REVIEW: Structure and Properties of Small Heat Shock Proteins (sHsp) and Their Interaction with Cytoskeleton Proteins
N. B. Gusev1*, N. V. Bogatcheva1, and S. B. Marston2
1Department of Biochemistry, School of Biology and Department of Biochemistry, School of Fundamental Medicine, Lomonosov Moscow State University, Moscow, 119992 Russia; fax: (095) 939-2747; E-mail: NBGusev@mail.ru2Department of Cardiac Medicine, National Heart and Lung Institute, Imperial College of Science, Technology and Medicine, Dovehouse Street London SW3 6LY, UK; E-mail: S.Marston@ic.ac.uk
* To whom correspondence should be addressed.
Received August 22, 2001
The modern classification of small heat shock proteins (sHsp) is presented and peculiarities of their primary structure and the mechanism of formation of oligomeric complexes are described. Data on phosphorylation of sHsp by different protein kinases are presented and the effect of phosphorylation on oligomeric state and chaperone activity of sHsp is discussed. Intracellular location of sHsp under normal and stress conditions is described and it is emphasized that under certain condition sHsp interact with different elements of cytoskeleton. The literature concerning the effect of sHsp on polymerization of actin in vitro isanalyzed. An attempt is made to compare effects of sHsp on polymerization of actin in vitro with the results obtained on living cells under normal conditions and after heat shock or hormone action. The literature concerning possible effects of sHsp on cell motility is also analyzed.
KEY WORDS: heat shock proteins, crystallins, phosphorylation, cytoskeleton, actin
The large group of heat shock proteins includes several classes of proteins different in molecular weight, properties, and structure. All these proteins are combined in one group since they participate in the proper folding of proteins under both normal conditions and especially under extreme conditions (such as heat shock, effect of organic compounds, strong oxidants, and other poisons). These proteins are also involved in renaturation of partially denatured proteins or complete elimination of fully denatured proteins. Heat shock proteins are usually divided into several classes according to their molecular weight [1]. Different classes of heat shock proteins have molecular weight of 100, 90, 70, 60, and 40 kD. Heat shock proteins having lower molecular weight are combined in the group of so-called small heat shock proteins. Heat shock proteins with molecular weight 100, 90, 70, and 60 kD possess ATP-binding sites. ATP hydrolysis is required for normal functioning of chaperones with molecular weight 70 and 60 kD, as well as for the heat shock proteins with molecular weigh 110 kD that are homologous to the proteins with molecular weight of 70 kD. The proteins with molecular weight 90 kD have low ATPase activity and it is suggested that the binding of ATP is important for regulation of interaction of Hsp90 with accessory proteins. Heat shock proteins with molecular weight 40 kD and small heat shock proteins do not have an ATP-binding site; however, the data indicate that ATP somehow affects the structure of small heat shock proteins and their interaction with protein substrate [2, 2a]. Small heat shock proteins may interact with partially denatured proteins, prevent their aggregation and under certain condition transfer partially denatured substrates to chaperones possessing ATPase activity [3, 4].
STRUCTURE OF SMALL HEAT SHOCK PROTEINS
The group of small heat shock proteins (sHsp) combines proteins with molecular weight in the range of 12 up to 43 kD that were isolated from archaeon, bacteria, plants, and animals [2]. All these proteins contain a rather conservative so-called alpha-crystallin domain, containing 80-100 residues located as a rule in the C-terminal part of these proteins. Homology of these domains vary from 20% between remote members of the family of small heat shock proteins isolated from bacteria and mammals up to 60% between closely related members of this family obtained from mammalian tissues [5].
It is supposed that vertebrates synthesize five major classes of small heat shock proteins. These are alphaA- and alphaB-crystallin, small heat shock proteins with molecular weight 25-27 kD (Hsp25/27), small heat shock proteins with molecular weight 20 kD (Hsp20), small heat shock proteins with molecular weight 17 kD (HspB3), and so-called activator of myotonic dystrophy protein kinase (MKBP, myosin dystrophy binding protein) that sometimes is designated as HspB2 [6-8]. Recently a new class of sHsp with molecular weight 22 kD was described in the literature [9, 10]. Several sites can be distinguished in the sHsp monomer (see Fig. 1). One can find not very conservative so-called WDPF domain on the very N-terminal end of some of sHsp monomer. This motif has a primary structure (W/F)(D/F)PF-X0-8-(W/F)(D/E)(P/F)F, where X-denotes non-conservative residues [11]. The WDPF domain is followed by a short variable sequence having on the C-terminal end a rather conservative site with the primary structure PSRLFDQXFGEXLL [7, 8]. The already mentioned alpha-crystallin domain consisting of 80-100 residues is located in the C-terminal part of the sHsp. This domain is usually followed by a short variable sequence having a rather high motility and flexibility. The difference in the molecular weight of sHsp is explained by the difference in the length of variable sites between the WDPF-domain and alpha-crystallin domains as well as by the length of the variable C-terminal tail.
Small heat shock proteins tend to form large oligomeric complexes. Oligomers of mammalian sHsp are very flexible, and this is probably the reason for the lack of X-ray crystallographic data for mammalian sHsp [12]. However there are data on the structure of small heat shock proteins from the hyperthermophilic archaeon Methanococcus jannaschii [13] and wheat [13a]. It was shown that the heat shock protein from M. jannaschii with molecular weight 16.5 kD (so-called MjHSP16.5) forms hollow spherical complexes consisting of 24 subunits. This oligomer is composed of dimers, which are formed by interaction of two alpha-crystallin domains of neighboring monomeric subunits of MjHSP16.5. The alpha-crystallin domain contains nine beta-sheets that interact with each other and form tight contacts with neighbor subunits. Recently published data on the structure of dimers of alphaB-crystallin obtained by synchrotron radiation X-ray scattering indicate that the crystallin domain possesses a rather large flexibility and may form different complexes being located inside the small heat shock protein from of M. jannaschii and alphaB-crystallin of mammals [12]. Anyhow, there are no doubts that the crystallin domain plays an important role in the oligomerization of small heat shock proteins.Fig. 1. Scheme of the structure of several representatives of the family of small heat shock proteins. The dark shaded area marks the so-called WDPF-domain, the light shaded area marks conservative region in the N-terminal part of sHsp and the black area denotes conservative alpha-crystallin domain. P, the sites of phosphorylation; zigzag, flexible C-terminal region. Figures above the scheme denote number of amino acid residues (after Lambert et al. [11] and Sugiyama et al. [8] with slight modifications).
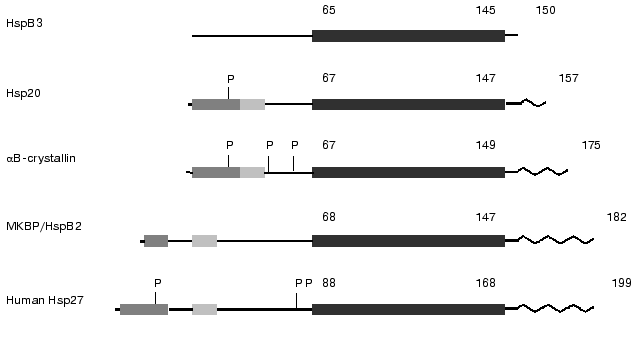
This viewpoint was supported in the series of investigations performed on crystallins and small heat shock proteins from Mycobacterium tuberculosis and mammals where site-directed mutagenesis was combined with ESR-labeling [5, 14]. It has been shown that certain sites of the crystallin domain indeed are involved in the formation of beta-sheets that interact with each other and directly participate in oligomer formation. In the case of human crystallins and small heat shock protein (Hsp27) some peculiarities of the primary structure of the alpha-crystallin domain lead to formation of stable dimers that aggregate to tetramers and form unstable oligomers consisting of 16-32 (and even more) subunits. It is worthwhile to mention that crystallins and small heat shock proteins with molecular weight of 25-27 kD can form mixed oligomers [2, 7]. Recent data indicate that heterologous oligomeric complexes containing different small heat shock proteins can exist in the living cell. At least three different heterologous complexes were described in the literature [8]. These were complexes simultaneously containing alphaB-crystallin and small heat shock proteins with molecular weight 25-27 kD (Hsp25/27); complexes consisting of alphaB-crystallin, Hsp25/27, and Hsp20; and complexes consisting of Hsp25/27 and Hsp20. The components of these complexes can be rather easily exchanged with each other [15]. Mammalian myosin dystrophy kinase binding protein (MKBP) having a molecular weight of 20 kD forms oligomeric complexes with molecular weight of about 150 kD [7]. However, this protein is also able to form heterooligomeric complexes with HspB3 [8]. It is supposed that in mammalian tissue there are several different forms of homo- and heterooligomeric complexes consisting of different small heat shock proteins [8]. There is no doubt that the alpha-crystallin domain plays a crucial role in formation of small oligomers consisting of 2-4 subunits. However, formation of large oligomers is to some extent dependent on the variable N- and C-terminal sequences of small heat shock proteins.
This suggestion was confirmed in investigations performed on small heat shock proteins of Caenorhabditis elegans. The genome of this worm contains 16 genes of small heat shock proteins, and depending on conditions this nematode can synthesize several small heat shock proteins with molecular weight between 12.2 and 25 kD [2, 16]. Small heat shock proteins with molecular weight 12 kD form only small oligomers (dimers and tetramers) and possess small chaperone activity. Deletion of the N-terminal residues of sHsp with molecular weight 16 kD also results in a decrease of chaperone activity and decrease of the size of oligomers [17]. It has been shown that deletion of residues 5-23 from the sequence of Chinese hamster small heat shock protein (Hsp27) prevents formation of large oligomers with molecular weight 700 kD without affecting formation of dimers [11]. It is worthwhile to mention that chimeric protein containing residues 5-109 of Chinese hamster Hsp27 and luciferase tends to aggregate with formation of the complexes with molecular weight 350 kD. Probably the crystallin domain provides for formation of only dimers and tetramers of sHsp [11]. Formation of large aggregates seems to be dependent on the presence of WDPF-domain located in the very N-terminal part of sHsp. Deletion of 33 residues located in the N-terminal part of sHsp beyond the WDPF-domain does not effect oligomerization and chaperone activity of sHsp [18, 19].
The data on the involvement of the flexible C-terminal tail of sHsp in formation of large oligomers are rather controversial. It has been shown that the negative charges of the flexible C-terminal tail of crystallin stabilize oligomer formation [20]. However, deletion of the last 18 residues of mouse Hsp25 does not affect the structure of monomers or oligomers of this protein. But this deletion influences the chaperone activity of Hsp25 with certain protein targets [21]. It is possible that the flexible C-terminal tail participates in the interaction of sHsp with the target proteins and affects general hydrophobicity of oligomeric complexes of sHsp. All these factors will affect the equilibrium between different oligomeric forms of small heat shock proteins.
PHOSPHORYLATION OF SMALL HEAT SHOCK PROTEINS AND EFFECT OF
PHOSPHORYLATION ON THEIR OLIGOMERIC STATE
In smooth muscle, stimulation of cyclic nucleotide dependent protein kinases and/or inhibition of phosphodiesterase results in phosphorylation of small heat shock proteins with molecular weight 20 kD [22-25]. cAMP- and cGMP-dependent protein kinases phosphorylate Ser16 of Hsp20 and induce dissociation of oligomers of this protein [23, 24]. It is supposed that phosphorylated Hsp20 predominantly interacts with monomeric actin (G-actin) whereas unphosphorylated Hsp20 predominantly interacts with F-actin [24]. It is important to mention that phosphorylation of the site that is located at the very N-terminal part of the molecule within the WDPF domain and far from the crystallin domain leads to the change in the oligomeric state. This is indirect evidence for the important role of the N-terminal part of Hsp20 in oligomer formation.
Stimulation of HeLa cells by arsenite, phorbol esters, or tumor necrosis factor (TNF) results in phosphorylation of Ser78, Ser82, and to a smaller extent Ser15 of Hsp27 [26]. All the above mentioned sites contain the consensus sequence RXXS. It is worthwhile to mention that the primary structure of sHsp in the vicinity of the sites of phosphorylation is rather conservative, however there are examples where this conservative sequence is not preserved in the vicinity of one or even two potential sites of phosphorylation. Ser15 is located at the N-terminal end in the WDPF-domain, whereas two other sites of phosphorylation are located in a variable part of the molecule close to the crystallin domain of small heat shock proteins [11] (Fig. 1).
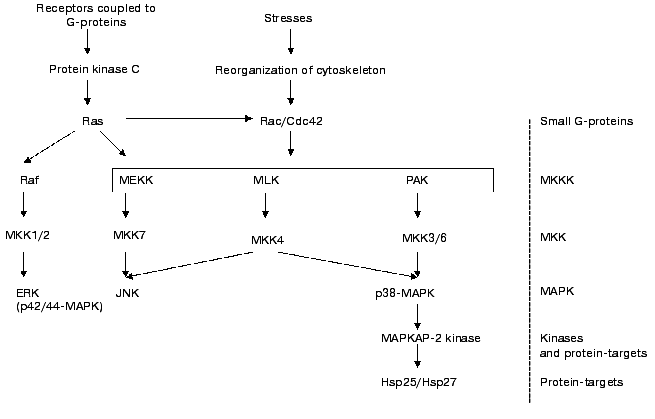
It is well known that certain hormones and biologically active compounds as well as different types of stresses activate the MAP-kinase cascade (see Fig. 2). Activation of p38 MAP-kinase results in increase of activity of so-called MAPKAP-2 kinase. This protein kinase phosphorylates the three above-mentioned sites in Hsp25/27 [27-31]. p42/p44 ERK kinase seems not to be involved in phosphorylation of small heat shock proteins [31, 32].
Recently it has been shown that certain isoforms of protein kinase C can be involved in phosphorylation of Hsp25/27 [33]. The delta isoforms of protein kinase C seem to phosphorylate the same sites of Hsp25/27 as MAPKAP-2/3. Platelet sHsp27 is phosphorylated by cGMP-dependent protein kinase that phosphorylates Thr143 [34]. Phosphorylation of Hsp27 by cGMP-dependent protein kinase does not affect phosphorylation of this protein by MAPKAP-2 kinase. At present little is known about enzymes involved in dephosphorylation of Hsp25/27. However it has been shown that under certain conditions Ca-calmodulin dependent protein phosphatase (calcineurin) is able to dephosphorylate Hsp25/27 [35].Fig. 2. Scheme of activation of MAP-cascade. Ras, Rac, and Cdc42 are small G-proteins. MKKK are kinases of kinase of MAP-kinase. They are poorly characterized and consist of MEKK (MAP-kinase and Extracellular responsive Kinase Kinases), Mixed Lineage Kinases (MLK), and kinases activated by p21 (PAK). These enzymes can phosphorylate and regulate activity of several mitogen activated kinases denoted as MKK 1/2, 3/6, 4, and 7. These enzymes phosphorylate Thr and Tyr of three different MAP-kinases. Namely Extracellular Responsive Kinase (ERK, p42/44 MAPK); kinase, phosphorylating the N-terminus of factor c-Jun (JNK); and MAP-kinase with molecular weight 38 kD (p38 MAP-kinase). This enzyme phosphorylates and activates MAPKAP-2 kinase which phosphorylates Hsp25/27 (after Sugden and Clerk [66] with simplifications).
It was found that eye lens alphaB-crystallin can also be phosphorylated and that cAMP-dependent protein kinase may participate in this process. Different stress factors induce phosphorylation of Ser19, Ser45, and Ser59 of alphaB-crystallin. Ser45 seems to be phosphorylated by p42/p44 kinase, whereas Ser59 is phosphorylated by MAPKAP-2 kinase [36]. As in the case with Hsp25/27, the sites of phosphorylation in crystallin are located either in the N-terminal part in the so-called WDPF-domain or in the variable sequence in the vicinity of the crystallin domain. Summing up, we conclude that crystallins and Hsp25/27 can be phosphorylated by different MAP-kinases at sites located either at the very N-terminal domain or in the vicinity of the conservative crystallin domain. In addition these proteins can be phosphorylated by cyclic-nucleotide dependent protein kinases (Fig. 1).
Phosphorylation of Hsp25/27 by MAPKAP-kinases or replacement of the corresponding Ser residues by an Asp [37] or Glu [11] leads to dissociation of large oligomers consisting of 24 (or even more) monomers to tetramers. In both cases phosphorylation or mutation of the site located in the N-terminal part (Ser15) induces only a small effect on the oligomerization, whereas phosphorylation of Ser78/Ser82 of human Hsp27 or Ser90 of Chinese hamster Hsp27 leads to dissociation of large oligomers of sHsp. Complete dissociation of oligomers was observed only after phosphorylation of all three potential sites of phosphorylation. Similar results were also observed in the case of alphaB-crystallin. Replacement of Ser, phosphorylated by MAPKAP-kinase, by Asp leads to dissociation of large 500 kD oligomers of alphaB-crystallin to smaller aggregates with molecular weight 100-300 kD [36].
Recently published results [36, 37] indicate that phosphorylation of Hsp25/27 accompanied by dissociation of large oligomers leads to decrease of chaperone activity. These data agree with the fact that dimers of Hsp25 do not possess chaperone activity, whereas large aggregates and granules formed after aggregation of large oligomers, have high chaperone activity [38]. It is worthwhile mentioning that heat shock leads to aggregation of mammalian sHsp25/27 and formation of large granules that retain high chaperone activity [38]. Amazingly, increase in the temperature induces the opposite effect in the case of Saccharomyces cerevisiae Hsp26 [39]. At low temperature this protein forms 24-mer oligomers that dissociate (not aggregate as in the case of mammalian sHsp) at elevated temperature, and this dissociation is accompanied by increase of chaperone activity [39]. This means that also highly homologous sHsp isolated from mammals and yeast have distinctly different properties and differently react to heat shock.
Unfortunately, at present it is not absolutely clear how phosphorylation and phosphorylation-induced dissociation of Hsp25/27 provides for increased tolerance of the cell to elevated temperature. For example, Arrigo et al. [40, 41] did not observe correlation between phosphorylation and the protective effect of Hsp25 in mouse murine L929 fibrosarcoma cells.
Summing up, we conclude that Hsp20 is predominantly phosphorylated by cyclic-nucleotide dependent proteins kinases, whereas Hsp25/27 and crystallins are predominantly phosphorylated by MAPKAP-kinases and certain isoforms of protein kinase C, although cyclic nucleotide dependent protein kinases may also phosphorylate Hsp25/27. Phosphorylation of certain sites results in partial dissociation of oligomeric sHsp and modifies chaperone activity and interaction of sHsp with target proteins.
INTERACTION OF SMALL HEAT SHOCK PROTEINS WITH CYTOSKELETAL
PROTEINS AND POSSIBLE INVOLVEMENT OF sHsp IN REGULATION OF MUSCLE AND
NON-MUSCLE MOTILITY
In early investigations performed on small heat shock protein from avian gizzard it was found that Hsp25 prevented actin polymerization by blocking the barbed end of the growing filament. In addition, Hsp25 prevented gel formation induced by addition of filamin and alpha-actinin to F-actin and enhances depolymerization of F-actin [42]. Very recently it has been shown that peptides of Hsp25 restricted by residues W43-R57 and I92-N106 interact with actin and inhibit actin polymerization. It is interesting that peptides of alphaB-crystallin homologous to the peptide of Hsp25 containing residues 92-106 did not affect polymerization of actin, whereas the same peptide containing an additional 11 residues at its N-terminal part significantly inhibited polymerization of actin [43].
At first the question of which types of oligomeric forms of small heat shock proteins affect actin polymerization was not analyzed. Later Benndorf et al. [44] found that unphosphorylated monomeric forms of Hsp25 effectively block actin polymerization, whereas phosphorylated monomers and unphosphorylated oligomers are ineffective in preventing actin polymerization. In some respect these data correlate with the fact that elongation of a short peptide of Hsp25 which is able to interact with actin and incorporation in this peptide residues mimicking the site of phosphorylation results in significant decrease of the interaction of this elongated peptide with actin [43]. The above mentioned results of Miron et al. [42] and Benndorf et al. [44] have become classic and are widely cited in the literature dealing with the interaction of Hsp25 with actin in vitro. However, there are still many unanswered questions. First, in both above-mentioned papers the process of actin polymerization was followed by fluorescent spectroscopy, viscosity, or electron microscopy. The method of cosedimentation was not used in either of these publications. Therefore, we have a feeling that the direct interaction of sHsp with F-actin was not unequivocally proved. Second, at present it is difficult to imagine how Benndorf et al. [44] were able to separate small and large oligomers of sHsp. It is well known that oligomeric forms of sHsp are in equilibrium [38] and therefore separation of oligomers would seem to be impossible. In addition, recently published data indicate that the large oligomers of sHsp dissociate to dimers and tetramers, but not to monomers [16, 38]. Third, in the investigation of Benndorf et al. [44] isolation of different isoforms of Hsp25 was achieved in the presence of rather high concentrations of Triton X-100 that can induce large (and probably irreversible) changes in the structure of Hsp25. It is interesting that Benndorf et al. [44] mentioned that in contrast to the tissue Hsp25, the recombinant protein did not affect polymerization of actin. The situation becomes even more controversial after a recent publication of Butt et al. [34]. It has been found that wild type recombinant Hsp25 does not affect actin polymerization, whereas after phosphorylation by MAPKAP-2 kinase Hsp25 starts to activate polymerization of actin. Thus, at present there is no consensus on the effect of Hsp25 on actin polymerization in vitro. It is traditionally accepted that Hsp25 is an inhibitor of actin polymerization and that phosphorylation of small heat shock proteins somehow affects their interaction with actin. Let us analyze experimental results obtained on living cells, i.e., under conditions close to in vivo conditions.
A rather large number of publications are devoted to analysis of intracellular localization of sHsp under different conditions. It is known that crystallins and MKBP are located both in the cytoplasm and on the thin filaments at the level of Z-lines [7] or on the intermediate filaments [45]. Under normal conditions Hsp25 is as a rule diffusely distributed in the cytoplasm [30, 46-49], although according to other publications it is colocalized with actin filaments [30, 50] and intermediate filaments [45]. Heat shock [47, 48] and certain hormones such as cholecystokinin [30], endothelin [49], angiotensin II [51], growth factors [52], and ceramides [49] induce redistribution of Hsp25 and a certain portion of Hsp25 migrates to membranes where it is colocalized with Rho A [49], Raf-1 [50], as well as with actin filaments and stress fibers [30, 47, 48, 50, 52, 53]. It is important to mention that if the heat shock is performed under depletion of ATP, the Hsp25 did not migrate from cytosol to actin filaments, but formed large aggregates (granules) located either in the nucleus [47, 48] or in the cytosol [46]. It is worthwhile to mention that heat shock induces aggregation of intermediate filaments and Hsp25/27 as well as alphaB-crystallin interacts with these aggregates of intermediate filaments [45]. Heat shock is also accompanied by migration of Hsp20 from the cytosol to the insoluble fraction [54].
The question arises how external signals can induce translocation of sHsp inside the cell. It was shown that expression of mutant Hsp25 with replacement of Ser phosphorylated by MAPKAP-kinases by Ala or Gly prevents translocation of small heat shock protein towards actin filaments [29]. If before stimulation the cells were treated by specific inhibitor of p38 MAP-kinase (SB 203580) and by this means blocked phosphorylation of Hsp25/27 [27], then stimulation of the cells was not accompanied by migration of small heat shock protein towards actin filaments [30]. Similar results were obtained if the cells were expressing mutant form of Rho [49]. In this case sHsp did nit migrate from the cytosol to the perimembrane region [49]. The data presented mean that different stresses or stimulation of the cells by different agonists lead to activation of cascade of MAP-kinases (or probably other protein kinases), phosphorylation of Hsp25/27, and its migration from the cytosol to cytoskeleton or to the perimembrane space. Unfortunately this attractive hypothesis was not confirmed in a number of other publications. It has been shown that phorbol esters activating protein kinase C and cascade of other protein kinases (including MAP-kinases) result in translocation to the perimembrane region of both wild type Hsp25 and its mutant that cannot be phosphorylated [55]. Heat shock of CHO cells increases the quantity of Hsp25 cosedimented with cell membranes. However, both phosphorylated and non-phosphorylated forms of sHsp migrate to the membrane [53]. Summing up, we conclude that phosphorylation somehow affects the interaction of sHsp with biological membranes, although many important questions remain unanswered. It is obvious that Hsp25 can interact both with phospholipids and proteins of biological membranes and that the interaction with membrane proteins can also depend on the extent of sHsp phosphorylation. Therefore, the quantity of sHsp bound to the membrane and its phosphorylation will depend on the total concentration of Hsp25 in the cell, the quantity and properties of protein targets inside of the membrane, and on the activity of membrane-bound protein kinases that are able to phosphorylate sHsp. Let us analyze the interaction of Hsp25 with different cytoskeleton proteins.
Heat shock or oxidative stress is accompanied by fragmentation of actin filaments and their dissociation from focal contacts fixed at the cell membrane [48, 52, 53, 56]. Similar effects were observed in response to the action of certain hormones [30]. Increased expression of intact Hsp25/27 protects actin filaments from fragmentation and preserves their contacts with the cell membrane [30, 47, 48, 52, 56, 57]. Wild type Hsp25 partially protected actin from depolymerization induced by cytochalasin D [58]. Mutant Hsp25/27 with replacement of Ser (which can be phosphorylated by MAPKAP-kinases) by Ala or Gly, was unable to protect actin from different stresses [30, 52, 56]. It is interesting that phosphorylation of Hsp25 is accompanied by polymerization of actin in the perimembrane region [52, 55], increased pinocytosis [52], increased secretion of certain hormones [59], and increased blebbing [56]. Inhibition of p38 MAP-kinase by specific inhibitors also prevented formation of actin filaments in the perimembrane region and membrane blebbing [30, 56].
The following hypothesis can be put forward in order to explain the effect of Hsp25 on actin cytoskeleton. We may suppose that under normal conditions the extent of Hsp25 phosphorylation is low and therefore it does not interact with membranes and remains in cytosol. Unphosphorylated Hsp25 interacts with the barbed end of actin filaments and prevents actin polymerization. Under different stress conditions or/and activation of protein kinase cascades induced by hormones Hsp25 becomes phosphorylated. This leads to redistribution of Hsp25 from cytosol to the membrane. In parallel phosphorylation of Hsp25 reduced its interaction with actin and therefore decreases its inhibitory effect on actin polymerization. This leads to increased rate of actin polymerization. This explanation does not contradict the largest portion of experimental results. However, many questions remain unanswered. First, as mentioned earlier, translocation of sHsp inside the cell does not always correlate with phosphorylation [53, 55]. Second, the question about the effect of Hsp25 on actin polymerization still remains controversial. Even more controversial is the question on the effect of phosphorylation of Hsp25 on its interaction with actin [34, 44]. Third, if phosphorylation decreases the interaction of Hsp25 with actin, why does Hsp25 (becoming predominantly phosphorylated) move to actin filaments after different type of stresses [30]. Fourth, if phosphorylation results in dissociation of Hsp25 from actin filaments, why are filament bundles formed in the perimembrane region close to the sites of Hsp25 binding [30].
The data mentioned indicate an important role of small heat shock proteins in formation and keeping of the cytoskeleton. The recently published data indicate that Hsp25 may participate in regulation of cell motility and smooth muscle contraction. Endothelin-1 stimulates contractile activity of vascular smooth muscle. It is supposed that endothelin stimulates phosphoinositide-3 kinase, this leading to translocation of Rho A from cytosol to membrane. These processes are accompanied by migration of Hsp25 from cytosol to the membrane and enhancement of contractile activity [49]. Antibodies to Hsp25 and a specific inhibitor of p38 MAP-kinase significantly diminished contraction induced by endothelin-1 in permeabilized cells [31]. Different growth factors and cytokines induce enhanced motility of smooth muscle cells that start to migrate and leave tissues where they are usually located. As a consequence of these events, different pathological processes (such as atherosclerosis, angiogenesis, hypertrophy and hyperplasia of smooth muscle) can start. Expression of unphosphorylatable forms of Hsp25 or specific inhibitors of p38 MAP-kinase completely prevents the effects of platelet growth factor or interleukin-1beta on the motility of smooth muscle cells [28].
The mechanism of the activating effect of Hsp25 on the contractile activity of smooth muscle cells remains enigmatic. One can suppose that small heat shock proteins affect contractile activity of smooth muscle not only because of their interaction with actin but also by affecting the interaction of actin-binding proteins with actin. There are indications that small heat shock protein interacts with tropomyosin, caldesmon, and smooth muscle myosin [50]. Under certain conditions Hsp25 interacts with alpha-actinin and vinculin [16], troponin T [60], and tubulin [61]. The content of small heat shock protein in certain tissues (especially in different types of muscle) is very high and is equal to 2 mg of Hsp25 per g of tissue protein [42]. At this high concentration Hsp25 may interact with a number of different actin-binding proteins and by this means strongly affect polymerization of actin and regulation of contractile activity.
Finishing this chapter it is worthwhile to mention the role of another small heat shock protein in regulation of smooth muscle contraction. It has been found that cyclic nucleotide dependent relaxation of smooth muscle correlates with phosphorylation of sHsp with molecular weight of 20 kD [22, 25, 62, 63]. Activation of cyclic-nucleotide dependent pathway inhibits contraction of carotid arteries induced by serotonin, and this is not accompanied by significant change in the extent of phosphorylation of myosin light chain [22]. It was also shown that contraction of renal arteries is accompanied by phosphorylation of myosin light chains and Hsp27, whereas relaxation is accompanied by decrease of the extent of phosphorylation of myosin light chains and Hsp27 and parallel increase in phosphorylation of Hsp20 [23]. Thus, it seems that phosphorylation of Hsp25/27 somehow activates smooth muscle contraction and increases motility of non-muscle cells, whereas phosphorylation of Hsp20 results in relaxation of different types of smooth muscles. Molecular mechanisms of these processes remain enigmatic; however, it is supposed that Hsp20 and Hsp25/27 may somehow interact with actin and actin-binding proteins and with the proteins of intermediate filaments and that this interaction somehow affects contractile activity of smooth muscles [24, 62].
PERSPECTIVES OF FUTURE INVESTIGATIONS
The literature concerning structure and properties of small heat shock proteins is very wide and cannot be overviewed in details in a small review. In addition to the above-mention review of MacRae [2] and review book containing information about all heat shock proteins [1], three interesting reviews were published very recently. A review of Clark and Muchowski [64] deals with potential role of sHsp in human diseases, a review of Gerthoffer and Gunst [65] is devoted to the analyzing of the role of sHsp in signal transduction and actin remodeling, and review of Narberhaus [67] describes general properties of small heat shock proteins. Summing up we conclude that there are many experimental results indicating that the small heat shock proteins play an important role in protection of cells from different stresses and in the transduction of the hormonal signal from receptors to the contractile apparatus and cytoskeleton. The largest progress was achieved in the field of cell biology. Distribution of sHsp, its phosphorylation, and participation of different protein kinases involved in phosphorylation of sHsp are thoroughly investigated. At the same time clear interpretation of the results obtained on the cell level is still impossible. Therefore detailed biochemical investigation performed on isolated proteins seems to be very important. The main efforts should be directed to the investigation of interaction of sHsp with monomeric and polymeric actin. It will be necessary to analyze interaction of Hsp25 with different actin-binding proteins and analysis of the effect of sHsp on the interaction of actin-binding proteins with actin. Analysis of the interaction of sHsp with biological membranes and membrane proteins is also necessary. Joint utilization of biochemical and cell-biological methods will provide new information important for a detailed description of the mechanism of action of sHsp in the cell.
This work was performed with financial support of the Russian Foundation for Basic Research, a joint project of the Russian Foundation of Basic Research and INTAS, and the Wellcome Trust.
REFERENCES
1.Gething, M.-J. (ed.) (1997) Guidebook to
Molecular Chaperones and Protein-Folding Catalysts, Oxford
University Press, Oxford.
2.MacRae, T. H. (2000) Cellular and Molecular Life
Sciences, 57, 899-913.
2a. Wang, K., and Spector, A. (2001) Eur. J. Biochem.,
268, 6335-6345.
3.Ehrnsperger, M., Gräber, S., Gaestel, M., and
Buchner, J. (1997) EMBO J., 16, 221-229.
4.Wang, X.-Y., Chen, X., Oh, H.-J., Repasky, E.,
Kazim, L., and Subjeck, J. (2000) FEBS Lett., 465,
98-102.
5.Berengian, A. R., Parfenova, M., and Mchaourab, H.
S. (1999) J. Biol. Chem., 274, 6305-6314.
6.Benjamin, I. J., and McMillan, D. R. (1998)
Circ. Res., 83, 117-132.
7.Suzuki, A., Sugiyama, Y., Hayashi, Y., Nyu, I. N.,
Yoshida, M., Nonaka, I., Ishiura, S., Arahata, K., and Ohno, S.
(1998) J. Cell Biol., 140, 1113-1124.
8.Sugiyama, Y., Suzuki, A., Kishikawa, M., Akutsu,
R., Hirose, T., Waye, M. M. Y., Tsui, S. K. W., Yoshida, S., and Ohno,
S. (2000) J. Biol. Chem., 275, 1095-1104.
9.Smith, C. C., Yu, Y. X., Kulka, M., and Aurelian,
L. (2000) J. Biol. Chem., 275, 25690-25699.
10.Benndorf, R., Sun, X., Gilmont, R. R., Biederman,
K. J., Molloy, M. P., Goodmurphy, C. W., Cheng, H., Andrews, P. C., and
Welsh, M. J. (2001) J. Biol. Chem., 276, 26753-26761.
11.Lambert, H., Charette, S., Bernier, A. F.,
Guimond, A., and Landry, J. (1999) J. Biol. Chem., 274,
9378-9385.
12.Feil, I. K., Malfois, M., Hedle, J., van der
Zandt, H., and Svergun, D. I. (2001) J. Biol. Chem.,276,
12024-12029.
13.Kim, K. K., Kim, R., and Kim, S.-H.
(1998) Nature (London), 394, 595-599.
13a. Van Montfort, R. L. M., Basha, E., Friedrich, K. L., Slingsby, C.,
and Vierling, E. (2001) Nature Struct. Biol., 8,
1025-1030.
14.Mchaourab, H. S., Berengian, A. R., and Koteiche,
H. A. (1997) Biochemistry, 36, 14627-14634.
15.Bova, M. P., Mchaourab, H. S., Han, Y., and Fung,
B. K.-K. (2000) J. Biol. Chem., 275, 1035-1042.
16.Ding, L., and Candido, E. P. (2000) J. Biol.
Chem., 275, 9510-9517.
17.Leroux, M. R., Melki, R., Gordon, B., Batelier,
G., and Candido, E. P. M. (1997) J. Biol. Chem., 272,
24646-24656.
18.Cooper, L. F., and Uoshima, K. (1994) J. Biol.
Chem., 269, 7869-7873.
19.Guo, Z., and Cooper, L. (2000) Biochem.
Biophys. Res. Commun., 270, 183-189.
20.Boelens, W. C., Croes, Y., de Ruwe, M., de Reu,
L., and de Jong, W. W. (1998) J. Biol. Chem., 273,
28085-28090.
21.Lindner, R. A., Carver, J. A., Ehrnsperger, M.,
Buchner, J., Esposito, G., Behlke, J., Lutsch, G., Kotlyarov, A., and
Gaestel, M. (2000) Eur. J. Biochem.,267, 1923-1932.
22.Woodrum, D. A., Brophy, C. M., Wingard, C. J.,
Beall, A., and Rasmussen, H. (1999) Am. J. Physiol. Heart Circ.
Physiol., 277, H931-H939.
23.Beall, A., Epstein, A., Woodrum, D., and Brophy,
C. M. (1999) Biochim. Biophys. Acta, 1449, 41-49.
24.Brophy, C. M., Dickinson, M., and Woodrum, D.
(1999) J. Biol. Chem., 274, 6324-6329.
25.Rembold, C. M., Foster, D. B., Strauss, J. D.,
Wingard, C. J., and van Eyk, J. E. (2000) J. Physiol.,
524, 865-878.
26.Landry, J., Lambert, H., Zhou, M., Lavoie, J. N.,
Hickey, E., Weber, L. A., and Anderson, C. W. (1992) J. Biol.
Chem., 267, 794-803.
27.Larsen, J. K., Yamboliev, I. A., Weber, L. A.,
and Gerthoffer, W. T. (1997) Am. J. Physiol., 273,
L930-L940.
28.Hedges, J. C., Dechert, M. A., Yamboliev, I. A.,
Martin, J. L., Hickey, E., Weber, L. A., and Gerthoffer, W. T. (1999)
J. Biol. Chem., 274, 24211-24219.
29.Schafer, C., Ross, S., Bragado, M. J.,
Groblewski, G. E., Ernst, S. A., and Williams, J. A. (1998) J. Biol.
Chem., 273, 24173-24180.
30.Schäfer, C., Clapp, P., Welsh, M. J.,
Benndorf, R., and Williams, J. A. (1999) Am. J. Physiol.,
277, C1032-C1043.
31.Yamboliev, I. A., Hedges, J. C., Mutnick, J.
L.-M., Adam, L. P., and Gerthoffer, W. T. (2000) Am. J. Physiol.
Heart Circ. Physiol., 278, H1899-H1907.
32.Kato, K., Ito, H., Iwamoto, I., Lida, K., and
Inaguma, Y. (2001) Cell Stress Chaperone, 6, 16-20.
33.Maizels, E. T., Peters, C. A., Kline, M., Cutler,
R. E., Shanmugam, M., and Hunzicker-Dunn, M. (1998) Biochem. J.,
332, 703-712.
34.Butt, E., Immler, D., Meyer, H. E., Kotlyarov,
A., Laaß, K., and Gaestel, M. (2001) J. Biol. Chem.,
276, 7108-7113.
35.Gaestel, M., Benndorf, R., Hayess, K., Priemer,
E., and Engel, K. (1992) J. Biol. Chem., 267,
21607-21611.
36.Ito, H., Kamei, K., Iwamoto, I., Inaguma, Y.,
Nohara, D., and Kato, K. (2001) J. Biol. Chem., 276,
5346-5352.
37.Rogalla, T., Ehrnsperger, M., Preville, X.,
Kotlyarov, A., Lutsch, G., Ducasse, C., Paul, C., Wieske, M., Arrigo,
A. P., Buchner, J., and Gaestel, M. (1999) J. Biol. Chem.,
274, 18947-18956.
38.Ehrnsperger, M., Lilie, H., Gaestel, M., and
Buchner, J. (1999) J. Biol. Chem., 274, 14867-14874.
39.Haslbeck, M., Walke, S., Stromer, T.,
Ehrnsperger, M., White, H. E., Chen, S., Saibil, H., and Buchner, J.
(1999) EMBO J., 18, 6744-6751.
40.Preville, X., Schultz, H., Knauf, U., and Arrigo,
A. P. (1998) J. Cell Biochem., 69, 436-452.
41.Preville, X., Gaestel, M., and Arrigo, A. P.
(1998) Cell Stress Chaperones, 3, 177-187.
42.Miron, T., Wilchek, M., and Geiger, B. (1988)
Eur. J. Biochem., 178, 543-553.
43.Wieske, M., Benndorf, R., Behlke, J., Dolling,
R., Grelle, G., Bielka, H., and Lutsch, G. (2001) Eur. J.
Biochem., 268, 2083-2090.
44.Benndorf, R., Hayeß, K., Ryazantsev, S.,
Wieske, M., Behlke, J., and Lutsch, G. (1994) J. Biol. Chem.,
269, 20780-20784.
45.Perng, M. D., Cairns, L., van den Ijssel, P.,
Prescott, A., Hutcheson, A. M., and Quinlan, R. A. (1999) J. Cell
Sci.,112, 2099-2112.
46.Miron, T., Vancompernolle, K., Vandekerckhove,
J., Wilchek, M., and Geiger, B. (1991) J. Cell Biol.,
114, 255-261.
47.Loktionova, S. A., Ilyinskaya, O. P., Gabai, V.
L., and Kabakov, A. E. (1996) FEBS Lett., 392,
100-104.
48.Loktionova, S. A., and Kabakov, A. E. (1998)
FEBS Lett., 433,294-300.
49.Wang, P., and Bitar, K. (1998) Am. J.
Physiol., 275, G1454-G1462.
50.Ibitayo, A. I., Sladick, J., Tuteja, S.,
Louis-Jacques, O., Yamada, H., Groblewski, G., Welsh, M., and Bitar, K.
(1999) Am. J. Physiol., 277, G445-G454.
51.Muller, E., Burger-Kentischer, A., Neuhofer, W.,
Frank, M. L., Marz, J., Thurau, K., and Beck, F. X. (1999) J. Cell
Physiol., 181, 462-469.
52.Lavoie, J. N., Hickey, E., Weber, L. A., and
Landry, J. (1993) J. Biol. Chem., 268, 24210-24214.
53.Lavoie, J. N., Lambert, H., Hickey, E., Weber, L.
A., and Landry, J. (1995) Mol. Cell. Biol.,15,
505-516.
54.Kato, K., Goto, S., Inaguma, Y., Hasegawa, K.,
Morishita, R., and Asano, T. (1994) J. Biol. Chem., 269,
15302-15309.
55.Pietrowicz, R. S., and Levin, E. G. (1997) J.
Biol. Chem., 272, 25920-25927.
56.Huot, J., Houle, F., Rousseau, S., Deschesnes, R.
G., Shah, G. M., and Landry, J. (1996) J. Cell Biol.,
143, 1361-1373.
57.Huot, J., Houle, F., Spitz, D. R., and Landry, J.
(1996) Cancer Res., 56, 273-279.
58.Lavoie, J. N., Gingras-Breton, G., Tanguay, R.
M., and Landry, J. (1993) J. Biol. Chem., 268,
3420-3429.
59.Pietrowicz, R. S., Martin, J. L., Dillman, W. H.,
and Levin, E. G. (1997) J. Biol. Chem., 272,
7042-7047.
60.Ehrnsperger, M., Hergersberg, C., Wienhues, U.,
Nicht, A., and Buchner, J. (1998) Analyt. Biochem., 259,
218-225.
61.Hino, M., Kurogi, K., Okubo, M. A., Murata-Hori,
M., and Hosoya, H. (2000) Biochem. Biophys. Res. Commun.,
271, 164-169.
62.Beall, A. C., Kato, K., Goldenting, J. R.,
Rasmussen, H., and Brophy, C. M. (1997) J. Biol. Chem.,
272, 11283-11287.
63.Beall, A., Bagwall, D., Woodrum, D., Stoming, T.
A., Kato, K., Suzuki, A., Rasmussen, H., and Brophy, C. M. (1999) J.
Biol. Chem., 274, 11344-11351.
64.Clark, J. I., and Muchowski, P. J. (2000)
Curr. Opin. Struct. Biol., 10, 52-59.
65.Gerthoffer, W. T., and Gunst, S. J. (2001) J.
Appl. Physiol., 91, 963-972.
66.Sugden, P. H., and Clerk, A. (1998) Cir.
Res., 83, 342-352.
67.Narberhaus, F. (2002) Microbiol. Mol. Biol.
Rev., 66, 64-93.