REVIEW: Bioinformatics and Molecular Modeling in Chemical Enzymology. Active Sites of Hydrolases
S. D. Varfolomeev*, I. V. Uporov, and E. V. Fedorov
School of Chemistry, Lomonosov Moscow State University, Moscow, Russia; fax: 939-3589; E-mail: sdvarf@enzyme.chem.msu.ru* To whom correspondence should be addressed.
Received April 26, 2002; Revision received June 17, 2002
Comparison and multiple alignments of amino acid sequences of a representative number of related enzymes demonstrate the existence of certain positions of amino acid residues which are permanently reproducible in all members of the whole family. The use of the bioinformatic approach revealed conservative residues in each of the related enzymes and ranked amino acid conservatism for the overall enzymatic catalysis. Glycine and aspartic acid residues were shown to be the most essential for structure and catalytic activity of enzymes. Amino acid residues forming catalytic subsite of the active site of enzymes are always highly conservative. Analysis revealed that aspartic acid carboxyl group is the most frequently employed nucleophilic (in deprotonated form) and electrophilic (in protonated form) agent involved in activation of molecules by the mechanism of general base and acidic catalyses in the catalytic sites of enzymes. Glycine is a unique amino acid possessing the highest possibilities for rotation along C-C and C-N bonds of the polypeptide chain. The conservative fixation of the glycine residue in polypeptide chains of related enzymes provides a possibility for directed assembly of amino acid residues into the catalytic subsite structure. It is possible that the conservative glycines provide known conformational mobility of the protein and the active site. Methods of molecular modeling were used for analysis of structural substitutions of conservative and non-conservative glycines and their effects on geometry of catalytic site of typical hydrolases. The substitution of glycine(s) for alanine significantly altered the catalytic site structures.
KEY WORDS: bioinformatics, molecular modeling, multiple alignment, glycine, aspartic acid, alpha-chymotrypsin, pepsin, alkaline phosphatase, inorganic pyrophosphatase, computer mutations, RMSD
Computer methods are widely employed in modern biochemistry and molecular biology for various purposes [1-3]. During the last decade two approaches, bioinformatics and molecular modeling, have become especially popular and intensively developing areas. The methodology of bioinformatics is based on the informational analysis of nucleotide and protein sequences [4-11]. Within a framework of adequate physical considerations molecular modeling allows to characterize protein structures and their changes induced by some treatments or local structural alterations. In combination with methods of molecular dynamics molecular, modeling represents a powerful experimental approach that provides understanding of numerous physical and chemical aspects of protein molecules. Graphic capacities of modern work stations, relative low cost and potency of modern processors, storage capacity of accumulated information, availability of a large number of resolved spatial protein structures, nucleic acids and their complexes are the background for rapid world-wide distribution of computer modeling methods among scientists. These methods of computer modeling are widely used for determination of structures of biological molecules (X-ray analysis and multimer nuclear magnetic resonance), theoretical studies of their interactions, studies of the interaction between membrane and membrane proteins, structural analysis of site-directed mutagenesis and prediction of new mutation sites, and protein structure prediction (method of homologous modeling).
Such wide employment of molecular modeling methods resulted in appearance of adequate software such as InsightII (Accelrys, Inc., San Diego, CA, USA; http://www.accelrys.com) and Sybyl (Tripos, Inc., St. Louis, MO, USA; http://www.tripos.com). These softwares are easy in use. They allow the construction of various biological molecules, edit their structure (by substituting some groups, changing conformation of molecular groups) followed by subsequent structural relaxation by means of methods of molecular mechanics and dynamics. These programs also allow the calculation of various characteristics of the constructed molecules (e.g., electrostatic potential, solvent accessible surface, etc.), study of interactions between various molecules, alignment of protein sequences, creation of homology-based structures, and the investigation of ligand docking to active sites of enzymes. Recently some freely available programs possessing some of the above mentioned functions also appeared. They require only an ordinary computer. From our viewpoint the freely available Swiss PDB Viewer [12] (http://www.expasy.ch/spdbv/) has the widest capacities for computer modeling.
Molecular mechanics is a theoretical basis for calculation of structure of biological macromolecules and their interaction. This approach employs potential energy of the system and global minimum of this function corresponds to the equilibrium structure. Various methods of dynamic programming are used in the search for this minimum. (Potential energy depends on Cartesian coordinates of all atoms constituting this system and the number of variables of this function may be about several thousands.) The contribution of each atom into potential energy depends on its valence and charge. Rules by which each atom of the biological (macro)molecule can be characterized in term of parameters of its contribution into potential energy of the system and means of corresponding constitute a so-called force field. Now CHARMM22 [13-16] and AMBER [17] are commonly accepted force fields. The former, developed by Professor M. Karplus and his colleagues at Harvard University, is a part of applied software package CHARMM; the latter was developed by the group of the late Professor P. Kollman at University of California is a part of applied software pocket AMBER [18, 19]. Both force fields may be applied to biological objects that consist of major macromolecules (proteins, nucleic acids, lipids, and carbohydrates) and they give similar results. Flexibility of application, effectiveness, and the range of analyzed objects of these software packages (CHARMM and AMBER) exceed any of the commercially available programs. However, the language of these programs is rather complex and so these programs require an experienced user.
The use of various relaxation procedures on protein structures is one of the most common approaches of computer modeling of biological macromolecules. In many cases an initial structure obtained by homology modeling method, or by amino acid substitution has certain sites in which atoms are closely positioned, valent bonds are very extended, or side chains exist in unusual conformations. For regularization of these non-equilibrium spatial structures the method of simulated annealing is employed. The latter consists of calculations by methods of molecular dynamics at extremely high temperature (up to several thousand degrees) followed by subsequent reduction of temperature (to room temperature). It is suggested that such calculations lead this system to the global minimum of potential energy. If structural changes are rather small (e.g., changes induced by substitution of one amino acid residue for another one with similar structure and these changes are not accompanied by formation of a region of steric overlapping) relaxation of the resultant structure requires only potential energy minimization. The employment of only minimization preserves native geometry of catalytic site of enzymes, whereas molecular dynamics alters active site structure even at room temperature.
We believe that the bioinformatic approaches and molecular modeling methods promote better understanding of the fundamentals of biocatalysis. In the present paper we have used methods of bioinformatics and molecular modeling as a single combined approach for functional description of active sites of hydrolases, representing the largest class of enzymes. Some aspects of this approach and principles of our methodology have been published in previous papers [20-22].
MULTIPLE ALIGNMENT OF AMINO ACID SEQUENCES ALLOWS RECOGNITION OF
THE CATALYTIC SITE OF AN ENZYME
Amino acid sequence determines structure and properties of each protein. Now good evidence exists that in almost endless variability of proteins some structural elements are rather conservative, and these elements mainly determine function of the protein molecule. This is especially demonstrative in the case of catalytic proteins. For example, in the case of hydrolases, which represent about one third of all enzymes (about 1100 of 3700 enzymes) listed in the enzyme classification, only four main types of sites forming the catalytic structure are known [22].
Consideration of active site structure of enzymes requires subdivision of the active site into two structural constituents [23-31]:
1) substrate-binding subsite, which is responsible for binding, fixation and certain orientation of substrate(s); it determines enzyme specificity;
2) catalytic subsite, which is responsible for chemical transformation of substrate molecule; this site usually employs general acid-base catalysis.
It is possible that within one large enzyme superfamily the substrate-binding subsite responsible for the enzyme specificity exists as quite variable protein structure corresponding to variations in the substrate structures. However, catalytic sites should represent rather conservative structural elements due to limited number of catalytic site types. To test this hypothesis we have employed the bioinformatic approach based on the comparison of amino acid sequences of proteins constituting one large family. We analyzed results of sequence alignment of a few large enzyme families from HSSP database (http://www.sander.embl-heidelberg.de/hssp/) [31]. These enzyme families were selected by the following criteria:
1) number of analyzed enzymes should exceed 100; this provides reasonable statistical significance of the results;
2) this analysis requires selection of enzymes with known active site structures and well documented catalytic mechanism.
Usually results of alignment are summarized as large tables obtained by superimposition of protein sequence on the reference sequence. Conservative sequence elements are recognized by visual comparison. Clearly, for comparison of more than 3-5 sequences this method is almost inapplicable and non-informative. However, it can be automated by characterizing the number of positions of conservative residues in the sequence. One of quantitative criteria of position conservatism for each residue in the protein sequence is the statistical criterion in the form of Shannon entropy. In information theory Shannon entropy is one of the most important functions [32-34]. This function was introduced as a measure of uncertainty, characterizing some event with certain probability. Within this framework the information can be defined as the measure of uncertainty quantity, which may be further specified after an experiment. The amount of information represents a difference between informational entropies before and after experiment. The informational entropy (Shannon entropy) is a very convenient function for comparison of related proteins with distinct amino acid sequences. The alignment procedure versus some reference protein represents sequence positioning one over another one followed by fixation of homologous sites, and recognition and eliminations of inserts. Such comparison of a large number of protein sequences allows the calculation of probability of localization of some amino acid residue in certain position. This probability is determined as relative frequency of the amino acid j in a given position i. The entropy function for all 20 amino acids in each position in the protein sequence can be calculated using the following formula:
![]()
This function tends to zero in the case of events with high (pji --> 1) and low (pji --> 0) probability. So calculation of Shannon entropy may recognize positions in the protein sequence which are common (absolutely conservative) for a given j amino acid in the whole protein family. This is a position in which probability of appearance of this amino acid is close to unity, whereas in all other cases probability tends to zero. High values of Shannon entropy are typical for positions in protein sequences characterized by high variability of amino acid residues, whereas low values are indicative for positions of conservative residues. Within pji --> 1 (absolute conservatism) Hj --> 0.
In this study we have used hydrolases as the object of research. Four mechanisms of water activation in reactions catalyzed by these enzymes are known [20]. They involve:
- aspartic (glutamic) acid carboxyl group, water activation by nucleophilic mechanism;
- histidine imidazole group, water activation by nucleophilic mechanism;
- complex with zinc or cobalt ions, water activation by electrophilic mechanism;
- complex with magnesium or manganese ions, water activation by electrophilic mechanism.
We have chosen the enzyme families representing these four mechanisms of water activation: pepsin family (activation by carboxyl group), chymotrypsin and subtilisin family (activation by imidazole group), alkaline phosphatase family (activation by complex with zinc or cobalt ions), pyrophosphatase family (activation by complex with magnesium or manganese ion).
Figure 1 shows alignment profiles for all representatives of these four hydrolase families.
Analysis of highly conservative amino acids (for which Hj ~ 0) revealed that during alignment amino acid residues forming catalytically active subsite always represent conservative elements of the protein sequence. The catalytic site of acidic pepsin type proteases includes carboxyl groups of Asp31 and Asp215. These residues are recognized during alignment of amino acid sequences of pepsin family as conservative positions characterized by minimal value of Shannon entropy (Fig. 1a).Fig. 1. Shannon entropy for multiple alignments of proteins of aspartyl proteases versus pepsin (a), imidazole-activating hydrolases versus alpha-chymotrypsin (b), family of Zn2+ (Co2+)-dependent hydrolases versus alkaline phosphatase (c), Mg2+ (Mn2+)-dependent hydrolases versus inorganic pyrophosphatase (d).
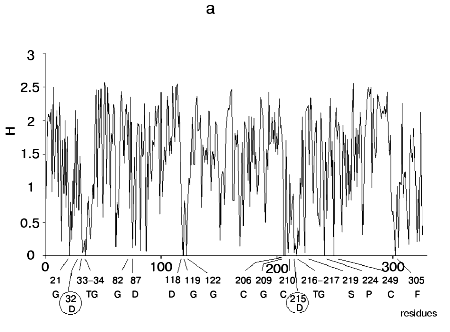
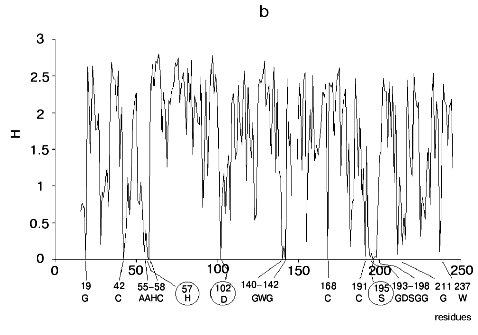
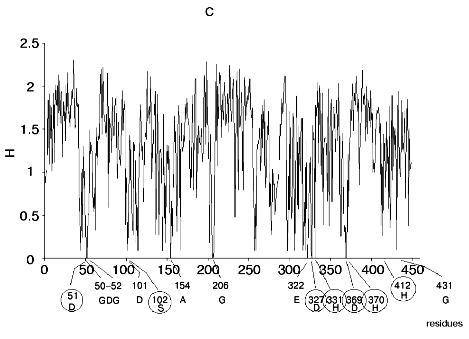
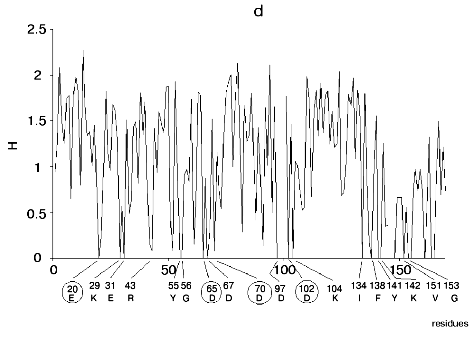
The catalytic site of the chymotrypsin family includes Ser195, His57, and Asp102 (Fig. 1b), whereas the catalytic site of the alkaline phosphatase family includes Asp51, Asp369, His370, Asp327, His412, His331, and Ser102 (Fig. 1c). Catalysis by inorganic phosphatase involves Glu20, Asp65, Asp70, and Asp102 (Fig. 1d).
All these residues are recognized as conservative ones during multiple alignment of amino acid sequences of corresponding protein families.
Thus, the bioinformatic approach allows recognition of side chains of amino acid residues forming a catalytic site of the enzymes and responsible for nucleophilic/electrophilic substrate conversion.
The comparison of enzyme amino acid sequences also revealed that Gly and Asp are the most frequently recognized as absolutely conservative residues [21, 22]. The finding that Gly is the most conservative residue was rather unexpected. Asp takes the second position in this list, and the sum of Gly and Asp represents about 50% of all conservative residues recognized.
Amino acid residues were ranked by their conservatism in these four protein families. For each amino acid we determined its frequency as the conservative element (Hj ~ 0), normalization on total number of conservative positions for all amino acid residues in these families. Figure 2a shows rating of amino acid conservatism. Figure 2b shows frequency of amino acid residues in proteins determined using Swiss-Prot database. Total frequency of conservative residues in enzymes completely differs from total frequency of amino acid residues in proteins, where Leu is the most frequent residues, whereas Gly takes only the fourth place in this list. (Frequency of Gly as the most conservative residue is 37%.)
Gly, Asp, Cys, Pro, and His are the most frequent conservative residues in enzymes. They represent about 70% of all conservative positions in these enzymes, whereas Met and Ile represent the most variable elements in the amino acid sequences.Fig. 2. Frequency of amino acid residues as conservative elements during multiple alignment of amino acid sequences in enzymes (a) [22], and frequency of amino acids in proteins of Swiss-Prot database (b).
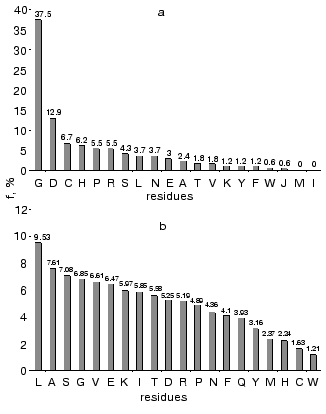
Thus, the most conservative residues can be separated into two principally different groups: 1) residues involved in substrate activation and acting as acids and bases (Asp and His); 2) residues forming active site architecture (Gly, Cys, Pro).
ASPARTIC ACID AND HISTIDINE IN THE ENZYMATIC CATALYTIC
CYCLE
The bioinformatic approach used in this study demonstrates a crucial role of aspartic acid and histidine residues in the functioning of the active site. Let us consider this role in more detail based on the analysis of mechanism of hydrolase action. As mentioned above, hydrolases represent the largest class of all known enzymes and their molecular mechanisms of catalysis have been well characterized. For most of hydrolases functional groups of amino acid residues constituting catalytic sites have also been identified and the interaction between these groups during catalytic cycle has been elucidated.
Based on the active site structure and mechanism of action all hydrolases can be arbitrary subdivided into the four main types:
1) hydrolases containing aspartic or glutamic acid residues in the active site (lysozyme-pepsin type);
2) hydrolases using imidazole group for water activation (the type of pancreatic ribonuclease, chymotrypsin, subtilisin, papain, lipase);
3) hydrolases using complexes with Zn2+ or Co2+ for activation of water and substrate (type of alkaline phosphatase, carboxypeptidase A, and organophosphate hydrolase);
4) hydrolases using Mg2+ and Mn2+ for activation of water and substrate (inorganic pyrophosphatase type).
Analysis of the catalytic mechanisms revealed that in most types of catalytic sites aspartic acid and histidine play principally important role [22].
Hydrolases of lysozyme-pepsin type. The catalytic subsite of this type of enzymes contains two or more carboxyl groups. These enzymes catalyze hydrolysis of ester, amide, and glycoside bonds. Usually the pH optimum of their catalytic activity is at relatively low pH values. Pepsin and lysozyme are typical members of this group. Protonated carboxyl group (acting as a general acid) is involved into substrate activation, whereas deprotonated form (acting as a base) activates water. Mechanism of concerted effect of two carboxyl groups acting as a nucleophile (water activator) and electrophile (substrate activator) has been considered in detail earlier [26, 27].
The protonated form of the Glu35 carboxyl group of lysozyme activates the reaction center; giving a proton it induces deficit of electron density. The deprotonated form of the carboxyl group of Asp52 stabilizes the oxocarbenium cation formed during reaction due to formation of a covalent complex [28].
Thus, in these reactions the ionic form of one carboxylic group acts as nucleophilic activator of water, whereas the protonated form of another carboxylic group operates as electrophilic activator of substrate.
Hydrolases of chymotrypsin type. Chymotrypsin, trypsin (serine proteases), papain (cysteine protease), and subtilisin (bacterial protease) are typical representatives of this type of enzymes. The catalytic mechanism includes intermediate acylation of hydroxyl of thiol group of the active site followed by formation of acyl-enzyme intermediates. In chymotrypsin type hydrolases a chain of proton transfer was identified. It includes imidazole, carboxyl group involved in activation of a water molecule or hydroxyl group of serine at the stage of acylation. The catalytic mechanism of this type of enzymes was exhaustively analyzed in several monographs [23, 27, 30]. In the case of alpha-chymotrypsin, imidazole of His57 coupled to Asp102 via a hydrogen bond is responsible for activation of water molecule. Activation of hydroxyl group of serine and water molecule involves a proton transport chain including imidazole and carboxyl group. It should be noted that isomorphic substitutions of catalytic groups for related ones are typical for the active sites of this type of enzymes. For example, Ser substitution for Thr insignificantly influence catalytic mechanism [35].
Ribonuclease is also related to an imidazole-activating hydrolase. Concerted interaction between imidazole groups of two histidine residues provides the catalytic mechanism for this type of enzymes. The imidazole group of His12 activates a carbohydrate hydroxyl group by proton transfer at the stage of cyclophosphate intermediate formation, whereas the imidazole group of His119 is involved in activation of water molecules at the stage of cyclophosphate hydrolysis [36].
Alkaline phosphatase and organophosphate hydrolase type. Active sites of enzymes of this group contain bivalent metal ions (Zn2+, Co2+, or Ni2+), which form uniform structural complexes with protein. Functional groups of histidine (imidazole) and aspartic (glutamic) acid (carboxyl group) are ligands for the metal ion. Carboxypeptidase A and thermolysin are typical representatives of this group. In the active site of these enzymes metal ions may act as: 1) electrophilic agent activating attacked reaction center; 2) electrophilic activator of water molecules and generator of hydroxyl cations. In the structure of the active sites of carboxypeptidase and thermolysin the metal ion complex acts as an electrophilic agent, which activates attacked reaction center of substrate. Deprotonated carboxyl group acts as a base during the activation of water molecules. Organophosphate hydrolase, the enzyme catalyzing hydrolysis of organophosphate pesticides and organophosphate compounds of chemical weapons (zarin, zoman, and VX) [37-43] is an example of another type of substrate activation. The catalytic mechanism of this type of enzymes involves concerted action of two ion complexes of Zn2+ or Co2+.
Pyrophosphatase type (Mg2+-Mn2+-dependent hydrolases). The metal complex in the active site of this type of enzymes acts as a potent electrophilic agent that activates the substrate by inducing electron density deficit at its reaction center. The metal complex may be also involved in electrophilic activation of water molecules. These functions of metal complexes are especially demonstrative in the catalytic mechanism of Mg2+ (Mn2+)-dependent enzymes such as inorganic pyrophosphatase [44-48]. The substrate is fixed in the active site by multiple ionic interactions involving positively charged groups of Lys193, Arg78, and Lys56. The main catalytic events involve two other metal ions, Mg2+ or Mn2+. Carboxyl groups of Asp65, Asp70, and Asp102, and three water molecules are the ligands for Mg2+ or Mn2+ high affinity site of complex formation. Low affinity site for metal ion is formed by carboxyl groups of aspartic acid, five water molecules, and carboxyl group of Glu20. This complex is positively charged. It is plausibly suggested that the high affinity site acts as electrophilic water activator, whereas the low affinity site functions as electrophilic substrate activator.
These examples clearly demonstrate the role of aspartic acid and histidine residue in the mechanisms of enzyme action. Concerted action of nucleophilic and electrophilic active site components responsible for high efficacy of biological catalysts is the basis for the functioning of active site of enzymes. Evidently, Asp plays the principal role in these processes. The ionized form of aspartic acid carboxyl group is a potent nucleophilic agent for activation of a water molecule and proton transport (pepsin, lysozyme, alpha-chymotrypsin). The Asp residue is principally important for metal complex formation at the active site of metal-dependent enzymes. The protonated form of carboxyl group of Asp residue is a proton donor (i.e., electrophilic agent).
It is interesting to analyze enzymes hydrolyzing certain bonds and enzymes forming these bonds. Exonucleases hydrolyzing DNA are enzymes with known active site structure [49-53]. The latter is a magnesium ion complex formed by carboxyl groups of aspartic and glutamic acids. This is the pyrophosphatase type active site. The active site of DNA-polymerase, the enzyme involved in DNA synthesis, shares high homology with the active site of this nuclease. These enzymes are also characterized by high structural homology of their active sites. Activation of the reaction center and nucleophile in DNA-polymerase involves the same functional groups, but in this enzyme sugar hydroxyl group is activated instead of water (by accepting proton) and nucleophilic attack at the reaction center of phosphate group provides elongation of the DNA chain.
Structural elements constituting active sites of hydrolases can also be found in enzymes of other classes. For example, a charge transfer chain including an imidazole-aspartic acid carboxyl group couple may be identified in active sites of oxidoreductases. In several studies structure of peroxidases and protein environment of heme were analyzed [54-56]. It was shown that in these enzymes imidazole-carboxyl group couple is directly involved in the catalytic site, where it plays the role of general nucleophilic agent activating hemin iron ion. It is also possible to identify the structural element of a charge transfer system, the complex of imidazole with carboxyl group, in active sites of dehydrogenases [57-59].
ROLE OF Gly IN FORMATION AND CONFORMATIONAL FLEXIBILITY OF ACTIVE
SITE STRUCTURE
Gly plays evident role in active site functioning. It is clear that conservative Gly residues do not play an important role in the activation of water molecules during the catalytic cycle because Gly does not have any substituent at the alpha-carbon atom and therefore it lacks pronounced chemical function.
Nevertheless, Gly residues are essential for protein structure [60-65]. This was demonstrated by mutation experiments where conservative Gly residues were substituted for any other amino acid. As a rule such substitutions resulted either in complete loss or significant decrease in catalytic activity (see [63] as an example).
Apparently, conservative Gly residues are principally important for the following functions.
1. Being a unique amino acid with the most energetically favorable rotation along C-N and C-C bonds of polypeptide chain (phi and iota Ramachandran angles) glycine may be a “junction point”, providing change in the direction of polypeptide chain during “assembly” of amino acid residues into functionally competent active site. Thus, the presence of conservative glycines may explain the structural paradox of enzymatic catalysis, when completely identical active sites are formed from completely distinct protein sequences. They just share several common features such as the existence of conservative glycines and factors stabilizing the assembled structure. In this connection we should mention that cysteine residues involved in disulfide bond formation (one of the most common stabilizing factors) take the third place in the rating of conservatism.
2. Conservative glycine residues may also function as a “hinge” responsible for known conformational flexibility of the active site. In many cases conservative glycines are located near catalytically active residues. For example, the following conservative motifs were found in hydrolases from various families: Asp215XGly217 (pepsin), Asp170XXGly173 (thermolysin), Asp32XGly, His63Gly64, Gly119XSer221 (subtilisin), Gly17XSer177 (trypsin), His76Gly77, Ser153XGly155, Gly75XAsp177 (lipases). In these enzymes Asp, Ser, and His residues are from the active site structures.
For some enzymes values of phi and iota angles for amino acid residues of the catalytic site are out of the energetically “relaxed limits”. This was found for amino acid residues of alpha-chymotrypsin (His57, Asp102, Ser195) by using the Ramachandran map of these residues. The active site of these enzymes is conformationally tensed (values of phi and iota angles are in energetically unfavorable region).
In enzymatic catalysis the conversion of initial substrate into reaction product(s) involves a series of intermediates possessing distinct structures. Conservative glycines of the active site may function as “relaxing” elements adapting active site conformation for the catalytic conversion of the next intermediate.
Cysteine and proline residues take top positions in the rating of amino acid conservatism. They are also essential for active site formation. Proline is a unique amino acid that unfolds the polypeptide chain.
Cysteine residues provide “required” active site structure by forming a disulfide bridge, which promotes fixation of catalytic residues (often located in different positions of a polypeptide chain) in the active site structure. For many enzymes disulfide bond formation is the final step terminating active site assembly.
MOLECULAR MODELING OF ACTIVE SITES WITH COMPUTER MUTATIONS OF
CONSERVATIVE AND NON-CONSERVATIVE GLYCINE RESIDUES
For elucidation of the role of conservative glycines in enzymatic catalysis, we simulated substitution of conservative and non-conservative glycine residues by means of molecular modeling (computer mutation). All conservative or non-conservative Gly residues in some proteins were substituted for Ala. The latter was chosen as the amino acid with the highest structural similarity to glycine. Using this approach we compared structural changes in the catalytically active groups (acids and bases recognize during multiple alignment) induced by this substitution.
We also calculated changes in enzyme structure induced by substitution of conservative Gly for Ala. For each hydrolase family we selected a representative with known spatial structure (see table) and determined positions of conservative Gly using the HSSP database [31] and number and positions of conservative and non-conservative Gly in it. Three spatial structures were analyzed for each selected enzyme (native structure, and structures with mutated conservative or non-conservative glycines, respectively). They were subjected to a structural relaxation procedure. The latter included 1000 steps of potential energy minimization of the structure. We believe that such gentle structure relaxation is adequate for detection of structural rearrangements induced by mutations of Gly because structural tensions induced by substitution of Gly for Ala are rather weak and may be abolished during a single minimization. This method preserves the structural characteristics of the catalytic site and characterizes mutation-induced spatial changes. After minimization structures were compared using the best superimposition of atoms of amino acid residues constituting catalytic site (see the fifth column of the table). The table shows the effect of substitutions of conservative and non-conservative Gly residues for Ala on root-mean-square deviation (RMSD) values (expressed per atom of the active site) for these atoms during superimposition of minimized native structure onto the relaxed mutant structures. Mutations, energy minimization, and analysis of the resultant structures were carried out using InsightII software. For structure minimization CVFF force field [66] was used.
Effect of substitution of conservative and non-conservative Gly residues
in various hydrolases on RMSD of atoms of catalytic site amino acid
residues
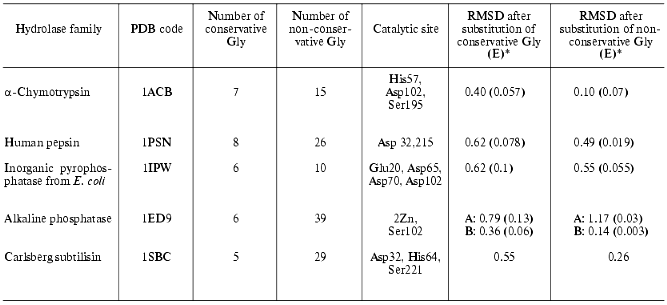
*In brackets RMSD values expressed per substitution of one
glycine are given (RMSD/n, where n is number of
substituted glycines).
Assuming that the minimized native structure has ideal mutual orientation of active site functional groups these RMSD values characterize the effect of amino acid substitution on spatial active site architecture (atom deviations from initial structure). The table shows that in the case of alpha-chymotrypsin, pepsin, inorganic pyrophosphatase, and subtilisin substitution of all conservative Gly for Ala resulted in significant changes of the active site geometry, whereas substitution of non-conservative Gly had minor effect. Taking into consideration subunit structure of alkaline phosphatase (which consists of A and B subunits) we have separately analyzed the effects of mutations in these subunits. The mutations caused distinct effects on the behavior of active sites of these subunits. Substitution of non-conservative glycines in A-subunit caused more pronounced changes than substitution of conservative glycines, whereas for B-subunit the situation is quite opposite.
Figure 3 illustrates the effect of computer substitution of Gly for Ala on structural changes of active sites of hydrolases. Figure 3 (a and b) shows the effect of the amino acid substitution on alpha-chymotrypsin active site structure (dark line designates native relaxed structures, light lines show the catalytic site structure after computer mutation of conservative (Fig. 3a) and non-conservative (Fig. 3b) glycines). It is clear that catalytic active site groups are more sensitive for mutation of conservative rather than non-conservative glycines. This difference is given quantitatively in the table.
It should be noted that number of non-conservative glycines is several times higher than that of conservative glycines (table). So, it might be expected that mutations of larger number of glycines should result in more pronounced changes of the active site structure. However, “in reality” for most hydrolases the mutation of limited number of conservative glycines caused significant changes in the geometry of the catalytic site and, consequently, catalytic properties of enzymes (see means in brackets).Fig. 3. Effects of computer substitutions of conservative and non-conservative glycines on structural changes in active sites of proteins. Substitutions of conservative (a) and non-conservative (b) glycines in alpha-chymotrypsin, conservative glycines in pepsin (c), conservative glycines in alkaline phosphatase (B-subunit), and conservative glycines in inorganic pyrophosphatase (d). Dark and light lines designate native relaxed and mutated structures, respectively.
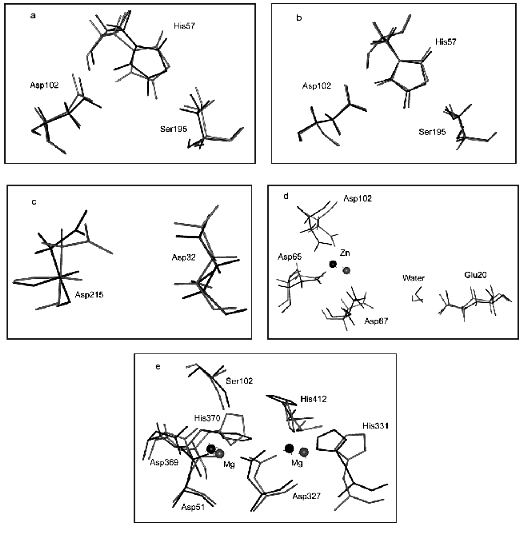
Figure 3 (c and d) illustrates changes in the architecture of the active site catalytic groups of pepsin, alkaline phosphatase, and inorganic pyrophosphatase, induced by computer mutations of conservative glycines.
All these results demonstrate higher sensitivity of catalytic subsite structure of hydrolases to mutations of conservative Gly. The nature of such sensitivity should be analyzed for each structure. Our criterion for evaluation of structural changes in the catalytic site of hydrolases is not specific with respect to certain structures. Using this criterion it is possible to demonstrate different effects of conservative and non-conservative amino acid residues on spatial structure of hydrolases and, consequently, on catalytic activity of these enzymes.
The authors are grateful to V. V. Poroikov, B. N. Sobolev, and A. E. Fomenko (Institute of Biomedical Chemistry, Russian Academy of Medical Sciences) and K. G. Gurevich for their help and discussion of results of the present study.
REFERENCES
1.Waterchmen, M. S. (ed) (1999) Mathematical
Methods for Analysis of DNA Sequences [Russian translation], Mir,
Moscow.
2.Gelfand, M. S., and Mironov, A. A. (1999) Mol.
Biol. (Moscow), 33, 969-984.
3.Pellegrini, M. (2001) Curr. Opin. Chem.
Biol., 5, 46-50.
4.Altshul, S. F., Madden, T. L., Schaffer, A. A.,
Zhang, J., Zhang, Z., Miller, W., and Lippman, D. J. (1999) Nucleic
Acids Res., 25, 3389-3402.
5.Bairoch, A., and Apweiler, R. (2000) Nucleic
Acids Res., 28, 45-48.
6.Bateman, A., Birney, E., Durbin, R., Eddy, S. R.,
Howe, K. L., and Sonnhammer, E. L. (2000) Nucleic Acids Res.,
28, 263-266.
7.Hofmann, K., Bucher, P., Falquet, L., and Bairoch,
A. (1999) Nucleic Acids Res., 27, 215-219.
8.Berman, H. M., Westbrook, J., Feng, Z., Gilliland,
G., Bhat, T. H., Weissing, H., Shindyalov, I. N., and Bourne, P. E.
(2000) Nucleic Acids Res., 28, 235-242.
9.Bairoch, A., and Apweiler, R. (2000) Nucleic
Acids Res., 28, 45-48.
10.Benson, D. A., Karsch-Mizrachi, I., Lipman, D.
J., Ostell, J., Kapp, B. A., and Wheeler, D. L. (2000) Nucleic Acids
Res., 28, 15-18.
11.Stoesser, G., Backer, W., van den Brock, A.,
Camon, E., Gareia-Pastor, M., Kanz, C., Kulikova, T., Lombard, V.,
Lopes, R., Parleison, H., Redaschi, N., Sterk, P., Stoehr, P., and
Tulu, M. A. (2001) Nucleic Acids Res., 29, 17-21.
12.Guex, N., and Peitsch, M. C. (1997)
Electrophoresis, 18, 2714-2723.
13.McKerell, A. D., Jr., Wiyrkiewicz-Kuczera, J.,
and Karplus, M. (1995) J. Am. Chem. Soc., 117,
11946-11975.
14.MacKerell, A. D., Jr., Bashford, D., Bellott, M.,
Dunbrack, R. L., Jr., Evanseck, J. D., Field, M. J., Fischer, S., Gao,
J., Guo, H., Ha, S., Joseph-McCarthy, D., Kuchnir, L., Kuczera, K.,
Lau, F. T. K., Schlenkrich, Smith, J. C., Stote, R., Straub, J.,
Watanabe, M., Wiyrkiewicz-Kuczera, J., Yin, D., and Karplus, M. (1998)
J. Phys. Chem., 102, 3586-3616.
15.Schlenkrich, M., Brickmann, J., MacKerell, A. D.,
Jr., and Karplus, M. (1996) in A Molecular Perspective from
Computation and Experiment (Merz, K. M., Jr., and Roux, B., eds.)
Birkhauser, pp. 31-81.
16.Ha, S. N., Giammona, A., Field, M., and Brady, J.
W. (1988) Carbohydr. Res., 180, 207-221.
17.Cornell, W. D., Cieplak, P., Bayly, C. I., Gould,
I. R., Merz, K. M., Jr., Ferguson, D. M., Spellmeyer, D. C., Fox, T.,
Caldwell, J. W., and Kollman, P. A. (1995) J. Am. Chem.
Soc.,117,5179-5197.
18.Case, D. A., Pearlman, D. A., Caldwell, J. W.,
Cheatham III, T. E., Ross, W. S., Simmerling, C. L., Darden, T. A.,
Merz, K. M., Stanton, R. V., Cheng, A. L., Vincent, J. J., Crowley, M.,
Tsui, V., Radmer, R. J., Duan, Y., Pitera, J., Massova, I., Seibel, G.
L., Singh, U. C., Weiner, P. K., and Kollman, P. A. (1999) AMBER
6, University of California, San Francisco.
19.Pearlman, D. A., Case, D. A., Caldwell, J. W.,
Ross, W. S., Cheatham III, T. E., DeBolt, S., Ferguson, D., Seibel, G.,
and Kollman, P. (1995) Comp. Phys. Commun.,91,1-41.
20.Varfolomeev, S. D., and Pozhitkov, A. E. (2000)
Vestnik Mosk. Univer., Ser. 2, Khimiya, 41,
147-156.
21.Varfolomeev, S. D., Gurevich, K. G., Poroikov, V.
V., Sobolev, B. N., and Fomenko, A. E. (2001) Dokl. RAN,
379, 548-550.
22.Varfolomeev, S. D., and Gurevich, K. G. (2001)
Izv. Akad. Nauk, Ser. Khim.,No. 10, 1629-1637.
23.Berezin, I. V., and Martinek, K. (1977)
Principles of Physical Chemistry of Enzymatic Catalysis [in
Russian], Vysshaya Shkola, Moscow.
24.Varfolomeev, S. D., and Zaitsev, S. V. (1982)
Kinetic Methods in Biochemical Studies [in Russian], Moscow
University Press, Moscow.
25.Klesov, A. A. (1979) Advances in Biological
Catalysis [in Russian], Moscow University Press, Moscow.
26.Poltorak, O. M., and Chukhrai, E. S. (1971)
Physico-Chemical Bases of Enzymatic Catalysis [in Russian],
Vysshaya Shkola, Moscow.
27.Antonov, V. K. (1991) Chemistry of Proteolysis
[in Russian], Nauka, Moscow.
28.Vocadlo, D. J., Davies, G. J., Laine, R., and
Withers, S. G. (2001) Nature, 412, 835-838.
29.Bender, M. L. (1971) Mechanisms of Homogeneous
Catalysis from Proton to Proteins, Wiley Interscience, New
York.
30.Jencks, W. P. (1972) Catalysis in Chemistry
and Enzymology [Russian translation], Mir, Moscow.
31.Sander, C., and Schneider, R. (1991)
Proteins, 9, 56-68.
32.Yaglom, A. M., and Yaglom, I. M. (1973)
Probability and Information [in Russian], Nauka, Moscow.
33.Shannon, C. E., and Weaver, W. (1949) The
Mathematical Theory of Communication, University of Illinois Press,
Illinois.
34.Wilson, A. G. (1970) Entropy in Urban and
Regional Modelling, Pion Limited, London.
35.Dodson, G., and Wlodawer, A. (1998) Trends
Biochem. Sci., 23, 347.
36.Knorre, D. G., and Myzina, S. D. (1998)
Biological Chemistry [in Russian], Vysshaya Shkola, Moscow.
37.Crimsky, J. K., Schetz, J. M., Pace, C. N., and
Wild, J. R. (1992) Biochemistry, 36, 14-66.
38.Lay, K., Dave, K. I., and Wild, R. (1994) J.
Biol. Chem., 269, 16579-16588.
39.Danilova, I. G., Ryabov, A. D., and Varfolomeev,
S. D. (1997) J. Mol. Cat., 118, 161-168.
40.Sergeeva, V. S., Efremenko, E. N., Kazankov, G.
M., Gladilin, A. K., and Varfolomeev, S. D. (1999) Biotechnol.
Techniques, 13, 479-485.
41.Omburo, G. A., Kuo, J. M., Mullins, L. S., and
Rushel, F. M. (1992) J. Biol. Chem., 267,
13278-13285.
42.Sergeeva, V. S., Efremenko, E. N., Kazankov, G.
M., and Varfolomeev, S. D. (2000) J. Mol. Cat. Part B:
Enzymatic, 10, 571-579.
43.Efremenko, E. N., and Sergeeva, V. S. (2001)
Izv. Akad. Nauk, Ser. Khim.,No. 10, 1743-1749.
44.Burton, P. H., Hall, D. C., and Josse, J. (1970)
J. Biol. Chem., 245, 4346-4355.
45.Avaeva, S. M., and Nazarova, T. I. (1985) Usp.
Biol. Khim., 26, 42-48.
46.Oganessyan, Yu. V., Kurilova, S. A., Vorobyova,
N. N., Nazarova, T. I., Popov, A. N., Lebedev, A. A., Avaeva, S. M.,
and Harutyunyan, E. H. (1974) FEBS Lett., 348,
301-304.
47.Harutyunyan, E. H., Oganessyan, B. Yu.,
Oganessyan, N. N., Terzyan, S. S., Popov, A. N., Rubinsky, S. V.,
Vainstein, B. K., Nazarova, T. H., Kurilova, S. A., Vorobieva, N. N.,
and Avaeva, S. M. (1996) Kristallografiya, 41, 84-90.
48.Harutyunyan, E. H., Oganessyan, V. Yu.,
Oganessyan, N. N., Avaeva, S. M., Nazarova, T. H., Vorobyova, N. N.,
Kurilova, S. A., and Huler, R. (1996) Biochemistry, 35,
7754-7761.
49.Steitz, T. A. (1999) J. Biol. Chem.,
274, 17395-17405.
50.Sam, D., and Perona, G. G. (1999)
Biochemistry, 38, 6576-6584.
51.Baldwin, G. S., Sessions, R. B., Erskine, S. G.,
and Halford, S. E. (1999) J. Mol. Biol., 288, 87-95.
52.Viadiu, H., and Aggarwal, K. (1998) Nature
Struct. Biol., 5, 910-912.
53.Holm, L., and Seadek, C. (1996) Science,
273, 595-607.
54.Gazaryan, I. G. (1992) Advances in Science and
Technology, Ser. Biotechnology [in Russian], Vol. 36,
VINITI, Moscow, pp. 4-42.
55.Finzel, B. C., Poulos, T. I., and Kraut, J.
(1984) J. Biol. Chem., 259, 13027-13037.
56.Kunishima, N., Fukuyama, K., Matsubara, H.,
Hatanaka, H., Shibano, Y., and Amachi, T. (1994) J. Mol. Biol.,
235, 331-344.
57.Birktoft, J. J., and Banaszak, L. J. (1983) J.
Biol. Chem., 258, 472-487.
58.Clarke, A. R., Wilks, H. M., Barstow, D. A.,
Atkinson, T., Chia, N. N., and Holbrook, J. J. (1988)
Biochemistry, 27, 1617-1625.
59.Taguchi, H., Ohta, T., and Matsuzawa, H. (1977)
J. Biochem. (Tokyo), 122, 802-810.
60.Esipova, N. G., and Tumanyan, V. G. (1972)
Mol. Biol. (Moscow), 6, 840-849.
61.Sasai, M. (1995) Proc. Natl. Acad. Sci.
USA, 92, 8438-8445.
62.Friedberg, I., Kaplan, T., and Margalit, H.
(2000) ISMB, 8, 162-170.
63.Cheng, Z. Q., and McFadden, B. A. (1998)
Protein Eng., 11, 457-465.
64.Martin, A. C., and Thornton, J. M. (1996) J.
Mol. Biol., 263, 800-810.
65.Efimov, A. V. (1999) FEBS Lett.,
355, 213-217.
66.Dauber-Osguthorpe, P., Roberts, V. A.,
Osguthorpe, D. J., Wolff, J., Genest, M., and Hagler, A. T. (1988)
Proteins: Structure, Function and Genetics, 4,
31-47.