microbial cells is satisfied by consuming from the environment first of all the most available forms--orthophosphate and its easily hydrolyzed phosphate esters containing phosphorus in its highest oxidation state. However, more and more data show the ability of bacteria to utilize the more reduced organophosphorus compounds as phosphorus sources, in particular, phosphonates characterized by a single carbon-to-phosphorus (C-P) bond. In contrast to the more labile O-P, N-P, and S-P linkages, it is extremely resistant to chemical hydrolysis, thermal decomposition [1], and photolysis [2]. Phosphonates are widespread among naturally occurring compounds in all kingdoms of wildlife, but only procaryotic microorganisms are able to cleave this bond. Interest in microbial degradation of phosphonates has grown in recent years due to ecological problems. The matter is that organophosphorus compounds with the C-P bond are widespread among man-made chemical substances--xenobiotics, which uncontrollably enter the environment and become a toxic factor. The search for the ways of phosphonate biodegradation to understand molecular mechanisms of this process is an urgent basic problem of physicochemical biology that would also contribute to the solution of practical problems of environmental protection biotechnology. Unfortunately, current research into the physiology and biochemistry of phosphonate biodegradation and the ecology of microorganisms degrading phosphonates is insufficient, so a systematized analysis of the data available is particularly important for specifying the prospects and ways of solving the above problem.
PHOSPHONATES AND PHOSPHONATE-DEGRADING MICROORGANISMS
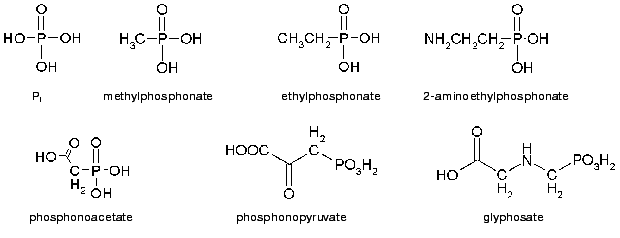
Phosphonates are a class of organophosphorus compounds with a C-P bond, which makes their molecules resistant to chemical hydrolysis and thermal decomposition [1]. Phosphonates occur widely among biogenic and man-made compounds. 2-Aminoethylphosphonic acid (2-AEP) found in flagellates from rumen protozoa [3] was the first known natural compound with the C-P bond. Further investigations established that 2-AEP is a constituent of lipids, named as phosphonolipids by analogy with phospholipids [4]. These compounds have been found in protozoa, flagellates, coelenterates, mollusks [5], the lower fungi [6], and even in man [7, 8]. Besides in lipids, 2-AEP was found to be a constituent of proteins [5, 9] and polysaccharides [10]. Other representatives of biogenic phosphonates are antibiotics synthesized by Streptomyces. They include phosphonomycin (1,2-cis-epoxyprolylphosphonic acid), an inhibitor of biosynthesis of UDP-N-acetylmuramic acid essential for microbial cell wall formation [11], and bialaphos (L-alanyl-L-alanyl-phosphinothricin) [12], an inhibitor of glutamine synthetase in Escherichia coli and plants[13, 14]. Biogenic phosphonates also include phosphonopyruvate [15] and phosphonoacetate (PA) [16]. It is suggested [17] that phosphonates, as well as phosphites, emerged at an early stage of the Earth's evolution and could be prebiotic phosphorus carriers. Phosphite radicals could serve as starting material for the formation of vinylphosphonic acid, which in turn could be an initial compound for the synthesis of phosphonoacetaldehyde, phosphonoacetic acid, and ethyl-, 1-hydroxyethyl-, and 2-hydroxyethylphosphonic acids. Phosphonoaldehyde could thus be formed in great amounts [18-20]. The presence of methyl-, ethyl-, and other alkylphosphonates in a sample of the Murchison meteorite [21] with the dominating methylphosphonate allowed the investigators to suggest that these compounds might also have occurred in high concentrations on the Earth in the initial period of life origin.
The first synthetic derivatives of phosphonic acids were obtained at the end of the 19th century, and their industrial synthesis became possible since 1905 after the discovery of the Arbuzov reaction [22]. At present, synthetic phosphonates are the basis of many xenobiotics and are widely used in different fields of human economic activity [16, 23-27]. They include the herbicide glyphosate, which is an inhibitor of 3-enolpyruvylshikimate-5-phosphatesynthase (EC 2.5.1.19)--an enzyme involved in the synthesis of aromatic amino acids [28]. Derivatives of ethyl- and phenylphosphonates are used as insecticides; alaphosphaline and phosphonomycin (biphosphonates) as antibiotics; cyclic esters of aromatic biphosphonates as polymer additives; Phyrol 67, an oligomer of vinylphosphonate-methylphosphonate, as a flame extinguisher; and polyaminopolyphosphonic acids as corrosion inhibitors [16]. Aminotri-(methylenephosphonic) and hydroxyethylidenediphosphonic acids are widely used as chelate additives to household detergents [24]. Phosphonic acids are used in scientific research [25, 26] and as drugs [27].
Man-made phosphonate derivatives include also toxic constituents of chemical weapon (VX, sarin, and soman) [29]. The first is alkylphosphonothiolate and the latter two are methylphosphonofluoride esters. The final products of their degradation are less toxic alkylphosphonic acids, which however are highly resistant to degradation in soil where they can be found decades later even more than 1 m deep [30]. When contaminating water reservoirs, they appear to be toxic for the inhabiting organisms: Daphnia magna, Selenastrum capricornutum, and flagellates [31]. Wide and uncontrollable use of phosphonate xenobiotics in economic activity resulted in significant contamination of the environment and thus created the problem of its protection and remediation. The structures of some of the simplest phosphonates are given in Fig. 1.
The structural diversity of phosphonate substrates requires different enzymatic systems for their degradation. The ability of microorganisms to utilize organophosphorus compounds with the C-P bond as the sole phosphorus source has been known for a long time [32-34]. The first evidence of biological cleavage of the C-P bond was obtained with E. coli [32]. This bacterium utilized methylphosphonic or ethylphosphonic acids as sole phosphorus sources. Probably, after depletion of easily assimilated phosphorus sources in the environment, in some cases it appeared to be available for microorganisms only as phosphites and phosphonates, and they therefore developed systems for catabolism (utilization) and conversion of the latter substances to more available phosphorus compounds. This is indirectly supported by molecular-genetic analysis of phosphite and hypophosphite oxidation in Pseudomonas stutzeri WM88 [35]. It suggests an interrelation between phosphonate metabolism and phosphite and hypophosphite metabolism because some genes of the operon encoding proteins responsible for the decomposition of these compounds are homologous to the genes responsible for the cleavage of the C-P bond in alkylphosphonates. Also, mutations disturbing phosphonate utilization affect also phosphite utilization [36, 37]. In addition to E. coli and Ps. stutzeri, utilization of phosphonates and phosphites was shown in Klebsiella aerogenes [38].Fig. 1. Structures of orthophosphate and some phosphonates.
Works published to date show wide occurrence of phosphonate-degrading microorganisms in nature, including Gram-positive and Gram-negative bacteria [33, 39-47] as well as some yeasts [40, 48, 49] and fungi [50-52]. However, only special strains but not certain groups of microorganisms are suggested to be capable of phosphonate degradation [40]. The attempt to close this gap was made, in particular, in the two papers cited below [39, 40]. Thus, the first work [39] presented the bacteria from five soil isolates that were able to degrade a wide group of structurally different phosphonates. These were first of all various strains of Pseudomonas and Bacillus megaterium decomposing 14 of 15 studied substrates, which is comparable with the broad substrate specificity revealed previously in Agrobacterium radiobacter [41]. This work showed for the first time that Gram-positive bacteria are able to directly degrade the C-P bond. However, neither of the isolates could degrade isopropylphosphonate or the phosphinate herbicide phosphinothricin. The second work assayed microorganisms from seven ecosystems and 19 laboratory microorganisms for the ability to grow on different natural and xenobiotic phosphonates as sole phosphorus sources. This study revealed the phosphonate degraders to occur among various bacterial species and systematic groups from both phosphonate polluted and non-polluted environmental sources, indicating their wider occurrence than had been supposed previously [53]. The work confirmed that eucaryotic organisms were incapable of phosphonate degradation and first revealed such ability in the photosynthetic organism Rhodobacter capsulatus and decomposition of polyphosphonic acids by microorganisms. Of course, the study of degrading ability of microorganisms is not limited to the above works.
Bacterial strains able to mineralize both natural and xenobiotic phosphonates as not only phosphorus but also nitrogen and carbon sources have been identified quite recently [46, 54]. These isolates possessed new inducible enzymes with high specificity to individual substrates and became good ground for departure from the previously adopted conception that phosphonates are utilized by organisms as the phosphorus source only [55]. There are two reports about utilization of phosphonates by yeast as a sole nitrogen source: natural 2-amino-3-phosphonopropionic acid (phosphonoalanine) by cells of Candida maltosa [48] and synthetic 4-aminobutylphosphonate by cells of Kluyveromyces fragilis [49]. In the latter case, phosphonate utilization did not depend on the phosphorus status of the cells, like it is in degradation of 2-AEP by cells of Ps. putida [46].
PHOSPHONATE METABOLISM
Biosynthesis of phosphonates has been studied by the example of 2-AEP to elucidate the mechanism of the C-P bond formation. It was shown that the first reaction in the pathway of the C-P bond formation is intramolecular restructuring of phosphoenolpyruvate to phosphonopyruvate (Fig. 2, III A), catalyzed by phosphoenolpyruvate-phosphomutase [15, 56]. Phosphonopyruvate under the action of enzyme phosphonopyruvate-decarboxylase is then converted to phosphonoacetaldehyde. Phosphonoacetaldehyde can be converted either to 2-hydroxypropylphosphonic acid during phosphonomycin biosynthesis or to hydroxymethylphosphonic acid during bialophos biosynthesis. Thus, phosphonopyruvate-decarboxylase is one of the key enzymes of biosynthesis of different phosphonates [57, 58]. In addition to the above, two more enzymes were found responsible for the formation of the C-P bond in bialophos: carboxyphosphonoenolpyruvatemutase [59, 60] and “P-methylating enzyme” [61, 62]. The finding of enzymes of C-P bond biosynthesis suggested that under appropriate conditions in a cell they could also realize back reactions, i.e., to participate in decomposition of the C-P bond. Indeed, as it was shown later, phosphoenolpyruvate-phosphomutase is involved in biodegradation of phosphonoalanine [46, 63]. It is suggested [64] that the ability to synthesize phosphonates could give microorganisms, due to the presence of the chemically stable and phosphatase-resistant C-P bond in these compounds, certain biological advantages for survival in a potentially phosphate-limited marine environment and in the struggle for ecological niches.
As regards catabolism of phosphonates, several enzymes are known to catalyze this process: phosphonatase (phosphonoacetaldehyde hydrolase, EC 3.11.1.1) involved in 2-AEP degradation [44], phosphonoacetate hydrolase degrading phosphonoacetate [45], phosphonopyruvate hydrolase [46], and C-P lyase [41] degrading the wide range of phosphonates in intact bacterial cells (Fig. 2).Fig. 2. Enzymes cleaving the C-P bond.
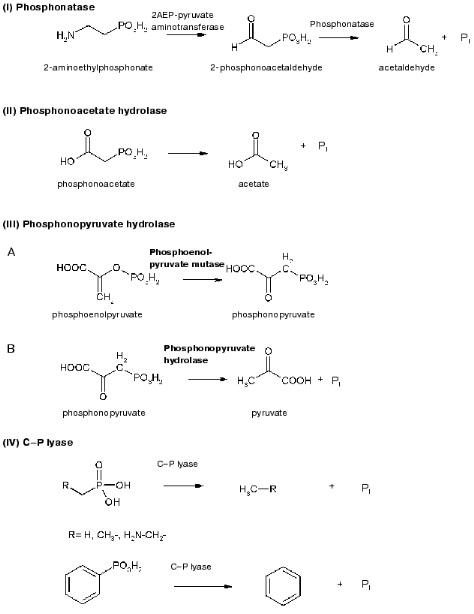
The first enzyme able to degrade the phosphorus-carbon salt, 2-phosphonoacetaldehyde hydrolase (phosphonatase), was identified and isolated from the Gram-positive bacterium B. cereus [44]. This bacterium was able to utilize 2-AEP as the sole phosphorus source and to degrade it to 2-phosphonoacetaldehyde using 2AEP-pyruvate aminotransferase (EC 2.6.1.37) [65] and then to degrade phosphonoacetaldehyde to acetaldehyde and orthophosphate [44, 66] in a reaction catalyzed by phosphonatase. The latter reaction cleaves the C-P bond and proceeds via the formation of a covalently bound imine intermediate with the carbonyl group and the side chain of lysine residue in the enzyme. Phosphonatase was isolated and purified, showed optimal activity at pH 8, needed Mg2+ for its activity, was inhibited by reagents degrading disulfide bonds, and consisted of two subunits with the molecular mass of 33-37 kD each [44]. Phosphonatase resembled alkaline phosphatase of E. coli in many properties [67] but was not able to degrade the wide range of phosphomonoesters hydrolyzed by phosphatase. The range of phosphonatase substrates was much less, and the enzyme was not a metalloenzyme in contrast to phosphatase. The pathway of degradation of 2-AEP was suggested to be present in all organisms capable of utilizing this substrate as a phosphorus source. Indeed, it was found in other microorganisms, e.g., Ps. aeruginosa [65], Salmonella typhimurium [68, 69], B. cereus [69], and Pseudomonas sp. 4ASW [70]. In addition to degradation of 2-AEP, phosphonatase can participate also in decomposition of glyphosate, as was shown in Arthrobacter atrocyanus [69]. The genes encoding phosphonatases from Salm. typhimurium [68, 69] and B. cereus [69] have been cloned and sequenced. Their analysis showed that phosphonatases belong to a new family of hydrolases having a highly conservative aspartate residue in their active center, to which the phosphoryl group from a lysine residue of the enzyme is being transferred [69]. The study of crystal structure of phosphonatase from B. cereus [71] showed that the enzyme is a homodimer. The occurrence of polar amino acid residues in active centers of dehalogenases, phosphonatases, phosphatases, and phosphomutases belonging to NAD-dependent superfamily indicates that active centers of these enzymes may be of common origin. The molecular and genetic characteristics of phosphonatase genes of Salm. typhimurium [69] and Enterobacter aerogenes [72] showed that they are members of the Pho regulon and, consequently, their expression is regulated by exogenous orthophosphate and proceeds only after Pi is depleted in the growth medium.
Phosphonoacetate hydrolase was found in Ps. fluorescens 23F [45]. This enzyme is induced by phosphonoacetate and does not need phosphorus starvation for its induction, and the products of its degradation--phosphate and acetate--are easily utilized by cell. The gene encoding phosphonoacetate hydrolase was cloned [73], and the enzyme was purified and characterized [74]. The enzyme consists of two identical subunits with the molecular mass of 40 kD. Analysis of the structural gene (phnA) of phosphonoacetate hydrolase showed five open reading frames [75]. 2-Phosphonopropionate was shown able also to induce this enzyme but is a bad substrate for it. Degradation of both substrates is supposed to require the expression of three genes: phnA, phnB, and phnR.
Burkholderia cepacia Pal6 is able to utilize L-phosphonoalanine as a source of nitrogen, carbon, or phosphorus, using the enzyme phosphonopyruvate hydrolase [46, 51] previously described as phosphoenolpyruvate phosphomutase. Phosphonopyruvate is a product of transamination of L-phosphonoalanine and is decomposed to pyruvate and Pi. Kinetic parameters of this enzyme were partially characterized, and the molecular mass of native protein was determined as equal to 232 kD [76]. These parameters are close to those of phosphoenolpyruvate phosphomutase purified from Ps. gladioli B-1 [77].
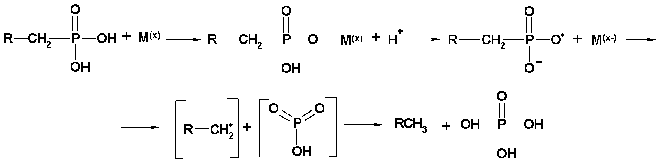
Finally, the enzyme C-P lyase is thought to catalyze the direct scission of inactivated C-P bonds to yield inorganic phosphate and the corresponding hydrocarbons. It is responsible for cleavage of alkylphoshponates, which are utilized as sole phosphorus sources by many bacteria. C-P lyase manifests its activity only in cells and has never been reliably found in cell-free extracts [78, 79] (testing of such activity in vitro turned out to be incorrect [80]). This considerably limits the possibility of understanding the mechanism of action of this enzyme. However, several hypothetical models for the mechanism of C-P lyase-catalyzed degradation of the C-P bond have been proposed [81, 82]. The first working hypothesis proposed that E. coli cells, growing in aerobic conditions, oxidize alkylphosphonic acids at the first carbon atom bound directly to phosphorus via incorporation of atomic or molecular oxygen in this position [81]. The resulting alpha-hydroperoxy, alpha-hydroxy, alpha-keto, or phophomonoesters could be easily utilized by cells. However, the study of degradation of alkylphosphonates by cells of this bacterium using isotopic-labeled material revealed no intermediates corresponding to the mechanism proposed. In fact, the main phenomenon of degradation of methylphosphonic acid by E. coli cells is predominant formation of methane as the final reaction product in a ratio of 1 : 1 to intracellular phosphorus formed from alkylphosphonates. Ethane, propane, butane, pentane, and hexane are formed by these cells using appropriate derivatives of phosphonic acid. A thorough study of degradation products of the above alkylphosphonates showed also the presence of ethene, propene, butene, etc., respectively. Results of the analysis of degradation products of alkylphosphonates do not thus fit the initially proposed mechanism and assume the possibility of another mechanism--a redox-dependent radical-based dephosphorylation, probably including also participation of transition metals in the reaction [82]. According to this mechanism (Fig. 3), the process is initiated by generation of a phosphonyl radical. Subsequent fragmentation of this reactive intermediate would lead to metaphosphate and alkyl moieties as the corresponding alkanes. Abstraction of a hydrogen atom by alkyl radical would yield the corresponding alkane. The formation of alkyl radical is a specific feature of alkylphosphonates degradation. The finding of not only alkanes but also alkenes among degradation products is evidence of the radical-based mechanism of degradation. It is yet unclear what is the final phosphorous product. It is assumed that monomer metaphosphoric acid forms orthophosphate when quickly reacting with water. However, the alternative assumption is also known, namely, that the catalysis of the C-P bond degradation involves ATP or other nucleotides [83]. Taking into account that alpha-1-ethylphosphono-ribose was among the products of degradation of alkylphosphonates, it is not improbable that it is precisely nucleotides that are acceptors of the phosphoryl group of alkylphosphonates. The radical-based mechanism of alkylphosphonate degradation is indirectly confirmed in a number of experimental works [83, 84]. Chemical modeling of such a process was performed using the reaction of alkylphosphonic acids with lead tetraacetate and their electrochemical oxidation using a platinum anode [82]. The formation of alkanes and alkenes as a result of bacterial degradation of alkylphosphonates and chemical degradation of alkylphosphonates with lead tetraacetate is an important link between biodegradation and the chemical model system, indicating the radical-based mechanism of phosphonate degradation via the C-P lyase pathway.
Fig. 3. Presumed mechanism of the C-P lyase action.
BACTERIA DEGRADING PHOSPHONATES BY THE C-P LYASE PATHWAY
With regard to the above, one can conclude that only microorganisms degrading alkylphosphonates with the formation of alkanes can be certainly attributed to microorganisms containing C-P lyase, though their substrate specificity may be not limited to alkylphosphonates. Some of the mentioned microorganisms are able to degrade other phosphonates as well, including glyphosate and 2-AEP (table).
Microorganisms degrading phosphonates by the C-P lyase pathway
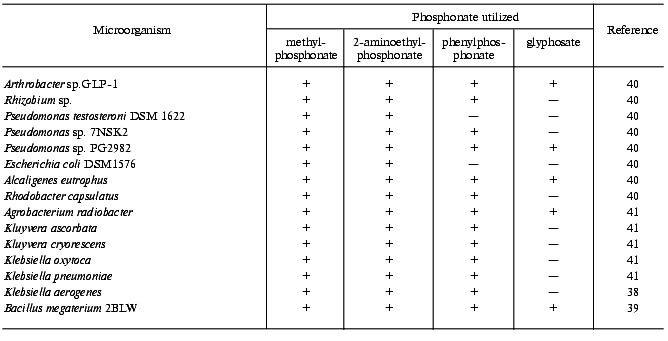
Most bacteria with exactly established degradation of phosphonates by the C-P lyase mechanism belong to the Gram-negative bacteria, but two representatives of the Gram-positive bacteria are also known: Arthrobacter sp. GLP-1 [42] and B. megaterium [39]. Among nearly forty identified soil isolates and collection bacterial strains tested for C-P lyase activity, only the mentioned Gram-positive bacteria possessed it. Other Gram-positive bacteria (Corynebacterium glutamicum MB-1789, Arthr. globiformis ATCC 8010, B. subtilis, B. cereus ATCC 13061, B. brevis ATCC 14579 [41], Arthr. globiformis DSM 20124, Arthr. paraffineus ATCC 21298, Arthr. paraffineus DSM 312, Arthr. oxydans DSM 420, Arthr. histidinolovorans DSM 20115, Arthr. variabilis DSM 20132, Arthrobacter sp. DSM 20389, Arthr. atrocyanus DSM 20217 [85], and Staphylococcus aureus [40]) showed no C-P lyase activity. One more representative of bacteria that degrade phosphonates by the C-P lyase pathway is Rhodobacter capsulatus, a phototrophic Gram-negative bacterium able to degrade the C-P bond under anaerobic conditions in the light [40]. The search for C-P lyase activity among fungi (Cladosporium herbarum, Fusarium culmorum, and Trichoderma viride [40]) gave no positive results.
Thus, the ability to degrade phosphonates by the C-P lyase mechanism occurs among Gram-negative bacteria much more often than among Gram-positive bacteria. These are representatives of the classes Arthrobacteriaceae, Bacillaceae, Rhodobacteriaceae, Alcaligenaceae, Pseudomonadaceae, Enterobacteriaceae (Escherichia, Enterobacter, Klebsiella, Kluyvera), and Rhizobiaceae (Rhizobium, Agrobacterium).
GENETIC CHARACTERIZATION OF PHOSPHONATE DEGRADATION BY THE C-P
LYASE PATHWAY
Recently, significant progress has been made in genetic characterization of the system of phosphonate degradation by microorganisms. Thus, mapping and molecular cloning of the phn genes responsible for C-P lyase activity, as well as mutation analysis, have been made in E. coli [37, 86-88]. Utilization of phosphonates by E. coli cells was shown to be encoded by a cluster of 14 genes--one of the largest transcription units of E. coli whose expression is controlled by Pho-regulon and exogenous orthophosphate [89]. Initially, the phn locus in the strain E. coli K12 was identified as a phosphate starvation-inducible gene, psiD, by constructing and analyzing Mud1 (bla lacZ) fusions and then renamed as phn after the insertion of Mu phage into the psiD locus had been shown to deprive the strain of the ability to grow in a medium containing methylphosphonic acid as the sole phosphorus source [87].
The fact that E. coli phn mutants cannot utilize alkylphosphonates as a sole phosphorus source resulted in the assumption that phn genes encode C-P lyase. The initial step of molecular-genetic study of this locus revealed a 15.6 kb fragment where 17 open reading frames denoted (in alphabetic order) the phnA to phnQ genes and five open reading frames in the opposite direction were identified [90].
Further analysis showed that the genes phnA, phnB, and phnQ do not code degradation of phosphonates [37]. Finally, the identified operon responsible for degradation of phosphonates in E. coli, 10.9 kb in size, consists of 14 genes phnCDEFGHIJKLMNOP localized close to 92.8 min of the chromosome map. It is evidently transcribed from the only promoter, immediately preceding the phnC gene. The genetic analysis [37] established the following.
1. Three gene products (PhnC, PhnD, and PhnE) are constituents of the alkylphosphonate transporter. The protein PhnD is hydrophilic and contains a signal sequence on its N-terminus. According to the mutation analysis of the analogous protein in Rhizobium meliloti, it is localized in the periplasm [91]. PhnC has two highly conservative sequences corresponding to the previously characterized nucleotide-binding domains. The search for homologous sequences using the database of the National Biomedical Research Foundation revealed a high homology between the above protein and some nucleotide-binding membrane domains of bacterial permeases, including those of PstB protein--a component of orthophosphate permease [92]. The protein PhnE is probably an integral membrane protein because it is highly hydrophobic. It has a certain similarity in sequence with integral membrane components RbsC and PstA of transport systems for ribose and inorganic phosphorus, depending on the corresponding metabolite-binding proteins.
2. Seven gene products (PhnG-PhnM) are assumed to participate in the catalysis of degradation of alkylphosphonates and are components of membrane associated C-P lyase complex. PhnM has a sequence that is similar to integral membrane components of the above transporters [37]. PhnL protein has nucleotide-binding sequences.
3. Two gene products (PhnN and PhnP) are not required for phosphonate use and may be accessory proteins for the C-P lyase; at the same time, the protein PhnN may have ATPase activity.
4. Two gene products (PhnF and PhnO) are unnecessary for the catalysis and play a regulatory role because they have a sequence in common with other regulatory proteins.
Thus, the molecular-genetic study suggests that the process of alkylphosphonate degradation involves a multi-component system with constituents localized in the membrane and periplasm, which previously was absolutely not considered in the search of C-P lyase activity in cell-free extracts and probably prevented identification of the enzyme.
Genes homologous to the phn genes of E. coli have also been found in Rhiz. meliloti [91, 93]. The expression of phn genes of Rhiz. meliloti cloned in plasmids revealed an enhanced synthesis of gene products 16, 22, 29 kD, corresponding to PhnG, PhnH, and PhnK components. These proteins are not subjected to posttranslational modification (signal peptide cleavage) and are expressed only in cells grown on methylphosphonate, aminomethylphosphonate, and glyphosate as sole phosphorus sources. It is interesting that the PhnI component of the C-P lyase complex of Rhiz. meliloti has a histidine residue on its C-terminus, in surroundings homologous to those of lipoxygenase of Oriza sativa, and may serve as a ligand for metals. Besides, PhnJ has four conservative cysteine residues, which can also be ligands for metals or sites for binding of sulfur-containing metal complexes. This suggests the involvement of metals in catalysis of alkylphosphonate degradation. The broader substrate specificity of degrading activity of Rhiz. meliloti [93] similar to those of Agrob. radiobacter [41] and Arthrobacter sp. [94] is a specific feature of this bacterium distinguishing it from E. coli. Agrob. radiobacter and Arthrobacter sp., however, use two different C-P lyases for this purpose, whereas Rhiz. meliloti uses only one. It was also established that Rhiz. meliloti contains an enzyme of 2-AEP degradation, genetically and biochemically different from C-P lyase, which obviously determines the wider substrate specificity of the organism. At the same time, phosphonoacetate (PA) hydrolase was not found in the organism. The presence of genes encoding the C-P lyase complex or proteins similar to its components has been shown to date not only in E. coli [61] and in Rhiz. meliloti [93], but also in Rhiz. leguminosarum [95], Mesorhizobium loti [96], Ps. aeruginosa PA01 [97], Ps. stutzeri WM88 [36], Kleb. aerogenes [98], and Enterob. aerogenes [72].
GENETIC AND PHYSIOLOGICAL REGULATION OF C-P LYASE
As mentioned above, the genes of the phn operon encoding degradation of phosphonates by the C-P lyase mechanism in E. coli are induced under phosphorus starvation. The promoter of this operon carries a sequence of 18 bp, CTGTtAgtcActTtTaAT, similar to the sense sequence found in promoters of the pho genes (called a PHO box) [99]. Thus, the genes of phosphonate assimilation are members of the Pho regulon. Their expression is regulated by exogenous orthophosphate and needs the expression of phoB and phoR genes [89] encoding the regulator and the sensor, respectively, of the two-component system of Pi signal transduction. The Pho regulon is a universal system that provides cell response to deficiency of exogenous orthophosphate, which was found in many taxonomic groups of microorganisms. The phosphate regulon of E. coli is several genes inducible under phosphorus starvation, whose products participate in primary phosphorus assimilation from the environment and are localized in different cell compartments [100]. They include an outer membrane pore protein PhoE facilitating the diffusion of phosphorus-containing compounds, hydrolytic enzymes of the periplasm cleaving organophosphorus compounds (alkaline (PhoA) and acid (AppA) phosphomonoesterases, 5´-nucleotidase (5´NUC), proteins involved in biosynthesis of cell wall polymers and utilization of polyphosphates (proteins PpX and PpK)), periplasmic Pi-binding protein PstS, and glycerophosphate-binding protein UgpB. The Pho regulon includes also the cytoplasmic membrane proteins involved in transport of orthophosphate (PstA, B, C) and other phosphorus-containing compounds (UgpE, A, C), as well as the cytoplasmic protein PhoU that does not participate in transport but mediates signal transduction in the Pho regulon [101]. The fact that C-P lyase belongs to the phosphate regulon has been proved by not only the presence of PHO box in the operator of phn locus. Cells were shown to be unable to produce C-P lyase and degrade methylphosphonate with the formation of methane in strains with non-inducible Pho regulon due to a mutation in PhoB transcription regulator [89, 102]. Mutations in the genes controlling biosynthesis of components of the Pho regulon repressor, on the contrary, result in production of C-P lyase, which degrades methylphosphonate in the presence of orthophosphate. The same is observed in recombinant E. coli strains, where phn operon is cloned under the control of a promoter not regulated by orthophosphate, e.g., lac promoter [103]. It is worth noting that the level of C-P lyase synthesis in mutants lacking the structural gene of alkaline phosphatase is comparable with that of the wild type strain and the production of this enzyme is not accompanied by phosphatase activity, demonstrating that alkaline phosphatase is not responsible for C-P lyase activity, while C-P lyase has no phosphatase activity [89, 102].
Regulation of transcription of pho genes involves the two-component regulatory system of Pi signal transduction: PhoB-PhoR in Gram-negative bacteria [104] and PhoP-PhoR in Gram-positive bacteria [105]. The proteins PhoB and PhoP are transcription regulators, which are directly bound with the promoters of pho genes. Protein PhoR is a transmembrane sensor. It receives Pi signal from the environment, is autophosphorylated, and participates in specific phosphorylation/dephosphorylation of the above transcriptional regulators. Phosphorylated regulators induce the transcription of pho genes. The genes of the Pho regulon are derepressed under orthophosphate deficiency in the medium, while Pi concentration in the cell remains rather high [106]. With excess of exogenous orthophosphate, expression of the Pho regulon genes is repressed due to formation of a repressor complex including proteins PhoR, PhoU, and all proteins of the Pst transport system (PstA, PstB, PstC, PstS). Components of other regulatory systems (under so-called cross regulation) [107], e.g., acetylphosphate, may be also inducers of the Pho regulon. Systems of such phosphate-independent control of expression of the Pho regulon genes are coupled with the steps of central cell metabolism and regulated by the carbon source. One of them functions during cell growth on glucose and needs the presence of sensor protein CreC (PhoM) [108]; another functions during cell growth on pyruvate and is coupled with acetylphosphate synthesis followed by PhoB activation directly [109] or via one of the sensor proteins [110]. Thus, the expression of the Pho regulon genes can be not only controlled by exogenous orthophosphate but also subjected to global control that combines protein regulators of this two-component system with other systems of signal transduction and with the total cell metabolism. The search for optimal conditions for degradation of phosphonates and biosynthesis of C-P lyase, which is a member of the Pho regulon, needs taking into account the above circumstance.
An important physiological feature of phosphonate degradation is necessary “adaptation” of cells to these compounds. In fact, even early physiological studies of phosphonate degradation showed [39] that degradation of and cell growth on these compounds as sole phosphorus sources begins after a prolonged latent phase. The duration of the latent phase preceding the growth on phosphonates varied strongly depending on the culture and the nature of the substrate, demonstrating multiple pathways of phosphonate degradation. Some bacteria, e.g., Pseudomonas sp. 4ASW, utilized phosphonates with the same rate as orthophosphate; however, the latent phase preceding their growth was significantly longer. Moreover, the duration of this latent phase showed a certain correlation with the subsequent maximal rate of growth. Phosphonates that are utilized after a short lag period (glyphosate, phenylphosphonate, aminomethylphosphonate, phosphonoacetate), maintain a comparatively high growth rate. The growth of B. megaterium on most phosphonates was characterized by a longer lag period and lower growth rate as compared with orthophosphate.
Regulation of direct degradation of the C-P bond was studied by the example of Pseudomonas sp. 4ASW. Orthophosphate (0.15-0.3 mM) + phenylphosphonate or 2-aminoethylphosphonate was added to orthophosphate-deficient medium to study cell growth and phosphonate utilization. It was shown that orthophosphate was preferably utilized in the presence of phenylphosphonate and orthophosphate simultaneously, but phenylphosphonate was utilized as well. The growth starts more quickly on orthophosphate and proceeds with the same rate with a combination of Pi and phenylphosphonate. With phenylphosphonate only, the growth rate is the same but after a longer lag period. Biomass yield is the same. In case of the above microorganism, no strict control of either transport or degradation of phosphonate by exogenous orthophosphate was observed, in contrast to Kluyv. ascorbata [41]. Regulation in Pseudomonas sp. 4ASW is similar to that in Agrob. radiobacter [41]. Cells of E. coli K-12 started to degrade methylphosphonic acid only in 48-70 h in the absence of other phosphorus sources with a rather slow growth [89, 102].
There are several possible explanations for the latent phase of phosphonate utilization. Phosphonate degradation probably includes some steps limiting the rate of degradation, e.g., phosphonate transport. It turned out to involve specific inducible transport systems, e.g., for 2-aminoethylphosphonate, usual transport systems of bound components [53], and systems inhibited by orthophosphate [42]. The readying of different transport systems needs different times. It is known, for example, that phosphonopeptides are transported to cells easier than free phosphonates [111]. This is probably the reason why 9-fold longer lag phase and reduced growth rate of Pseudomonas sp. 4ASW was revealed when 1-aminoethylphosphonate, as compared with its peptide analog, alaphosphaline, was used as a sole phosphate source [39]. Another probable reason for the latent phase of utilizing of phosphonates, particularly alkylphosphonates, may be formation of “adaptive” mutants [89, 102]. Deficiency of some nutrient substances and particularly in the presence of alternative nutrients may result in formation of adaptive mutants surviving under new conditions of nutrition. Accumulation of such mutants also needs time [112]. Selection of the expected mutants was recently carried out for the first time [102] by the example of several E. coli strains. Analysis of the dynamics of cell population during the adaptation to methylphosphonate showed its heterogeneity--the presence of cells yielding small and large colonies. The latter become more numerous in the process of adaptation and this increase correlates with the higher efficiency of methylphosphonate degradation. Colonies of different sizes were isolated and the corresponding cells were selected followed by their physiological and biochemical characterization. The growth rate of adapted cells on orthophosphate was the same as of the initial cells, but they differed in the rate of growth on methylphosphonate. The rate of growth of adapted cells was an order of magnitude higher as compared with the initial non-adapted cells. Besides, the adapted cells contained higher amounts of some proteins in different cell compartments [102], and these proteins were probably associated with the C-P lyase complex. Subcellular fractions of adapted cells, however, showed no C-P lyase activity, which confirms the impossibility or difficulty of detecting such activity in cell-free extracts. The mechanism of adaptation also remained unclear. One possible adaptation mechanism suggests a genetic analysis of the genes of the C-P lyase complex in E. coli K-12 [77]. The gene phnE was shown to carry an insertion of eight nucleotides resulting in mutations with a reading frame shift [113]. The strains of E. coli K-12 grow on a medium with methylphosphonic acid as the sole source of phosphorus when a spontaneous activation of the phn operon takes place followed by the loss of this insertion.
The study of different bacteria [39, 40, 102] showed a number of other physiological features of phosphonate degradation. Thus, intensive utilization of phosphonates had never been revealed. In most cases, the completeness of degradation was 20-50% of theoretically possible value, and phosphonate was always taken from the medium as consistent with the culture growth though not linearly. Utilization of 2-AEP was more rapid and almost completed in 7 days. Glyphosate was utilized with about the same rate, but the completeness of utilization was 40-45%. Polyphosphonates with a complex branched structure were degraded most intensively indicating that even quasi-complex degradation can be achieved through microbial action. The maximal utilization of methylphosphonic acid by E. coli cells in most cases did not exceed 30% of theoretically possible value at 2.5 mM methylphosphonate concentration in the medium [102]. This percentage increased with the lowering of methylphosphonate concentration in the medium, but the maximal substrate degradation remained the same, i.e., 400 nmol per unit of OD. This limitation, as well as the absence of growth on some phosphonates, was not due to their toxicity, since the inoculation of cells on Pi medium resulted in the control level of culture growth [40, 102]. An important factor determining the rate of phosphonate degradation is aeration. Biodegradation of phosphonates under anaerobic conditions, i.e., without molecular oxygen, was first shown by the example of photosynthetic Rhodob. capsulatus [40]. Using E. coli strains, it was demonstrated that in some of them the degradation of methylphosphonic acid is inhibited by free oxygen [102-104] while in others microaerobic conditions are required for this process and free nitrogen inhibits it. Thus, the role of aeration is still not clear.
Wide occurrence of phosphonates among biogenic and abiogenic (natural and man-made) organophosphorus compounds makes the question about catabolism of these compounds topical and at the same time potentially resolvable. A wide range of microorganisms, mainly bacteria, was shown able to degrade different phosphonates. The diversity of phosphonate structures, occurrence among them of compounds with activated C-P bond (aminophosphonates, acetylphosphonates) and compounds with non-activated, more stable C-P bond (alkylphosphonates) determine the great number of pathways of their catabolism. However, only the pathways of activated C-P bond degradation have been characterized rather completely, and enzymes catalyzing this degradation have been identified and characterized. The most problematic aspect of phosphonate biodegradation (from both fundamental and biotechnological points of view) is degradation of the most stable C-P bond of alkylphosphonates. Neither the mechanism of degradation of this bond nor the appropriate enzyme (or polyenzyme complex) C-P lyase have been finally determined and characterized so far. Meanwhile, the significant progress in genetic characterization of the system of alkylphosphonate degradation opens quite optimistic prospects for physiological and biochemical characterization of this system and answers to yet unsolved questions about the mechanism of this process. The search and characterization of C-P lyase, based on genetic knowledge of its probable structure, regulation, and localization, seems to be of the greatest priority in solution of the problem of alkylphosphonate biodegradation. At the same time, the more complete physiological characterization of alkylphosphonate degradation system (the mechanism of cell adaptation to alkylphosphonates, optimal conditions of biosynthesis and manifestation of C-P lyase activity in living organisms and other parameters of biodegradation) will promote more comprehensive biochemical characterization of the system. Taken together, the fundamental knowledge of the mechanism of biodegradation of organophosphorus compounds with the C-P bond will serve, in turn, as a basis of biotechnology for environmental protection and biodegradation of toxic organophosphorus compounds with stable C-P bond, which uncontrollably enter the environment as pesticides, herbicides, and other products of economic activity.
This work was supported by the Russian Foundation for Basic Research (grant Nos. 00-0448187 and 00-15-97851).
REFERENCES
1.Freedman, L. D., and Doak, G. O. (1957) Chem.
Rev., 57, 479-523.
2.Murai, T., and Tomizawa, C. (1976) J. Environ.
Sci. Health, B11, 185-197.
3.Horiguchi, M., and Kandatsu, M. (1959)
Nature, 184, 901-902.
4.Baer, E., and Stanacev, N. Z. (1964) J. Biol.
Chem., 239, 3209.
5.Kittredge, J. S., and Roberts, E. (1969)
Science, 164, 37-42.
6.Wassef, M. K., and Hendrix, J. W. (1976)
Biochim. Biophys. Acta, 486, 172-178.
7.Hasegawa, S., Tamari, M., and Kametaka, M. (1976)
J. Biochem. (Tokyo), 80, 531-535.
8.Tan, S. A., and Tan, L. G. (1989) Clin. Physiol.
Biochem., 7, 303-309.
9.Smith, J. D., and Lepak, N. M. (1982) Arch.
Biochem. Biophys., 213, 565-572.
10.Korn, E. D., Dearborn, D. G., Fales, H. M., and
Sokolovski, E. A. (1973) J. Biol. Chem., 248,
2257-2259.
11.Hendlin, D., Stapley, E. O., Jackson, M.,
Wallick, H., Miller, A. K., Wolf, F. J., Miller, T. W., Chaiet, L.,
Kanan, F. M., Foltz, E. L., Woodruff, H. B., Mata, J., Hernandez, S.,
and Mochales, S. (1969) Science, 166, 122-123.
12.Bayer, E., Gugel, K. H., Hagele, K., Hagenmaier,
H., Jessipow, S., Konig, W. A., and Zahner, Z. (1972) Helvetica
Chimica Acta, 55, 224-239.
13.Calanduoni, J. A., and Villafranca, J. J. (1986)
Bioorg. Chem., 14, 163-169.
14.Lee, P. J., Joy, K. W., Ramos, J. L., and
Guerrero, M. G. (1984) Phytochemistry, 23, 1-6.
15.Siedel, H. M., Freeman, S., Seto, H., and
Knowles, J. R. (1988) Nature, 335, 457-458.
16.Hilderbrand, R. L., and Henderson, T. G. (1983)
in The Role of Phosphonates in Living Systems (Hilderbrand, R.
L., ed.) CRC Press, Boca Raton, Florida, pp. 5-30.
17.De Graaf, R. M., Visscher, J., and Schwartz, A.
W. (1997) Mol. Evol., 44, 237-241.
18.De Graaf, R. M., Visscher, J., and Schwartz, A.
W. (1998) Origins Life Evol. Biosphere, 28, 271-282.
19.Glindemann, D., De Graaf, R. M., and Schwartz, A.
W. (1999) Origins Life Evol. Biosphere, 29, 555-561.
20.De Graaf, R. M., and Schwartz, A. W. (2000)
Origins Life Evol. Biosphere, 30, 405-410.
21.Cooper, G. W., Onwo, W. M., and Cronin, J. R.
(1992) Geochim. Cosmochim. Acta, 56, 4109-4115.
22.Nifantyev, E. E. (1971) Chemistry of
Organophosphorus Compounds [in Russian], MGU Publishers,
Moscow.
23.Fest, C., and Schmidt, K.-J. (1988)
Organophosphorus Pesticides, Springer-Verlag, Berlin.
24.Egli, T. (1988) Microbiol. Sci., 5,
36-41.
25.Tyhach, R. J., Engel, R., and Tropp, B. (1976)
J. Biol. Chem., 251, 6717-6723.
26.Viktorova, L. S., Arzumanov, A. A., Shirokova, E.
A., Yasko, M. V., Aleksandrova, L. A., Shipitsyn, A. V., Skoblov, A.
Yu., and Krayevsky, A. A. (1998) Mol. Biol. (Moscow), 32,
162-171.
27.Fleisch, W. (1991) Drugs, 42,
919-944.
28.Jaworski, E. G. (1972) J. Agric. Food
Chem., 20, 1195-1198.
29.Munro, N. B., Talmage, S. S., Griffin, G. D.,
Waters, L. C., Watson, A. P., King, J. F., and Hauschild, V. (1999)
Environ. Health Perspect., 107, 933-974.
30.Small, M. J. (1984) Tech Rpt8304; AD A149515.
Fort Detrick, MD: US Army Medical Bioengineering Research and
Development Laboratory.
31.Williams, R. T., Miller, W. R., III, and
MacGillivray, A. R. (1987) CRDEC-CR-87103: NTIS AD-A184 959/5. Aberdeen
Proving Ground, MD: US Army Armament Munitions Chemical Command,
Chemical Research, Development and Engineering Center.
32.Zeleznick, L. D., Myers, T. C., and Titchener, E.
B. (1963) Biochim. Biophys. Acta, 78, 546-547.
33.Cook, A. M., Daughton, C. G., and Alexander, M.
(1978) J. Bacteriol., 133, 85-90.
34.Harkness, D. R. (1966) J. Bacteriol.,
92, 623-627.
35.Metcalf, W. W., and Wolfe, R. S. (1998) J.
Bacteriol., 180, 5547-5558.
36.Metcalf, W. W., and Wanner, B. L. (1991) J.
Bacteriol., 173, 587-600.
37.Metcalf, W. W., and Wanner, B. L. (1993) J.
Bacteriol., 175, 3430-3442.
38.Imazu, K., Tanaka, S., Kuroda, A., Anbe, Y.,
Kato, J., and Ohtake, H. (1998) Appl. Environ. Microbiol.,
64, 3754-3758.
39.Quinn, J. P., Peden, J. M. M., and Dick, R. E.
(1989) Appl. Microbiol. Biotechnol.,31, 283-287.
40.Schowanek, D., and Verstraete, W. (1990) Appl.
Environ. Microbiol., 56, 895-903.
41.Wackett, L. P., Shames, S. L., Venditti, C. P.,
and Walsh, C. T. (1987) J. Bacteriol., 169, 710-717.
42.Pipke, R., Schulz, A., and Amrhein, N. (1987)
Appl. Environ. Microbiol., 53, 974-978.
43.McGrath, J. W., Hammerschmidt, F., and Quinn, J.
P. (1998) Appl. Environ. Microbiol.,64, 356-358.
44.La Nauze, J. M., Rosenberg, H., and Shaw, D. C.
(1970) Biochim. Biophys. Acta, 212, 332-350.
45.McMullan, G., and Quinn, J. P. (1994) J.
Bacteriol., 176, 320-324.
46.Ternan, N. G., and Quinn, J. P. (1998)
Biochem. Biophys. Res. Commun., 248, 378-381.
47.Obojska, A., Lejczak, B., and Kubrak, M. (1999)
Appl. Microbiol. Biotechnol., 51, 872-876.
48.Bode, R., and Birnbaum, D. (1989) Biochem.
Physiol. Pflanzen., 184, 163-170.
49.Ternan, N. G., and McMullan, G. (2000) FEMS
Microbiol. Lett., 184, 237-400.
50.Zboinska, E., Maliszewska, I., Lejczak, B., and
Kafarski, P. (1992) Lett. Appl. Microbiol., 15,
269-272.
51.Bujacz, B., Wieczorek, P., Krzysko-Lupicka, T.,
Golab, Z., Lejczak, B., and Kafarski, P. (1995) Appl. Environ.
Microbiol., 61, 2905-2910.
52.Krzysko-Lupicka, T., Strof, W., Kubs, K.,
Skorupa, M., Wieczorek, P., Lejczak, B., and Kafarski, P. (1997)
Appl. Microbiol. Biotechnol., 48, 549-552.
53.Smith, J. D. (1983) in The Role of
Phosphonates in Living Systems (Hilderbrand, R. L., ed.) CRC Press,
Boca Raton, pp. 31-35.
54.Ternan, N. G., and Quinn, J. P. (1998) Syst.
Appl. Microbiol.,21, 346-352.
55.Ternan, N. G., McGraff, J. W., McMullan, G., and
Quinn, J. P. (1998) World J. Microbiol. Biotechnol., 14,
635-647.
56.Bowman, E. D., McQueeny, M. S., Barry, R. J., and
Dunaway-Mariano, D. (1990) Biochemistry,29,
7059-7063.
57.Nakashita, H., Watanabe, K., Hara, O., Hidaka,
T., and Seto, H. (1997) J. Antibiotics, 50, 212-219.
58.Nakashita, H., Kozuka, K., Hidaka, T., Hara, O.,
and Seto, H. (2000) Biochim. Biophys. Acta, 1490,
159-162.
59.Hidaka, T., Imai, S., Hara, O., Anzai, H.,
Murakami, T., Nagaoka, K., and Seto, H. (1990) J. Bacteriol.,
172, 3066-3072.
60.Pollack, S. J., Freeman, S., Pompliano, D. L.,
and Knowles, J. R. (1992) Eur. J. Biochem., 209,
735-743.
61.Kamigiri, K., Hidaka, T., Imai, S., Murakami, T.,
and Seto, H. (1992) J. Antibiotics, 45,781-787.
62.Nakashita, H., Hidaka, T., Kuzuyama, T., and
Seto, H. (1995) Gene, 158, 149-150.
63.Nakashita, H., Shimazu, A., Hidaka, T., and Seto,
H. (1992) J. Bacteriol., 174, 6857-6861.
64.Kitteredge, J. S., Roberts, E., and Simonsen, D.
G. (1962) Biochemistry, 1, 624-628.
65.Dumora, C., Lacoste, A.-M., and Cassaigne, A.
(1983) Eur. J. Biochem., 133, 119-125.
66.McMullan, G., Harrington, F., and Quinn, J. P.
(1992) Appl. Environ. Microbiol., 58, 1364-1366.
67.Garen, A., and Levinthal, C. (1960) Biochim.
Biophys. Acta, 38, 470-483.
68.Jiang, W., Metcalf, W. W., Lee, K. S., and
Wanner, B. L. (1995) J. Bacteriol., 177, 6411-6421.
69.Baker, A. S., Ciocci, M. J., Metcalf, W. W., Kim,
J., Babbitt, P. C., Wanner, B. L., Martin, B. M., and Dunaway-Mariano,
D. (1998) Biochemistry, 37, 9305-9315.
70.La Nauze, J. M., Coggins, J. R., and Dixon, H. B.
F. (1977) Biochem. J., 165, 409-411.
71.Morais, M. C., Zhang, W., Baker, A. S., Zhang,
G., Dunaway-Mariano, D., and Allen, K. N. (2000)
Biochemistry, 39, 10385-10396.
72.Lee, K. S., Metcalf, W. W., and Wanner, B. L.
(1992) J. Bacteriol., 174,2501-2510.
73.Kulakova, A. N., Kulakov, L. A., and Quinn, J. P.
(1997) Gene, 195, 49-53.
74.McGrath, J. W., Wisdom, G. B., McMullan, G.,
Larkin, M. J., and Quinn, J. P. (1995) Eur. J. Biochem.,
234, 225-230.
75.Kulakova, A. N., Kulakov, L. A., Akulenko, N. V.,
Ksenzenko, V. N., Hamilton, J. T., and Quinn, J. P. (2001) J.
Bacteriol., 183, 3268-3275.
76.Ternan, N. G., Hamilton, J. T., and Quinn, J. P.
(2000) Arch. Microbiol., 173, 35-41.
77.Nakashita, H., Shimazu, A., Hidaka, T., and Seto,
H. (1992) J. Bacteriol., 174, 6857-6861.
78.Murata, K., Higaki, N., and Kimura, A. (1988)
Biochem. Biophys. Res. Commun., 157, 190-195.
79.Murata, K., Higaki, N., and Kimura, A. (1989)
J. Bacteriol., 171, 4504-4506.
80.McMullan, G., Watkins, R., Harper, D. B., and
Quinn, J. P. (1991) Biochem. Int., 25,271-279.
81.Cordeiro, M. L., Pompliano, D. L., and Frost, J.
W. (1986) J. Am. Chem. Soc., 108, 332-334.
82.Frost, J. W., Loo, S., Cordeiro, M. L., and Li,
D. (1987) J. Am. Chem. Soc., 109, 2166-2171.
83.Shames, S. L., Wackett, L. P., LaBarge, M. S.,
Kuczkowski, R. L., and Walsh, C. T. (1987) Bioorg. Chem.,
15, 366-373.
84.Avila, L. Z., Draths, K. M., and Frost, J. W.
(1991) Bioorg. Med. Chem. Lett., 1, 51-54.
85.Pipke, R., and Amrhein, N. (1988) Appl.
Environ. Microbiol., 54, 1293-1296.
86.Metcalf, W. W., Steed, P. M., and Wanner, B. L.
(1990) J. Bacteriol., 172, 3191-3200.
87.Wanner, B. L., and Boline, J. A. (1990) J.
Bacteriol., 172, 1186-1196.
88.Wanner, B. L., and Metcalf, W. W. (1992) FEMS
Microbiol. Lett., 79, 133-139.
89.Wackett, L. P., Wanner, B. L., Venditti, C. P.,
and Walsh, C. T. (1987) J. Bacteriol., 169,
1753-1756.
90.Chen, C. M., Ye, Q. Z., Zhu, Z. M., Wanner, B.
L., and Walsh, C. T. (1990) J. Biol. Chem.,265,
4461-4471.
91.Bardin, S., Dan, S., Osteras, M., and Finan, T.
M. (1996) J. Bacteriol., 178, 4540-4547.
92.Surin, B. P., Rosenberg, H., and Cox, G. B.
(1985) J. Bacteriol., 161, 189-198.
93.Parker, G. F., Higgins, T. P., Hawkes, T., and
Robson, R. L. (1999) J. Bacteriol., 181, 389-395.
94.Kertesz, M., Elgorriga, A., and Amrhein, N.
(1991) Biodegradation, 2, 53-59.
95.Stevens, J. B., de Luca, N. G., Beringer, J. E.,
Ringer, J. P., Yeoman, K. H., and Johnston, A. W. (2000) Mol. Plant
Microbe Interact., 13, 228-231.
96.Kaneko, T., Nakamura, Y., Sato, S., Asamizu, E.,
Kato, T., Sasamoto, S., Watanabe, A., Idesawa, K., Ishikawa, A.,
Kawashima, K., Kimura, T., Kishida, Y., Kiyokawa, C., Kohara, M.,
Matsumoto, M., Matsuno, A., Mochizuki, Y., Nakayama, S., Nakazaki, N.,
Shimpo, S., Sugimoto, M., Takeuchi, C., Yamada, M., and Tabata, S.
(2000) DNA Res., 7, 331-338.
97.Stover, C. K., Pham, X. Q., Erwin, A. L.,
Mizoguchi, S. D., Warrener, P., Hickey, M. J., Brinkman, F. S.,
Hufnagle, W. O., Kowalik, D. J., Lagrou, M., Garber, R. L., Goltry, L.,
Tolentino, E., Westbrock-Wadman, S., Yuan, Y., Brody, L. L., Coulter,
S. N., Folger, K. R., Kas, A., Larbig, K., Lim, R., Smith, K., Spencer,
D., Wong, G. K., Wu, Z., and Paulsen, I. T. (2000) Nature,
406, 959-964.
98.Ohtake, H., Wu, H., Imazu, K., Anbe, Y., Kato,
J., and Kuroda, A. (1994) Resour. Conserv. Recycl., 18,
125-134.
99.Makino, K., Shinagawa, H., Amemura, M., and
Nakata, A. (1986) J. Mol. Biol., 190, 37-44.
100.Wanner, B. L. (1990) in The Molecular Basis
of Bacterial Metabolism (Hauska, G., and Thauer, R., eds.)
Springer-Verlag, Heidelberg, pp. 152-163.
101.Steed, P. M., and Wanner, B. L. (1993) J.
Bacteriol., 175, 6797-6809.
102.Matys, S. V., Laurinavichus, K. S., Krupyanko,
V. I., and Nesmeyanova, M. A. (2001) Process Biochem.,
36, 821-827.
103.Yakovleva, G. M., Kim, S. K., and Wanner, B. L.
(1998) Appl. Microbiol. Biotechnol., 49,573-578.
104.Wanner, B. L. (1993) J. Cell Biochem.,
51, 47-54.
105.Hulett, F. M., Sun, G., and Liu, W. (1994)
in Phosphate in Microorganisms: Cellular and Molecular Biology
(Torriani-Gorrini, A., Yagil, E., and Silver, S., eds.) ASM Press,
Washington, pp. 50-54.
106.Torriani, A., and Ludtke, D. N. (1985) in
The Escherichia coli and Salmonella typhimurium: Cellular and
Molecular Biology (Neitdhardt, F. C., Ingraham, J. L., Low, K. B.,
Magasanik, B., Schaechter, M., and Umbarger, H. E., eds.) Vol. 2, ASM
Press, Washington, pp. 1326-1333.
107.Wanner, B. L. (1992) J. Bacteriol.,
174, 2053-2058.
108.Amemura, M., Makino, K., Shinagawa, A.,
Kobayashi, A., and Nakato, A. (1990) J. Mol. Biol., 184,
241-259.
109.Wanner, B. L., and Wilmes-Riesenberg, M. R.
(1992) J. Bacteriol., 174, 2124-2130.
110.Kim, S. K., Wilmes-Riesenberg, M. R., and
Wanner, B. L. (1996) Mol. Microbiol., 22, 135-147.
111.Allen, J. G., Atherton, F. R., Hall, M. J.,
Hassall, C. H., Holmes, S. W., Lambert, R. W., Nisbet, L. J., and
Ringrose, P. S. (1978) Nature, 272, 56-58.
112.Radicella, J. P., Park, P. U., and Fox, M. S.
(1995) Science, 268, 418-420.
113.Makino, K., Kim, S.-K., Shinagawa, H., Amemura,
M., and Nakata, A. (1991) J. Bacteriol., 173,
2665-2672.
114.Matys, S. V., Laurinavichius, K. S., and
Nesmeyanova, M. A. (1996) Mikrobiologiya, 65,
481-487.