REVIEW: Pyridoxal 5´-Phosphate as a Catalytic and Conformational Cofactor of Muscle Glycogen Phosphorylase b
N. B. Livanova*, N. A. Chebotareva, T. B. Eronina, and B. I. Kurganov
Bach Institute of Biochemistry, Russian Academy of Sciences, Leninskii pr. 33, Moscow, 119071 Russia; fax: (095) 954-2732; E-mail: livanova@inbi.ras.ru* To whom correspondence should be addressed.
Received April 17, 2002; Revision received May 28, 2002
This review summarizes data on structure of muscle glycogen phosphorylase b and the role of the cofactor pyridoxal 5´-phosphate in catalysis and stabilizing the native conformation of the enzyme. Specific attention is paid to the stabilizing role of pyridoxal 5´-phosphate upon denaturation of phosphorylase b. Stability of holoenzyme, apoenzyme, and enzyme reduced by sodium borohydride is compared.
KEY WORDS: glycogen phosphorylase b, mechanism of reaction, pyridoxal 5´-phosphate, regulation, thermal inactivation, quaternary structure
Abbreviations: DSC) differential scanning calorimetry; GuHCl) guanidine hydrochloride; Pha) glycogen phosphorylase a; Phb) glycogen phosphorylase b; PLP) pyridoxal 5´-phosphate.
Glycogen phosphorylase (1,4-alpha-D-glucan:
orthophosphate-alpha-D-glucosyltransferase; EC 2.4.1.1) is a key
enzyme of glycogen metabolism in skeletal muscle. Phosphorylase
catalyzes the first step in the pathway of degradation of the principal
storage form of carbohydrate in muscle--glycogen:
(alpha-1,4-glycoside)n + Pi (alpha-1,4-glycoside)n-1+ alpha-D-glucose 1-phosphate,
where n is the number of glucose residues. The phosphorolytic cleavage of the alpha-1,4-glucosidic bonds proceeds from the non-reducing ends of the glycogen chain, and phosphorylase extends the splitting up to the fourth residue from a point of branching. The reaction catalyzed by phosphorylase is reversible and the ratio of the equilibrium concentrations [glucose 1-phosphate]/[Pi] is 0.28 at pH 6.8. However, in vivo the enzyme acts only in the direction of glycogen phosphorolysis because in the cell the inorganic phosphate concentration significantly exceeds that of glucose 1-phosphate. The physiological role of phosphorylase in skeletal muscle is that it provides an energy source by generating glucose 1-phosphate via the phosphorylase reaction.
Phosphorylase activity in muscles was discovered in 1936 by Parnas [1] and by Cori and Cori [2] and since that time during more than six decades phosphorylase continues to be the subject of very intensive research throughout the world. Many years of phosphorylase study brought a number of fundamental discoveries founding the development of several new promising lines in enzymology. Thus, phosphorylase was the first enzyme shown to undergo allosteric activation by an effector (AMP) which has no structural similarity to the substrate [3, 4]; phosphorylase was the first enzyme demonstrated to be regulated by transitions at the level of its quaternary structure [5, 6]; covalent modification via phosphorylation was also described for the first time for muscle phosphorylase [7]; phosphorylase was unique among pyridoxal enzymes where a phosphate but not an aldimine group of the cofactor participates in the catalysis [8]. The properties of glycogen phosphorylase have been elucidated in several reviews [8-12].
Phosphorylase comprising up to 2% of the total amount of soluble protein in muscle tissue exists in resting muscle as an inactive (without AMP) non-phosphorylated form b. Phosphorylase b is regulated by several effectors, among which AMP and IMP are known as activators [3], ATP and ADP [13, 14], purines [15], flavins [16], D-glucose and UDP-glucose [17, 18] as inhibitors.
Phb in solution without any additions exists as a dimer and Pha under high concentration of the enzyme as a tetramer [19]. A dimer of phosphorylase consists of two symmetrically positioned monomers with the full set of the binding sites for specific ligands. However, the active conformation the enzyme develops only in the dimeric form; tetramers are inactive [20]. Monomers show no catalytic activity as well [21]. Each monomer contains 842 amino acid residues and one residue of PLP and has a relative molecular mass 97,432 daltons. The amino acid sequence of the monomer polypeptide chain has been established [22, 23].
A schematic ribbon diagram of the Phb monomer is shown in Fig. 1 [24]. Each monomer represents a protein of alpha-beta type with the polypeptide chain folded into a rather compact globule with radius of 3 nm, consisting of two separate domains: N-terminal (residues 10-484) and C-terminal (residues 485-842). The catalytic site, identified by binding of the substrates glucose 1-phosphate, Pi, and glycogen and of the competitive inhibitor glucose, is located in a deep crevice between the two domains in the center of the molecule. The cofactor necessary for the activity, PLP, also constitutes part of the active site. The aldehyde group of PLP is bound as a Schiff base with Lys680 [25].
The allosteric regulatory site is positioned at the subunit interface in the dimer and its distance from the active site is approximately 3.3 nm [26]. At this site, binding of both allosteric activators (AMP, IMP) and inhibitors (ATP, glucose 6-phosphate, UDP-glucose) occurs. Glucose 6-phosphate is a potent physiological inhibitor of Phb, being a classic feedback inhibitor. It is constantly generated in muscle from the product of the phosphorylase reaction, glucose 1-phosphate, by phosphoglucomutase, and ensures that Phb is inactive in resting muscle.Fig. 1. Schematic diagram of the Phb subunit in the T-state [24]. View down the crystallographic Y-axis. alpha-Helices and beta-strands are shown as cylinders and arrows, respectively.
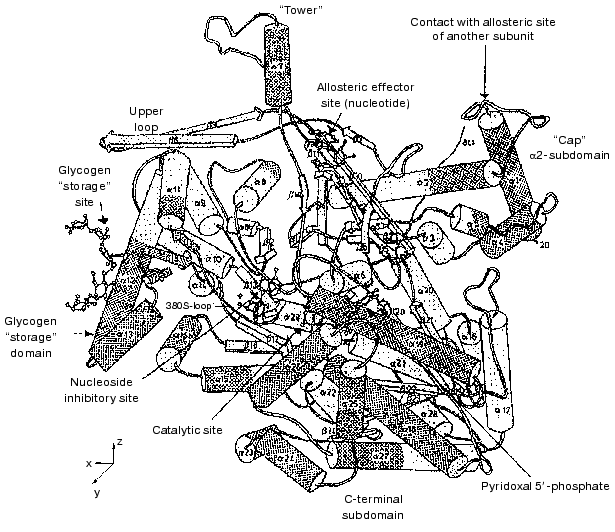
In the region of the subunit interface, at a distance of 1.2 nm from the nucleotide site, the phosphorylation site is identified as Ser14 [27]. In Fig. 1 it is not indicated because the N-terminal tail (residues 1 to 18) is disordered and could not be localized it in the molecule of Phb by crystallographic methods. After phosphorylation of Ser14 by phosphorylase kinase, the phosphate group interacts with Arg69 of the same subunit, and with Arg43 of the neighboring subunit, and the appearance of a dianion causes the ordering of the basic N-tail.
At the entrance into the tunnel leading to the catalytic site, at a distance of approximately 1 nm from this site, the nucleoside (inhibitor) site is located. In the T-state of phosphorylase, various heterocyclic compounds (adenine, adenosine, caffeine, flavins) are able to bind to this site [10, 28, 29]. Upon binding to this site, the inhibitors are intercalated between the aromatic side chains of Phe285, which belongs to the N-terminal domain, and Tyr613, which belongs to the C-terminal domain, and close the entrance to the active site. AMP and IMP are also able to bind to the nucleoside site with low affinity, in this case acting as inhibitors.
A special “glycogen storage site” is located on the surface of the protein globule at the side of the dimer-dimer interface at a distance of approximately 3 nm from the active site [10]. This is, probably, the site of phosphorylase binding to glycogen granules. It should be noted that in skeletal muscle up to 70% of the whole amount of phosphorylase, existing in a dimeric form as a complex with glycogen, is localized in these granules in addition to other enzymes of glycogen metabolism [30].
The subunit interface of the dimer is composed of two main contact regions: one is formed between the “cap” region, a loop connecting the alpha1 and alpha2 helices of one subunit with the beta7 strand, and the alpha2 helix of the opposite subunit (this area includes AMP- and Ser-P binding sites). Antiparallel association of the two symmetry-related “tower”-helices-alpha7 forms the other contact [27].
The dephosphorylated form b displays more pronounced allosteric regulation by effectors. The existence of the active conformation, R, induced by activator AMP and by substrates, and the less active one, T, induced by allosteric inhibitors, were first postulated in 1967 by Helmreich et al. [31]. Later, the molecular mechanism of the reversible transition R *** T was elaborated in detail by Johnson's group [27].
ROLE OF PLP AS A CATALYTIC COFACTOR
Pyridoxal 5´-phosphate was discovered in 1957 by Baranowski et al. [32] in rabbit muscle glycogen phosphorylase a in a stoichiometric ratio of 1 mole per mole of subunit. All glycogen phosphorylase prepared from different sources of animal, plant, and microbial origin contain PLP in the same stoichiometric ratio [33]. PLP participates as a cofactor in several enzymatic reactions of amino acid metabolism (transamination, decarboxylation, beta-elimination, etc.). As a basis of all these reactions, so unlike on first sight, is the common mechanism discovered first by Braunstein and Shemyakin [34-36] and by Snell and coworkers [37]. According to this mechanism, the main intermediate step in the reactions of such a type is the formation of Schiff base between pyridoxal phosphate (or pyridoxamine phosphate) and proper amino acid (or keto acid). In such “classic” pyridoxal enzymes cofactor is bound as a Schiff base (by aldimine bond) with epsilon-amino group of corresponding lysine residue of the protein molecule. The presence of this bond is a necessary condition to display the catalytic activity. Reduction of aldimine bond with sodium borohydride causes complete and irreversible loss of activity. Phosphorylase in this sense is not typical pyridoxal enzyme. Though PLP in phosphorylase as in other pyridoxal enzymes is connected by aldimine bond with epsilon-amino group of Lys680 [25], the existence of this bond is not essential for the phosphorylase reaction, and its reduction by sodium borohydride irreversibly fixes PLP on the enzyme with retention of 60% activity of the native enzyme [38]. Thus, the aldehyde group of PLP in position 4, which is necessary for the binding of cofactor, is not involved in catalysis. This is not surprising because phosphorylase catalyzes the phosphorolytic cleavage of glycogen, and it does not participate in amino acid metabolism. The resolution of Phb under mild conditions (preincubation in the protein structure deforming imidazole citrate buffer with subsequent removal of PLP by L-cysteine) causes the formation of completely inactive apoenzyme, which easy dissociates into monomers [39].
This raised the question of whether PLP functions as a structural ligand stabilizing the active dimeric structure of Phb, or it is involved in catalysis. The use of different chemical analogs of PLP with modification in every position around the pyridine ring showed that PLP not only supports the native structure of enzyme, but also participates in catalysis [8, 33, 40-42]. Partial or complete restoration of enzymatic activity occurred under reconstruction with analogs modified in positions 2, 3, and 6 of the pyridine ring of PLP.
None of the analogs modified in position 4 restored enzymatic activity. Once bound, however, the formyl group can be modified, e.g., by sodium borohydride reduction that covalently fixes the cofactor to the protein with retention of activity. This means that formyl group in position 4 takes part in the binding of cofactor with protein, but not in catalysis. The modification of pyridine ring in position 1, and all modifications of the 5´-phosphate group yielded inactive phosphorylase.
It was shown by the studies of Graves's laboratory [43, 44] that the phosphate moieties of PLP and the substrate phosphates are sufficiently close together in phosphorylase to interact directly.
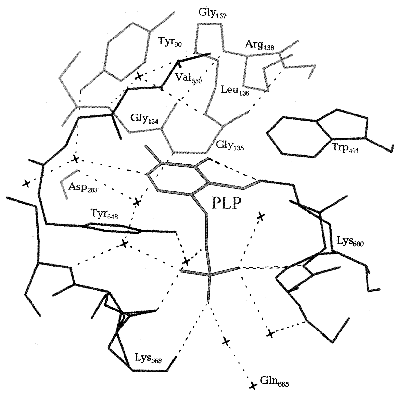
The exact arrangement of PLP in phosphorylase molecule and the nature of amino acid residues of the protein molecule interacting with the distinct structural elements of cofactor became evident after X-ray analysis of Johnson et al. [45]. The cofactor is buried in a highly hydrophobic environment, and its aldehyde group is binding as a Schiff base with Lys680. PLP is stabilized in a fixed conformation through many noncovalent interactions of the side groups of cofactor with the amino acid residues of both C- and N-terminal domains (Fig. 2). Among such interactions the interaction of the pyridine ring of cofactor with the neighboring three aromatic rings Tyr90, Trp491, and Tyr648 residues should be noted, and also the numerous Van-der-Waals interactions of C2´-methyl group and C6´-atom of the pyridine ring with residues Gly134, Gly135, and Leu136. Besides that, three oxygen atoms of the phosphate group of the cofactor form many hydrogen bonds both directly with the terminal groups of the amino acid residues (Lys568, Thr676, Gly677), and via water molecules with the far removed groups.
Upon the allosteric transition T --> R the affinity of the catalytic site for phosphate increases 15-fold [46, 47]. The sequence of events upon this transition begins with antiparallel interaction of two symmetrical tower alpha-helices of the two neighboring subunits. The tower alpha-helix is immediately connected with loop 280S which is then displaced and opens the entrance to the active site, simultaneously blocking access to the inhibitory site. In addition, displacement of this loop causes a shift of the side chain of Arg569 of the active site by 0.7 nm. The basic residue Arg569 replaces the acidic residue Asp283. This replacement ensures the increase in affinity to the phosphate group of the substrate and makes possible a closer contact between the substrate anion and anion of the cofactor, PLP, which constitutes an important part of the catalytic mechanism. As was shown by X-ray analysis [27] and NMR spectroscopy [48], upon transition from T- into R-state, and upon activation by phosphorylation the phosphate group of PLP is converted from monoanion to dianion, and it reverts to a monoanion upon ligation of the substrate phosphate. PLP is located inside a structurally conservative area, and therefore the cofactor does not change its position upon the T --> R transition [24].Fig. 2. Scheme of the active site of Phb. The molecules of water are shown by crosses [45].
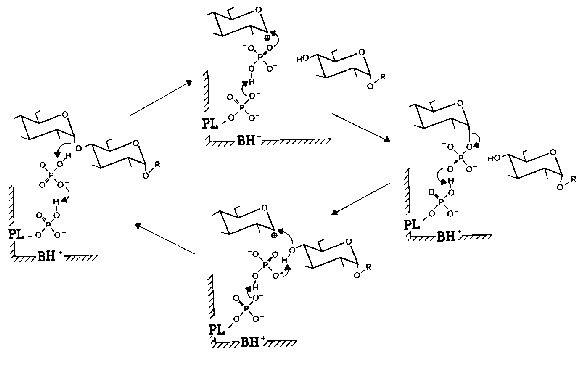
Nowadays is commonly accepted the mechanism of phosphorylase reaction proposed by Helmreich and coworkers [33, 49-51]. According to this mechanism, 5´-phosphate group of PLP is functioning in phosphorylase in the form of dianion as a proton donor-acceptor according to the general scheme of acid-base catalysis. Figure 3 presents in detail the mechanism of PLP participation in the reaction catalyzed by muscle phosphorylase. When the reaction proceeds in the forward direction (phosphorolysis of alpha-1,4-glycosidic bond) the first step is the protonation of the glycoside oxygen by orthophosphate. The oxocarbonium ion formed is stabilized by phosphate-anion. The subsequent covalent binding of phosphate causes the formation of glucose 1-phosphate. When the reaction proceeds in the reverse direction (synthesis of oligosaccharides) the protonation of the phosphate group of glucose 1-phosphate destabilizes the glycoside bond, and that favors the formation of the couple glycosyloxocarbonium ion-phosphate-anion. Phosphate-anion substantially facilitates the nucleophilic attack of the terminal glycosylic residue to the carbonium ion. This sequence of the reactions causes the formation of alpha-1,4-glycosidic bond and increase in the number of glycosidic residues in oligosaccharide primer. Thus, the function of PLP in the reaction catalyzed by phosphorylase is absolutely different compared to other PLP-dependent enzymes.
Fig. 3. Mechanism of PLP participation in the enzymatic reaction catalyzed by muscle Phb [51]. The reaction scheme accounts for the reversibility of phosphorolysis of oligosaccharides (R) in the presence of orthophosphate (upper half) and primer-dependent synthesis in the presence of glucose 1-phosphate (lower half). PL) Enzyme-bound pyridoxal; BH+) protonatable general base contributed by the enzyme protein.
ROLE OF PLP AS A CONFORMATIONAL COFACTOR
Quaternary structure of holoenzyme, apoenzyme, and enzyme reconstructed with PLP analogs. Phb in solution without any additions exists as a dimer which attains the active conformation in the presence of allosteric activator--AMP [19]. At rather high concentration of the enzyme in the presence of AMP the active dimer is transformed into the inactive tetramer; the formation of the tetramer is favored by low temperature [52]. The addition of high molecular weight substrate glycogen causes the dissociation of tetramer into dimers [8]. The equilibrium dimer *** tetramer is moved in favor of dimer in the presence of inhibitors--glucose [8, 52] and FMN [16, 53, 54]. The dimers of holoenzyme dissociate into monomers only under rather harsh conditions (in the presence of PCMB [5]).
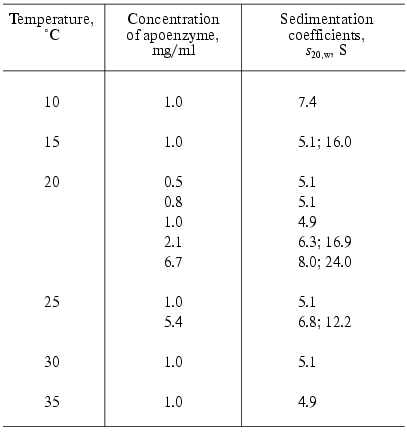
Apophosphorylase b (apoPhb) was firstly obtained in 1957 by Cori and Illingworth [55] by precipitation of the enzyme with ammonium sulfate at pH 3.4. The main part of the enzyme was denatured under such conditions. But it was possible to reactivate part of the apoenzyme leaving in solution by incubation at pH 6.0 with the equimolar quantity of PLP. In 1966 Shaltiel et al. [56] proposed the new, mild method of resolution of PLP from Phb, including two steps: the exposure of PLP on the surface of the protein globule by incubation with the structure deforming reagent, imidazole citrate buffer at pH 6.2, and subsequent removal of PLP by L-cysteine. The apoenzyme obtained in such a way was practically inactive, but was reconstructed and reactivated by incubation with equimolar amount of PLP by approximately 95%. The study of the properties of apoPhb showed that it is much more labile than the holoenzyme and presents a typical association-dissociation system with the state of aggregation very sensitive to the change of physicochemical conditions [39, 57, 58]. Decrease in temperature and increase in the protein concentration cause the aggregation of apoenzyme. In beta-glycerophosphate buffer in temperature intervals 15-35°C at the protein concentration until 1 mg/ml apoenzyme exists as a monomer (sedimentation coefficient s20,w = 5.1-5.3 S), at 10°C as a dimer (s20,w = 7.4 S) (Table 1) [58], and at 1°C as a mixture of dimers, tetramers, and aggregates [39]. It should be noted that holoPhb in the absence of AMP exists exclusively as a dimer independent of temperature.
Table 1. Sedimentation properties of
apophosphorylase b (0.05 M beta-glycerophosphate
buffer, pH 6.8) [58]
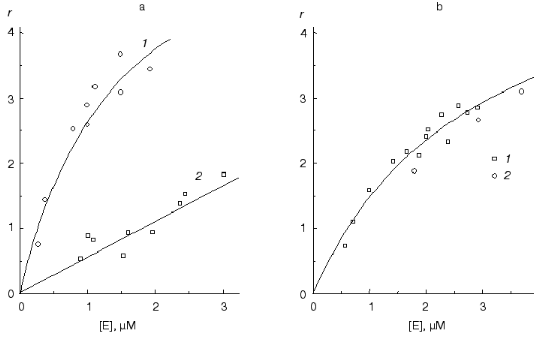
ApoPhb retains the ability to interact with AMP because the addition of this nucleotide results in the association and crystallization of the apoenzyme and protects it from denaturation in storage. ApoPhb retains the ability to transform into apoPha on incubation with phosphorylase kinase, i.e., the phosphorylation site, Ser14, does not change significantly its position in the apoenzyme as compared to holoPhb [8, 39]. However, apo- and holoenzyme have different affinity for the high molecular weight substrate, glycogen, and to the allosteric inhibitor, FMN. To calculate the dissociation constant (Kdiss) of the complex of phosphorylase with glycogen, we used the following equation: r = rmax/(1 + Kdiss/[E]), where r is the degree of saturation of glycogen by Phb expressed as moles of Phb bound with 1 g of glycogen, rmax is the adsorption capacity of glycogen, and [E] is the equilibrium concentration of enzyme. The adsorption capacity of glycogen was taken to be identical both for holo- and apoenzyme (rmax = 3.6*10-6 moles of bound enzyme dimers per 1 g of glycogen or 0.71 g of enzyme per 1 g of glycogen [57, 59]). The affinity of apoenzyme to glycogen is 5-fold lower than the affinity of holoenzyme to this ligand (Fig. 4a) [57, 59]. As shown in Table 2, the affinity of apoPhb for FMN is 35-fold lower than the affinity of holoenzyme to this ligand. The data presented show that the removal of the cofactor causes structural rearrangements in the Phb molecule, in particular, in the contact area of monomers in a dimer, in the region of the glycogen storage site, and in the region of the allosteric inhibitor site. Some additional data, testifying to differences in the structure of holo- and apoPhb, were obtained using hydrogen-tritium exchange method [60] and modification of SH-groups [61]. The reconstruction of holoenzyme from apoenzyme and PLP causes the restoration of phosphorylase affinity for glycogen (Fig. 4b) and for FMN (Table 2).
Table 2. Affinity of native Phb, apoPhb, and reconstructed Phb for FMN (20°C, 50 mM glycyl-glycine buffer, pH 6.8) [57]Fig. 4. Binding isotherms of glycogen with the native Phb, apoPhb, and Phb reconstructed with PLP. The data are presented in coordinates {r; [E]}, where r is the amount of Phb (moles of the dimer) adsorbed by 1 g of glycogen (the average molecular mass of glycogen is 5.5*106 daltons). a) Native Phb (1), apoPhb (2) in 0.05 M beta-glycerophosphate buffer, pH 6.8, 20°C; b) native Phb (1), Phb reconstructed with PLP (2) in 0.05 M glycyl-glycine buffer, pH 6.8, 20°C [57].

The analytical ultracentrifugation study of transitions on the level of quaternary structure and affinity for several ligand (glycogen and FMN) with the enzyme reconstructed with PLP analogs modified in 5´-position of the cofactor, allowed us to obtain some additional data concerning the role of PLP in phosphorylase as a conformational cofactor [57, 59]. The analogs studied contained the following substituents in 5´-position instead of -CH2OP(O)(OH)2: -CH2CH2COOH (analog I), trans-CH=CHCOOH (analog II), -C.COOH (analog III). As would be expected, the forms of the enzyme reconstructed with these analogs did not show any activity. These forms differed significantly from the apoenzyme and the enzyme reconstructed with PLP in their capability for association. It is known that when the allosteric activator site of Phb is saturated with AMP the enzyme represents an associating system of the type dimer *** tetramer [53]. At 17°C the association constant, Kass, for the native Phb is equal to 2.4*104 M-1 (Table 3) [62]. The values of Kass for the native Phb and for Phb reconstructed with PLP are not different from each other. For the enzyme reconstructed with analogs I, II, and III, the equilibrium dimer *** tetramer is strongly shifted toward the formation of the tetrameric form (Table 3) [57]. The nature of the substituents in the 5´-position has practically no influence on the affinity of Phb for glycogen [57, 59]. We observed an absolutely different picture for binding of the allosteric inhibitor, FMN. As shown in Table 2, the affinity of the enzyme reconstructed with analogs I, II, and III for FMN is noticeably lower than that for the native enzyme for this effector, with analog III being as low as for apoenzyme.
Table 3. Influence of the reconstruction on
Phb association, induced by AMP (17°C, 50 mM
glycyl-glycine buffer, pH 6.8) [57]

All these data indicate that PLP plays an important role in maintaining the quaternary structure and particular conformation of the enzyme, important for the interaction with the substrate and allosteric inhibitors.
Thermal stability of holoenzyme, apoenzyme, and enzyme reduced by sodium borohydride. One of the most important physicochemical characteristics of proteins is their stability at elevated temperatures. First the decrease in thermal stability of apoPhb as compared to the holoenzyme was discovered by Hedrick et al. [39]. These authors showed that holoPhb is completely inactivated after 60-min incubation at 55°C, whereas the apoenzyme loses its ability for the restoration of the activity under the reconstruction with PLP after 60-min incubation at 45°C.
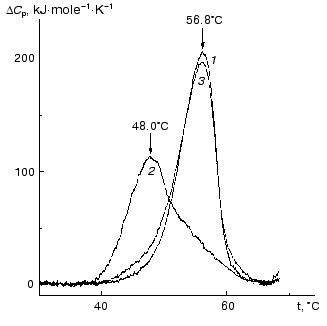
The kinetics of thermal denaturation of holoPhb, apoPhb, and Phb reduced by NaBH4 has been studied in detail [63-66]. The data presented showed that thermal denaturation proceeds according to a dissociative mechanism, i.e., includes a step of reverse dissociation of the dimer into the monomers with subsequent irreversible denaturation of the monomers. To characterize the role of cofactor of Phb, PLP, in sustaining the native conformation of the enzyme, the authors carried out a comparative analysis of thermal stability of the native Phb (holoform) and its apoform using differential scanning calorimetry (DSC). As can be seen from Fig. 5, a maximum on the excess heat capacity DeltaCp versus temperature profile for the apoform of Phb (48°C) is shifted markedly toward lower temperatures in comparison with that for the holoform (56.8°C). Thermal stability of the PLP-reconstructed enzyme (curve 3) is practically identical to that for the native enzyme. As mentioned above, the removal of PLP from Phb results in dissociation of the dimeric form into monomers [39, 58, 67]. Therefore, one can assume that the main reason for the lesser stability of the apoform of the enzyme is its easy transformation into the monomer form. Nevertheless, it is evident that PLP has a general stabilizing effect on the native Phb.
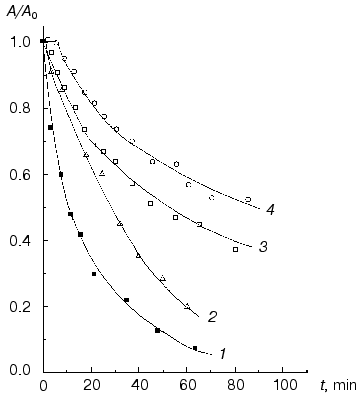
The study of thermal stability of Phb reduced by sodium borohydride gives an additional verification of PLP participation in stabilization of the native conformation of Phb. As seen from Fig. 6, thermal inactivation of Phb reduced by sodium borohydride proceeds much faster in comparison with the native enzyme. In both cases thermal inactivation is accelerated under dilution of the enzyme, i.e., it is believed that for the reduced enzyme it proceeds according to the dissociative mechanism, as for the native enzyme. A second circumstance which is worth mentioning is that if at the high concentration of the enzyme (5.2 mg/ml) at the initial part of the curve of thermal inactivation of the native enzyme a well pronounced lag period occurs, no lag period occurs on the corresponding curve for the reduced Phb. The lag period on the curve of thermal inactivation is connected with the presence of the step of predenaturational conformational changes in the dimer [63, 65]. Thus, the absence of lag period on the curve of thermal inactivation for the reduced form of Phb is a significant indication that during denaturation of this form the step of predenaturational conformational changes could be neglected. It is known that reduction of Phb by sodium borohydride causes substantial weakening of the intersubunit contacts in the dimer. Therefore, the reduced Phb partially dissociates into monomers at the relatively low concentrations of the enzyme (0.01 mg/ml) [67].Fig. 5. Excess heat capacity (DeltaCp) versus temperature profiles for holoform (1), apoform (2), and reconstructed form (3) of Phb (30 mM Hepes-KOH, pH 7.0). Enzyme concentration was 0.7 mg/ml for all the enzyme preparations. The scanning rate was 1°C/min [63].
Sedimentation analysis of NaBH4-reduced Phb (0.92 mg/ml) at 53°C indicates that the reduced enzyme moves as an unfolded monomer (s20,w = 3.6 S), whereas the native enzyme at the same temperature and protein concentration sediments mainly as a dimeric form [62]. Thus, at elevated temperatures NaBH4-reduced Phb dissociates into monomers more easily than the native enzyme.Fig. 6. Dependence of the relative activity (A/A0) on time (80 mM Hepes-NaOH, pH 6.8, 53°C) for the NaBH4-reduced form of Phb at enzyme concentration 1.6 (1) and 5.2 mg/ml (2) and for the native Phb at concentration 1.6 (3) and 5.2 mg/ml (4) [63].
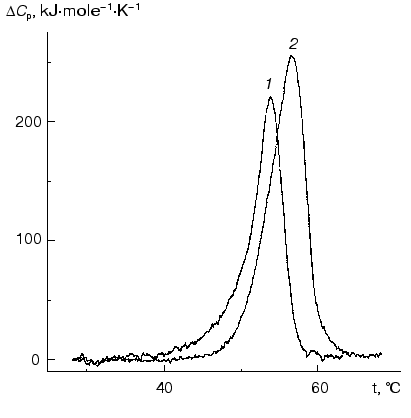
The lower thermal stability of sodium borohydride reduced Phb, as compared to the native enzyme, was also supported by DSC data. As can be seen from Fig. 7, the maximum on the DeltaCp versus temperature profile for the reduced form of Phb occurs at 54.2°C, i.e., at a lower temperature than for the native enzyme (56.8°C).
Stability of holoenzyme and enzyme reduced by sodium borohydride relative to guanidine hydrochloride (GuHCl). The data on Phb denaturation in the presence of GuHCl are also indicative of the lower stability of the bonds between the subunits in the dimer after reduction by sodium borohydride. Dissociation into monomers in the case of reduced Phb occurs under lower concentration of GuHCl as compared to the native enzyme. According to the data of analytical ultracentrifugation, after incubation with 0.2 M GuHCl for 2 h the native enzyme remains in the dimeric form (20°C, pH 6.8; 0.4 mg/ml of Phb) but 30% of the reduced enzyme exists under the same conditions as a monomer. The fact that the character of the curves of inactivation of the native Phb and Phb reduced with NaBH4 changes upon the variation of the protein concentration points to the dissociative mechanism of the unfolding of the protein molecule ([68], Chebotareva, Eronina, unpublished results).Fig. 7. Excess heat capacity (DeltaCp) versus temperature profiles for NaBH4-reduced (1) and for the native (2) forms of Phb (enzyme concentration for both preparations was 1 mg/ml) [63]. Hepes-KOH, 30 mM, pH 7.0. The scanning rate was 1°C/min.
Summing up of the preceding it is possible to conclude that PLP taking part in the catalysis and being a conformational cofactor of Phb plays a key role in the functioning of this enzyme. Since mammalian muscle contains phosphorylase in relatively high concentrations (approximately 30 mg per 100 ml of intracellular fluid), and since muscle tissue makes up 40% of the body mass, it is not difficult to calculate that more PLP is bound to muscle phosphorylase than is present in all other pyridoxal phosphate-dependent enzymes together [33].
Now it is possible to consider that the mechanism of participation of PLP in the reaction catalyzed by phosphorylase is revealed. It was established that the main catalytic group of PLP functioning as a donor-acceptor of a proton according to general scheme of acid-base catalysis is the phosphate group in 5´-position. It is important to note that phosphorylase is a glucosyl but not a phosphoryltransferase.
The direct participation of PLP in catalysis is accompanied by its role as a conformational cofactor of phosphorylase, stabilizing the active dimeric structure of the enzyme and regulating the affinity of Phb to the substrates and effectors.
This work was supported by grants 02-04-49099, 02-04-48704, and 00-15-97787 from the Russian Foundation for Basic Research.
We are very thankful to B. A. Kornilaev for help in preparation of the figures.
REFERENCES
1.Parnas, J. K., and Baranowski, T. (1935) Compt.
Rend. Soc. Biol., 120, 307-311.
2.Cori, C. F., and Cori, G. T. (1936) Proc. Soc.
Exp. Biol. Med.,34, 202-205.
3.Cori, G. T., Colowick, S. P., and Cori, C. F.
(1938) J. Biol. Chem., 123, 381-389.
4.Cori, G. T., and Cori, C. F. (1945) J. Biol.
Chem., 158, 324-332.
5.Madsen, N. B., and Cori, C. F. (1956) J. Biol.
Chem., 223, 1055-1066.
6.Wang, J. H., Shonka, M. L., and Graves, D. J.
(1965) Biochemistry, 4, 2296-2301.
7.Krebs, E. G., and Fischer, E. H. (1956) Biochim.
Biophys. Acta, 20, 150-156.
8.Graves, D. J., and Wang, J. H. (1972) in The
Enzymes (Boyer, P. D., ed.) Vol. 7, Academic Press, New York, pp.
435-482.
9.Dombradi, V. (1981) Int. J. Biochem.,
13, 125-139.
10.Oikonomakos, N. G., Acharya, K. R., and Johnson,
L. N. (1992) in Post-translational Modifications of Proteins
(Harding, J. J., and Grabbe, M. J. C., eds.) CRC Press, Boca Raton, pp.
81-150.
11.Kurganov, B. I. (1995) in Proteins and
Peptides (Ivanov, V. T., and Lipkin, V. M., eds.) Nauka, Moscow,
pp. 109-117.
12.Livanova, N. B., and Kornilaev, B. A. (1996)
Biochemistry (Moscow),61, 1432-1442.
13.Morgan, H. E., and Parmeggiani, A. (1964) J.
Biol. Chem., 239, 2440-2445.
14.Okazaki, T., Nakazawa, A., and Hayashi, O. (1968)
J. Biol. Chem., 243, 5266-5271.
15.Silonova, G. V., and Lisovskaya, N. P. (1967)
Doklady AN SSSR, 174, 718-721.
16.Klinov, S. V., Chebotareva, N. A., Kurganov, B.
I., Litvak, Zh. I., Zhilina, T. A., Glebova, G. D., Pekel, N. D., and
Beresovskii, V. M. (1984) Bioorg. Khim.,10,
1161-1170.
17.Cori, G. T., Colowick, S. P., and Cori, C. F.
(1939) J. Biol. Chem., 127, 771-782.
18.Kasvinsky, P. J., Madsen, N. B., and Fletterick,
R. J. (1978) J. Biol. Chem., 253, 1290-1296.
19.Cohen, P., Duewer, T., and Fischer, E. H. (1971)
Biochemistry, 10, 2683-2694.
20.Silonova, G. V., and Kurganov, B. I. (1970)
Mol. Biol. (Moscow), 4, 445-459.
21.Feldmann, K., Zeisel, H., and Helmreich, E.
(1972) Proc. Natl. Acad. Sci. USA, 69, 2278-2282.
22.Titani, K., Koide, A., Hermann, J., Ericsson, L.
H., Kumar, S., Wade, R. D., Walsh, K. A., Neurath, H., and Fischer, E.
H. (1977) Proc. Natl. Acad. Sci. USA, 74, 4762-4766.
23.Nakano, K., Hwang, P. K., and Fletteric, R. J.
(1986) FEBS Lett., 204, 283-287.
24.Barford, D., and Johnson, L. N. (1992) Protein
Sci., 1, 472-493.
25.Johnson, G. F., Tu, J. I., Bartlett, M. L., and
Graves, D. J. (1970) J. Biol. Chem., 245, 5560-5568.
26.Johnson, L. N., and Barford, D. (1990) J.
Biol. Chem., 265, 2409-2412.
27.Barford, D., and Johnson, L. N. (1989)
Nature, 340, 609-616.
28.Kasvinsky, P. J., Shechosky, S., and Fletterick,
R. J. (1978) J. Biol. Chem., 253, 9102-9106.
29.Sprang, S., Fletterick, R., Stern, M., Yang, D.,
Madsen, N. B., and Sturtevant, J. (1982) Biochemistry,
21, 2036-2048.
30.Meyer, F., Heilmeyer, L. M. G., Jr., Haschke, R.
H., and Fischer, E. H. (1970) J. Biol. Chem., 245,
6642-6648.
31.Helmreich, E., Michaelides, M. C., and Cori, C.
F. (1967) Biochemistry, 6, 3695-3710.
32.Baranowski, T., Illingworth, B., Brown, D. H.,
and Cori, C. F. (1957) Biochim. Biophys. Acta, 25,
16-21.
33.Helmreich, E. J. M. (1992) BioFactors,
3, 159-172.
34.Braunstein, A. E., and Shemyakin, M. M. (1952)
Doklady AN SSSR, 85, 1115-1118.
35.Braunstein, A. E., and Shemyakin, M. M. (1953)
Biokhimiya, 18, 393-411.
36.Braunstein, A. E. (1973) The Enzymes
(Boyer, P. D., ed.) Vol. 9, Academic Press, N. Y., pp. 379-481.
37.Snell, E. E. (1958) Vitamins and Hormones,
16,77-125.
38.Fisher, E. H., Kent, A., Snyder, E., and Krebs,
E. G. (1958) J. Am. Chem. Soc., 80, 2906.
39.Hedrick, J. L., Shaltiel, S., and Fischer, E. H.
(1966) Biochemistry, 5, 2117-2125.
40.Feldman, K., Zeisel, H., and Helmreich, E. J. M.
(1976) Eur. J. Biochem., 65, 285-291.
41.Shaltiel, S., Hedrick, J. L., Pocker, A., and
Fischer, E. H. (1969) Biochemistry, 8, 5189-5196.
42.Graves, D. J., Parrish, R. F., Uhing, R. J., and
Korytnyk, W. (1976) in Regulatory Mechanism of Carbohydrate
Metabolism (Essmann, V., ed.) Pergamon Press, Oxford, pp.
195-204.
43.Parrish, R. F., Uhing, R. J., and Graves, D. J.
(1977) Biochemistry, 16,4824-4831.
44.Chang, Y. C., McCalmont, T., and Graves, D. J.
(1983) Biochemistry, 22, 4987-4993.
45.Johnson, L. N., Hajdu, J., Acharya, K. R.,
Stuart, D. I., McLaughlin, P. I., and Barford, D. (1989) in
Allosteric Enzymes (Herve, G., ed.) CRC Press, Boca Raton, pp.
81-127.
46.Helmreich, E., and Cori, C. F. (1964) Proc.
Natl. Acad. Sci. USA, 51, 131-138.
47.Kastenschmidt, L. L., Kastenschmidt, J., and
Helmreich, E. (1968) Biochemistry, 7, 4543-4556.
48.Feldmann, K., and Hull, W. E. (1977) Proc.
Natl. Acad. Sci. USA, 74, 836-860.
49.Feldmann, K., Hörl, M., Klein, H. W., and
Helmreich, E. J. M. (1978) Proc. FEBS, 42, 205-218.
50.Helmreich, E. J. M., and Klein, H. W. (1980)
Angew. Chem. Int. Ed. Engl., 19, 441-455.
51.Palm, D., Klein, H. W., Schinzel, R., Buehner,
M., and Helmreich, E. J. M. (1990) Biochemistry,29,
1099-1106.
52.Lisovskaya, N. P., and Silonova, G. V. (1970)
Biokhimiya, 35, 448-457.
53.Kurganov, B. I., Klinov, S. V., and Chebotareva,
N. A. (1994) Uspekhi Biol. Khim., 34, 83-110.
54.Chebotareva, N. A., Klinov, S. V., and Kurganov,
B. I. (2001) Biotech. Genet. Eng. Rev., 18, 265-297.
55.Cori, C. F., and Illingworth, B. (1957) Proc.
Natl. Acad. Sci. USA, 43, 547-552.
56.Shaltiel, Sh., Hedrick, J. L., and Fischer, E. H.
(1966) Biochemistry, 5, 2108-2116.
57.Chebotareva, N. A., Sugrobova, N. P., Bulanova,
L. N., Poznanskaya, A. A., Kurganov, B. I., and Gunar, V. I. (1995)
Biochemistry (Moscow), 60, 1551-1558.
58.Gunar, V. I., Sugrobova, N. P., Chebotareva, N.
A., Stepanova, S. V., Poznanskaya, A. A., and Kurganov, B. I. (1990)
in Enzymes Dependent on Pyridoxal Phosphate and Other Carbonyl
Compounds as Cofactors (Fukui, T., Kagamiyama, H., Soda, K., and
Wada, H., eds.) Pergamon Press, Oxford, pp. 417-419.
59.Chebotareva, N. A., Sugrobova, N. P.,
Poznanskaya, A. A., Bulanova, L. N., Lyubarev, A. E., and Gunar, V. I.
(1994) Doklady AN SSSR, 335, 255-257.
60.Weisshaar, H. D., and Palm, D. (1972)
Biochemistry, 11, 2146-2154.
61.Avramovic-Zikic, O., Smillie, L. B., and Madsen,
N. B. (1970) J. Biol. Chem., 245, 1558-1565.
62.Chebotareva, N. A., Kurganov, B. I., Lyubarev, A.
E., Davidov, D. R., and Pekel, N. D. (1991) Biochimie,
73, 1329-1343.
63.Kurganov, B. I., Kornilaev, B. A., Chebotareva,
N. A., Malikov, V. P., Orlov, V. N., Lyubarev, A. E., and Livanova, N.
B. (2000) Biochemistry, 39, 13144-13152.
64.Kornilaev, B. A., Kurganov, B. I., Eronina, T.
B., and Livanova, N. B. (1996) Biochemistry (Moscow), 61,
628-634.
65.Kornilaev, B. A., Kurganov, B. I., Eronina, T.
B., Chebotareva, N. A., Livanova, N. B., Orlov, V. N., and Chernyak, V.
Ya. (1997) Mol. Biol. (Moscow), 31, 98-107.
66.Kornilaev, B. A., Kurganov, B. I., Livanova, N.
B., Eronina, T. B., Orlov, V. N., Chernyak, V. Ya., and Poglazov, B. F.
(1997) Doklady RAN,352, 256-258.
67.DeVincenzi, D. L., and Hedrick, J. L. (1970)
Biochemistry, 9,2048-2058.
68.Eronina, T. B., Chebotareva, N. A., Livanova, N.
B., and Kurganov, B. I. (2001) Biochemistry (Moscow), 66,
449-455.