REVIEW: Structural-Functional Analysis of Bacteriophage T7 RNA Polymerase
V. L. Tunitskaya and S. N. Kochetkov*
Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, Moscow, 119991 Russia; fax: (095) 135-1405; E-mail: kochet@eimb.ru* To whom correspondence should be addressed.
Received April 18, 2002
This review summarizes our results of the structural and functional studies of bacteriophage T7 DNA-dependent RNA polymerase (T7 RNAP). Particular features of this enzyme (the single-subunit composition, relatively low molecular weight) make it the most convenient model for investigating the physicochemical aspects of transcription. The review discusses the main properties of T7 RNAP, interaction between the enzyme and promoter, principle stages of T7-transcription, and also the results of structural and functional studies by affinity modification and both random and site-directed mutagenesis techniques.
KEY WORDS: bacteriophage T7, DNA-dependent RNA polymerase, structural-functional topography, affinity modification, mutagenesis
DNA-dependent RNA polymerase is responsible for one of the key processes in the living cell--the transcription, e.g., synthesis of the RNA replica on the DNA template. The transcription in prokaryotic and eukaryotic cells is performed using the complex multi-subunit RNA polymerases. The exception is RNA polymerases of certain phages such as T7, T3, SP6, and K11, and also mitochondrial RNA polymerases [1-3]. The common feature of all these enzymes is their simpler structure compared to prokaryotic and eukaryotic RNA polymerases. They all are single-subunit proteins, able to perform the complete transcriptional cycle in the absence of additional protein factors. This family of RNA polymerases is also characterized by higher (compared to bacterial RNA polymerases) rate of RNA synthesis and specificity towards their promoters. All these properties make the phage polymerases a rather convenient model for the investigation of the physicochemical aspects of transcription. The most extensively studied enzyme in this group is bacteriophage T7 RNA polymerase (T7 RNAP).
T7 RNAP was first isolated from T7-infected E. coli cells in 1969 [4]. The primary structure of T7 RNAP was determined during the 1980s [5]. Now, based on the X-ray analysis of four T7 RNAP crystal structures [6-9], a model of the initiation complex has been published [10].
The data of X-ray analysis suggest significant similarity between three-dimensional structure of T7 RNAP and a number of different (even evolutionally distant) DNA polymerases [2, 11, 12]. Most of these enzymes have a modular structure, where various functional activities are grouped into specific protein domains. Three-dimensional structures of polymerization domains are similar in all single-subunit DNA- and RNA polymerases, including T7 RNAP; they all resemble a right hand (Fig. 1), and therefore the corresponding subdomains are referred to as “palm”, “thumb”, and “fingers”, accordingly. These subdomains form the deep polynucleotide-binding cleft with the large amount of charged amino acid residues located on its walls and bottom. The length of this cleft in T7 RNAP molecule is 60 Å, the width is 15-25 Å, and the depth is 25-40 Å, which provides enough space for almost two complete turns of double helix DNA molecule.
Besides the polymerization domain, the structure of T7 RNAP contains a minor N-terminal domain whose functional role is discussed below.Fig. 1. Three-dimensional structure of T7 RNAP [9].
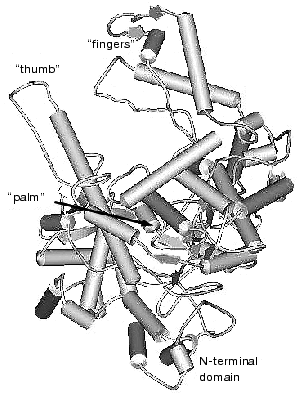
There is no obvious homology in primary structure of different single-subunit DNA- and RNA polymerases; however, the presence of three conservative motifs (A, B, C) was revealed [13]. Motifs A and C (Fig. 2) are present in absolutely all DNA- and RNA polymerases and contain invariant residues of aspartic acid. Motif B is only specific for some of DNA polymerases and all RNA polymerases and contains invariant residues of lysine, tyrosine, and glycine. These conservative structural motifs are positioned within spatial structures of polymerases in a completely analogous patterns--motifs A and C are in palm subdomain, motif B is in fingers subdomain (Fig. 1). These motifs form the active site of polymerases and in particular of T7 RNAP.
Hence, the X-ray analysis reveals the three-dimensional structure of T7 RNAP in detail and allows comparison with other polymerization enzymes. However, as it is commonly known, this technique provides complete information only about the static state of the enzyme or its complex. In order to study the dynamic changes during the enzymatic act, e.g., to clarify the functional topology of T7 RNAP, other methodology was applied, such as obtaining and investigation of different mutant forms of the enzyme and also affinity modification of the functionally important residues. These two approaches were in many cases complementary to each other, thus establishing a number of concepts concerning the mechanism of the functioning of the enzyme.Fig. 2. Structural motifs in single-subunit RNA- and DNA polymerases. Invariant residues are marked in bold; h is a hydrophobic amino acid residue.
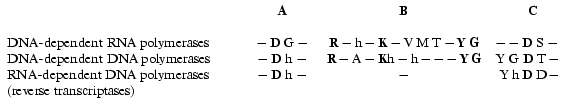
I. PREPARATION AND MAIN PROPERTIES OF THE ENZYME
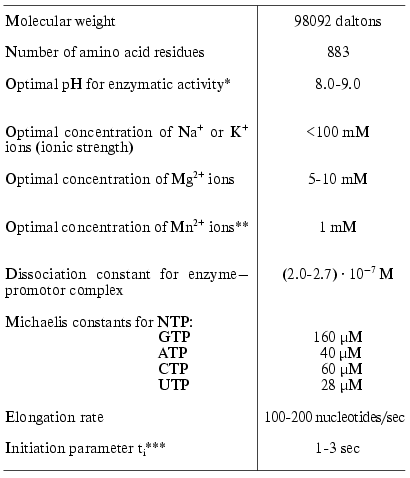
For the preparation of large amounts of both wild type T7 RNAP and its mutant forms, a plasmid vector pACT7 containing gene 1 of T7 bacteriophage controlled by thermoinducible promoter PR of phage lambda was constructed in our laboratory [14]. The vector provided a high level of RNA polymerase expression in E. coli cells (more than 50% of total cell protein). The same construction was used for the expression of T7 RNAP mutant forms. The isolation of the enzyme and measurement of its activity is thoroughly described in [14, 15]. The main properties of T7 RNAP are presented in Table 1 [16].
Table 1. The main characteristics of T7
RNAP
*Standard enzyme activity of T7 RNAP was determined as
described in [14].
**Enzyme activity after replacement of Mg2+ with
Mn2+ ions reduced 5-10 times.
***See comments in the text.
As a comment to Table 1, it should be noted that the formal kinetic analysis of the reactions catalyzed by RNA- and DNA polymerases (even using the simplified scheme) is very complicated due to the presence of five substrates and numerous stages [17]. Therefore, the presented values of Michaelis constants determined from the experiment are the apparent values. These parameters, however, may be used for comparison of different mutants or modified forms of the enzyme. As additional parameters characterizing the course of the reaction we also used the elongation rate (or coupling time for one nucleotide monomer) and ti-parameter characterizing the diffusion, initiation, and the termination stages [18] (hereafter called initiation parameter, see Table 1). To determine these parameters we proposed a conceptually new and simple experimental approach [18].
II. N-TERMINAL DOMAIN AND INTERDOMAIN LINKER
T7 RNAP consists of two domains. The first, large C-terminal domain, retains the polymerization activity and contains (in “palm” and “fingers” subdomains) the main functionally important structural motifs of single-subunit DNA- and RNA polymerases (A, B, and C). Besides, there is a small and relatively distinct N-terminal domain present in T7 RNAP structure, whose functional role is yet unclear. Domains are connected through an unstructured region (interdomain link) [6-9]. The investigation of thermal denaturation of T7 RNAP by scanning microcalorimetry technique revealed that the domains interact cooperatively, and the small domain increases the thermostability of the large one [19].
After limited trypsinolysis and removal of the N-terminal domain, the large C-terminal domain of T7 RNAP retains its polymerase activity, which indicates that the active site of the enzyme is located within this domain. However, the products of enzymatic reaction in this case are short transcripts with length not exceeding 8-10 bases (so-called abortive transcription). This observation indicated that the N-terminal domain is probably involved in binding of the 5´-end of the elongating RNA-product after it reaches a certain length, providing the transition from non-processive (abortive) to the processive mechanism of polymerization [20-22].
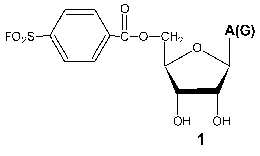
Investigation of the interaction of T7 RNAP with 5´-O-(4-fluorosulfonylbenzoyl)-guanosine (FSBG) and 5´-O-(4-fluorosulfonylbenzoyl)-adenosine (FSBA) (1) revealed that these agents are affinity modifiers of the enzyme the proof of which is the competition between analogs and NTP. Modification led to the formation of a covalent complex with protein to reagent molar ratio 1 : 1. The modification target was Lys172. This residue is located in the interdomain link, just in the site of the proteolytic cleavage during the domain separation. At the same time, this modification also led to the complete loss of the enzymatic activity, whereas the analysis of competitive interactions of reaction substrates and inhibitor gave evidence for the binding of the latter near the enzyme active site [15, 23]. These data indicate the spatial proximity of the active site and N-terminal domain during the enzymatic reaction and so giving a reason for more detailed investigation of the role of the interdomain link in functioning of the enzyme.
Measuring the rate of the limited T7 RNAP proteolysis in the presence of enzymatic reaction components [24] showed that both RNA and DNA significantly reduced the proteolysis rate; moreover, the DNA containing T7 promoter had a more profound protective effect. Meanwhile, the presence of NTP did not influence the rate of proteolysis. It can be assumed that when bound to the enzyme, polynucleotides interact with both domains and the interdomain link, thus protecting proteolysis sites. The fact, that this protection is performed not only by RNA-product but also by DNA (including promoter containing DNA) indicates that the role of the N-terminal domain is not confined to product binding.
The further studies of the interdomain link consisted in the oligonucleotide-directed mutagenesis of the Lys172 residue and the investigation of the properties of the mutants obtained (Table 2) [18, 25].
Table 2. Properties of the T7 RNAP mutant
forms containing the substitutions in the N-terminal domain and
interdomain linker* [25]
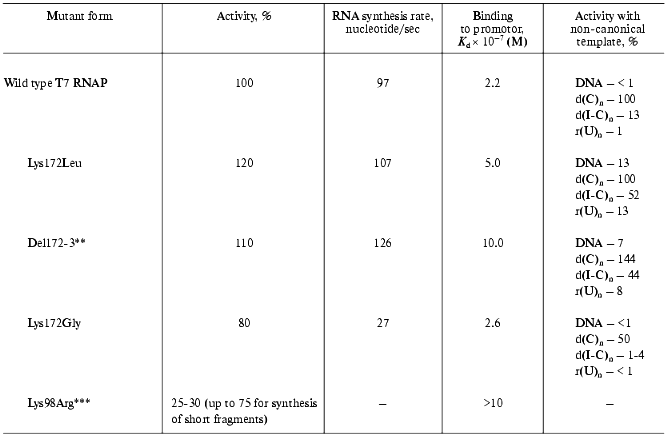
*Michaelis constants for rNTP are close to that for the wild
type enzyme.
**Deletion of Lys172Arg173 amino acid residues.
***Preliminary results.
All of the tested mutants retained their enzymatic activity. However, the nature of amino acid substitution influenced on the enzymatic reaction in a different way. For instance, the Lys172Gly substitution led to the significant decrease in RNA-synthesis rate, meanwhile binding rate with the promoter remained constant. Lys172Leu substitution and Lys172Arg173 deletion (del172,173) did not result in the decrease in activity, however, a weaker binding to the promoter was observed for these mutant forms along with wider specificity towards the template. These mutants displayed a noticeable activity in “promoterless” systems (in other words, they used either DNA without T7-promotor or a number of homo- and heteropolymers, including single-stranded ones as templates).
There is another interesting feature of the mutant (del172,173). It is known that when the promoter-containing templates with extending 3´-end are used, T7 RNAP of the wild type synthesizes (along with the main transcript of a proper length) longer aberrant transcripts. Earlier, Schenborn and Mierendorf [26] interpreted this phenomenon as an ability of the enzyme to “turn back” after reaching the end of the template and to carry out the synthesis on the complementary strain. At the same time, the “deletion” mutant did not exhibit this feature at all [18]. Probably, the altered structure of the interdomain link does not allow the enzyme to adopt the conformation facilitating the “turn-around”. This property of the mutant proved to be useful in the practical application: obtaining of RNA-transcripts on the templates with extending 3´-end could now be done without forming undesirable by-products.
The above-mentioned data show that the interdomain link interacts with the DNA template during the enzymatic reaction. Apparently, this interaction only takes place in the case of sufficiently elongated polynucleotides, because according to X-ray data of T7 RNAP complex with minimal promoter (17-22 b.p.) [8, 9] there are no such contacts.
More evidence of N-terminal T7 RNAP domain participating in the interaction with promoter and thus influencing the reaction was obtained from the investigation of enzyme interaction with synthetic oligonucleotides. Their nucleotide sequence corresponded to the consensus sequence of T7 promoter and contained active phosphate groups (the activation was achieved by the substitution of corresponding phosphodiester bond with trisubstituted pyrophosphate). The oligonucleotide containing an activated phosphate at (-14) position of the promoter formed a covalent bond with the Lys98 residue of the protein [27]. According to the X-ray data of T7 RNAP complex with a promoter [8, 9], this residue is located near the 5´-end (AT-enriched) region of the latter. The substitution of this residue with arginine led to a slight decrease in enzymatic activity of the resulting mutant, which was more appreciable for the elongated than for the abortive transcripts (Table 2). Furthermore, despite the homological nature of the substitution, this mutant form significantly lacked its binding activity with the promoter.
Hence, study of the N-terminal region of the enzyme suggested the participation of the minor domain and the interdomain link not only in the binding of the RNA-product, but also in interaction with the promoter-containing area of the DNA-template. Besides, there are grounds to believe that the amino acid sequence of the interdomain link determines the enzyme spatial conformation, allowing artifact synthesis of the transcripts exceeding the length of the transcribing template site.
III. REVEALING OF FUNCTIONALLY IMPORTANT T7 RNAP SITES BY THE
RANDOM MUTAGENESIS TECHNIQUE
Based on analysis of amino acid sequences of different single-subunit DNA- and RNA polymerases and also on the X-ray data, it seemed to be promising to address the homological motif A, B, and C. In order to search for other functionally important amino acid residues of T7 RNAP, we have developed a genetic system for obtaining point mutants with a defined lack of enzymatic activity described in [28, 29] and based on the principle of random mutagenesis. This technique is promising for protein analysis also because it allows detecting functionally important amino acid residues whose role is often not possible to elucidate by other means. The subsequent localization of mutations along with the analysis of primary structure and homologies between different DNA- and RNA polymerases showed the critical significance of certain amino acid residues for the enzyme functioning.
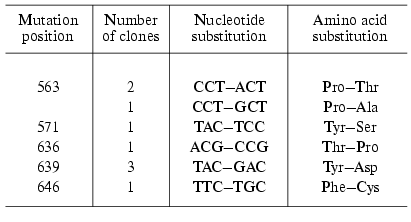
Four of 13 clones obtained by the “random mutagenesis” technique and potentially containing inactive enzyme did not exhibit the expression of the protein of corresponding molecular weight. Using the other clones the mutant forms of T7 RNAP were isolated and the positions of point amino acid substitutions were determined (Table 3). As the table illustrates, the mutations determined by this technique are located in two areas of the enzyme: 636-646 (partially matching with motif B mentioned above) and 563-571 [29]. Among the mutants obtained the major part was completely identical (repeated mutation in the same position) or contained different substitutions in the same position. This quite unexpected result indicates that these T7 RNAP regions are crucially important for enzyme functioning and due to yet unknown factors are most prone to mutations [29]. The remainder of this work was dedicated to the detailed investigation of the role of these regions.
Table 3. Random mutagenesis of T7 RNAP [28, 29]
IV. INVESTIGATION OF THE 563-571 REGION OF T7 RNAP
The collection of “random” mutations in this locus was enlarged and complemented by oligonucleotide-directed mutagenesis. The main characteristics of the examined mutants are presented in Table 4 [30, 31].
Table 4. Properties of the T7 RNAP mutant
forms containing point substitutions in 563-571 region* [30, 31]
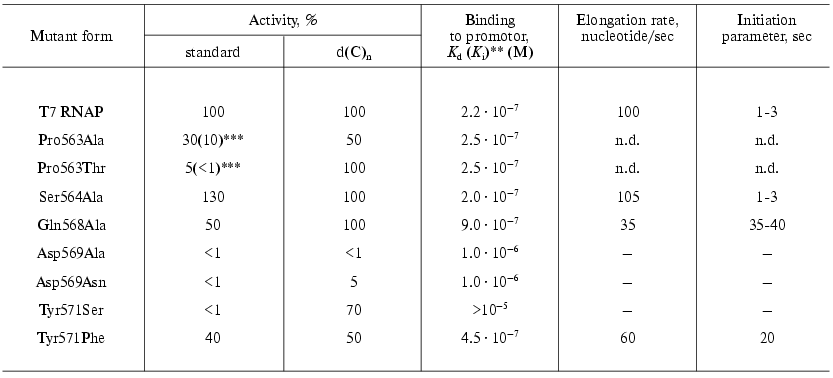
*Michaelis constants for rNTP are the same as for the wild
type enzyme with exception of Tyr571Phe, for whose these values are two
times higher.
**For inactive mutants the inhibition constants of wild type
enzyme activity by mutant was measured instead of enzyme-promoter
dissociation constant.
***Activity decreases under storage; activity 3-4 days after the
preparation is shown in the parenthesis.
The data presented indicate that this protein region is involved either directly or indirectly in the interaction with a promoter. For instance, the Tyr571Ser substitution caused the complete loss of specific binding and activity with the promoter-containing template [30]. However, at the same time this mutant retained a considerable activity with nonspecific single-stranded templates--poly(dC) and poly[d(I-C)], which indicates the maintenance of its ability for the catalytic act itself. It is essential that the substitution of Tyr571 with homological Phe did not affect so dramatically the enzyme-promoter interaction and mutant activity [31].
The binding parameters for the mutants containing substitutions at position 569 also were changed; these mutants lost the ability for RNA-synthesis on nonspecific templates.
The dissociation constant of the enzyme-promoter complex for mutants with Pro563 substitutions did not differ from that for the wild type enzyme. However, the initially low activity of these mutants was rapidly lost during the storage of the preparations; moreover, this property was distinctive only regarding the promoter-dependent synthesis; the activity on single-stranded homopolymer template was retained (Table 4). This suggests that this residue is not directly involved in binding with the promoter; however, the presence of proline at position 563 forms the local structure of this protein region and determines the correct interaction with the promoter of adjacent residues Asp569 and Tyr571. The substitution of proline with other residues which are significantly different in their spatial parameters results in the rapid denaturation of this protein region and loss of binding with promoter. The Ser564Ala substitution was negligible for the enzyme functioning [31].
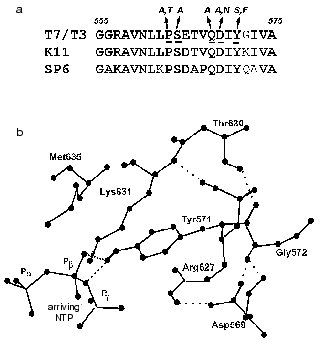
It should be noted that this region detected by us in T7 RNAP structure is located rather close to the enzyme active site (first of all to motif A), but according to X-ray data is not included in it and does not interact with a promoter [8, 9]. However, the analysis of X-ray data reveals possible hydrogen bonds of Asp569 and Tyr571 residues, contributing to the stabilization of the promoter complex (Fig. 3). The disruption of these bonds as a result of mutagenesis may lead to the distortion of complex formation and thus to the loss of enzymatic activity.
Fig. 3. a) Amino acid sequence of T7 RNAP 555-575 region and the corresponding regions of T3, K11, and SP6 RNA polymerases. The same and homologous amino acid residues are marked in bold, amino acids obtained by random and site-directed mutagenesis are italicized (see Table 4). b) Three-dimensional structure of this region according to [9].
V. STUDIES OF MOTIF B
The structure of T7 RNAP motif B is presented in Fig. 4. X-Ray data [6-9] show that this motif is located in the “finger” subdomain, close to motifs A and C, with both these motifs likely to from an active site. Most of this fragment is alpha-helix, and (according to the helix turn) the side radicals of four amino acid residues (Arg627, Lys631, Met635, and Tyr639) are directed towards the substrate-binding cleft. Typically, both Lys631 and Tyr639 are invariant in most of single-subunit DNA- and RNA polymerases, Arg627 is present in most of the enzymes, whereas the residue corresponding to Met635 is different for diverse enzymes [1, 2] (in particular, DNA polymerases contain an aromatic amino acid residue in this position, in many cases governing the specificity towards the carbohydrate residue dNTP [32]). Another structural dissimilarity of motif B in DNA- and RNA polymerases is the presence of regular repeatability of hydroxyl-containing amino acid residues, which are absent in DNA polymerases [33].
There were no systematic studies of this motif's role when the present work began. The research starting point was an affinity modification of the invariant Lys631 residue by GMP o-formylphenyl ester [34, 35], and also random mutagenesis T7 RNAP studies discussed above [28, 29], resulting in the detection of sufficient number of mutations in motif B, crucial for T7 RNAP activity. Site-directed mutagenesis was performed in order to determine the role of these and other residues, and the properties of mutant forms are summarized in Table 5.Fig. 4. a) Amino acid sequence of motif B in T7 RNAP, in T3, K11, and SP6 RNA polymerases, and in T7, Klenow fragment, and Taq DNA polymerases. Invariant amino acid residues are underlined; b) amino acid substitutions in motif B of T7 RNAP (Table 5); c) three-dimensional structure of this region according to [9]. Amino acid residues substituted within this investigation are marked.
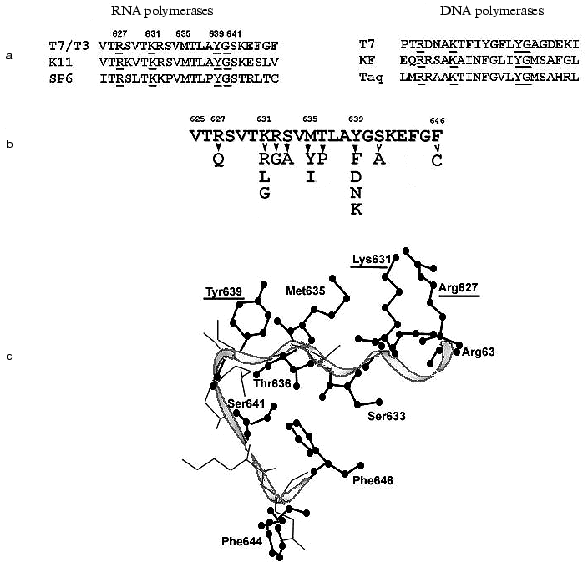
Table 5. Properties of T7 RNAP mutant forms
containing point mutations in motif B (627-646)
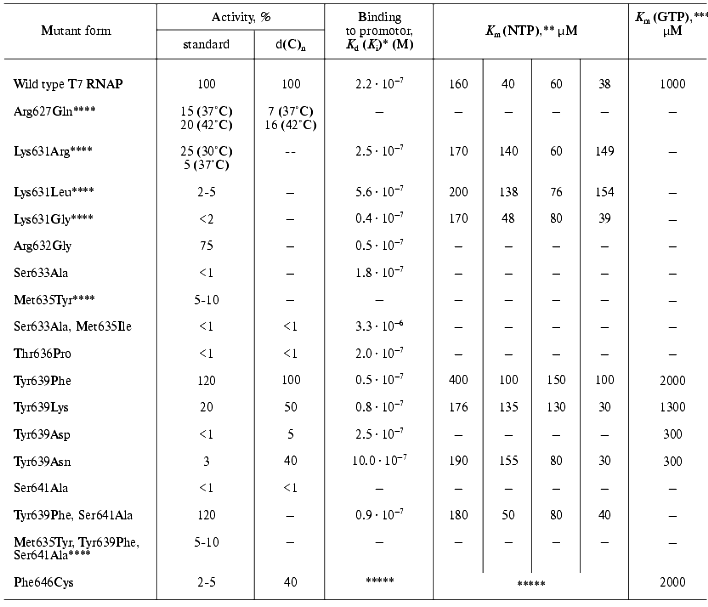
*For inactive mutants the inhibition constants of wild type
enzyme activity by mutant was measured instead of enzyme-promoter
dissociation constant.
**Template was pGEM-2 plasmid (promoter-containing).
***Template was d(C)n.
****Mutants are unstable under storage.
*****Non-linear binding kinetics.
The presented data show that practically all mutations in the examined range (even those resulting in enzyme inactivation) are not accompanied by significant changes in protein-promoter interaction. Indeed, according to X-ray data structural elements of T7 RNAP directly responsible for binding with the promoter are located in other regions of the molecule [7-9]. The only exception is mutant Phe646Cys, for which two facts indicating its involvement in the interaction with the template were discovered: first, the significant distortion of enzyme binding parameters, not only with nucleoside triphosphate but also with the promoter; second, the maintenance of rather high activity on single-stranded homopolymer template, and at the same time the complete loss of the promoter-dependent activity [28]. The second feature (as Table 5 illustrates) is not typical for the mutants of this group.
The data of Table 5 also suggest that most of the mutations in the N-terminal region of motif B lead to noticeable changes in enzyme activity, and, in some cases, to the complete inactivation. At the same time, the character and increment of these changes significantly differ, indicating that the role of amino acid residues also differs during the different stages of the enzymatic process. For instance, the activity of Arg627Gln mutant was 15% of the control under normal conditions, but it appeared to be slightly more thermostable; during measuring at 42°C the activity of the mutant reached 20% of that of wild type enzyme. At the same time, the isolation and purification as well as storage of this mutant were associated with significant difficulties connected to the increased proteolytic lability of the protein. The activity of the mutant at position 631 was also unstable; on one hand, its initial value significantly depended on the nature of the substituting residue, but on the other hand, this activity dropped practically to zero during one week period of storage, this loss of activity being not accompanied by the proteolysis of protein molecule [35-37]. For Lys631Arg and Lys631Gly contrary to Arg627Gln, a significant decrease in optimal reaction temperature (to 18-20°C), and practically complete inactivation at 40°C could be demonstrated. The analysis of CD spectra showed major changes in secondary structure of these mutants (alpha-beta-transfer), analogous to those observed in wild type T7 RNAP only at temperature increased to 50°C [36]. The mutation Met635Tyr caused a decrease in enzymatic activity to 5-10%, even this remaining activity (as in the previous case) being lost during 5 days without associated proteolysis [33]. Substitution of Ser633Ala resulted in a complete loss of enzymatic activity of the mutant [33].
Based on the data obtained, it can be assumed that the N-terminal part of motif B (where point substitutions are accompanied by different conformational rearrangements attended by changes in the reaction temperature parameters and leading to structural and proteolytic lability) plays a major role in the formation of T7 RNAP active center, contributing to the fixation of the spatial arrangement of functionally important amino acids involved in catalysis. (However, the mutation Arg632Gly did not lead to any changes in enzyme properties compared to the wild form [35].)
Regarding the mutations in the C-terminal region of the motif, a few remarks can be made. First, the mutation Thr636Pro (random mutagenesis) results in complete loss of activity. At the same time, binding to the promoter remains practically unchanged, though there are significant changes in kinetic parameters of the interaction with GTP (“starting” NTP) [37]. Probably the loss of the mutant activity is caused not only by the critical role of Thr636 (according to X-ray data it is turned toward the active center) but mostly by the “bad” type of the random mutation. The proline residue is known to disturb the local secondary structure near its location. Meanwhile the fact of inactivation during this substitution indicates the importance of this structure (alpha-helix, in this case) for the enzyme functioning.
The most important results were obtained while studying point substitutions of the invariant residue Tyr639. The significant role of this residue was also emphasized in other publications [38, 39]. The results of random mutagenesis (see above) also showed that most of the mutations lethal for enzymatic activity was found namely in this position [28, 29]. The studies of random mutants along with the site-specific mutant forms of T7 RNAP revealed the dependence of activity on the nature of substituting amino acid residue; from the absolute loss of activity in the case of Tyr639Asp and Tyr639Asn and to its complete retaining in the case of Tyr639Phe [37, 40, 41].
Evidence for the participation of Tyr639 in the formation of the initiation region of the transcription complex was obtained during the investigation of the interaction of T7 RNAP with synthetic promoters containing an additional ribose (2) residue at 2´-position at different positions of the oligonucleotide. After the periodate oxidation of this residue, the reactive dialdehyde group capable of covalent interaction with spatially close lysine residue of the protein arises [42, 43]. Neither of the modified promoters formed a covalent complex with the wild type enzyme. However, this complex was detected using the Phe639Lys mutant (retaining a partial enzymatic activity) and a promoter with the reactive group at position (+2) regarding the transcription start; other mutants in this position were not targets for this lysine-specific dialdehyde group. These data indicate the spatial proximity of the T7 RNAP 639 residue and the adjacent post-promoter sequence of the template, which is directly involved in the catalytic act, defining the correct arrangement of the complementary ribonucleotide.
Hence, if the “beginning” of motif B probably determines a spatial arrangement of transcription complex components under the enzymatic act, the “end” and especially the Tyr639 residue are directly involved in the enzymatic act at the stage of complementary nucleotide detection and/or forming of phosphodiester bond.
VI. UNIQUE PROPERTIES OF Tyr639Phe MUTANT. “DOUBLE”
MUTANT Tyr639Phe, Ser641Ala
Along with the activity characteristic for a wild type enzyme, the Tyr639Phe mutant exhibited the ability to use deoxynucleoside phosphates as substrates to a minor extent [44, 45]. The introduction of an additional substitution Ser641Ala into this mutant resulted in a significant increase in this ability [46, 47]. The detailed study of this abnormal behavior of the “double” mutant revealed a connection between the efficiency of deoxynucleotide inclusion into the growing chain and the distance between the transcription start and position of the nucleotide. The synthesis initiation and inclusion of deoxyribonucleotides into the growing strain at position 1-3 were ineffective. Thereafter, at increasing distance from the transcription start, the inclusion of deoxynucleotide links by the mutant enzyme was more and more facilitated; after position 7-8 (corresponding to the transition from the abortive to processive mechanism) the difference in inclusion efficiency of ribo- and deoxy-links was practically eliminated. However, if Km for the corresponding “substituted” nucleotide is used as a reference of inclusion efficiency instead of obtained product estimation, this value (depending on the distance from the first deoxyribonucleotide inclusion into the growing chain) steadily decreased and reached the Km value for the corresponding ribonucleotide only when the first inclusion of deoxy-link took place at position 21-22 from the transcription start [45, 48].
The discovery of this unique property of the “double” mutant Tyr639Phe, Ser641Ala allowed its use for the preparative synthesis of hybrid DNA/RNA-polynucleotide products (so-called mixed polynucleotides) [49]. Regarding the difficulties emerging during the inclusion of deoxyribonucleotides at initial stages of the synthesis, the promoter-containing plasmid pTZR-7G was constructed [48]. When this plasmid was used, the corresponding transcript contained the first seven GMP residues. At the same time, the inclusion of remaining deoxyribonucleotides (the first of which only has the eighth position from the start) was significantly facilitated, which allowed obtaining “mixed” ribo- and deoxynucleotide-containing polymers over 100 nucleotides length with high yield. For instance, the yield of the mixed polynucleotide containing one ribonucleotide (GMP) and three deoxyribonucleotides was approximately 30% of the corresponding “wild type” polynucleotide [48]. Mixed polynucleotides, obtained using T7 RNAP “double” mutant were later successfully employed as templates for research on protein-nucleic interaction, performed by the HIV-1 reversed transcriptase [49] and certain aminoacyl-tRNA-synthetases [50].
Another unique property of the “double mutant” was its ability to perform polynucleotide synthesis in a primer elongation mode specific for DNA polymerases. Moreover, the efficiency of this synthesis significantly increased if the primer nucleotide sequence coincided with the consensus sequence of T7-promotor [51]. The extension of primer, however, was limited to 5-7 nucleotides. This fact may be explained on the assumption that the transition from the abortive to processive transcription in T7 RNAP is accompanied by fixing of 5´-end of the growing RNA-chain free end by the N-terminal domain of the enzyme (see above). In the case of primer elongation reaction, the free 5 end of the product is missing: the primer remains bound to the template. The fixation on the enzyme, resulting in conformation changes and transition to the processive mechanism does not occur. So, the reaction does not achieve the stable elongation mode and, as the tendency to abort prevails, the length of the product is restricted to 8 nucleotides.
VII. C-TERMINAL AREA OF ENZYME. His811 RESIDUE
Motif C is present in the C-terminal area of T7 RNAP both in DNA- and RNA polymerases. His811 and Asp812 are invariant amino acid residues in this motif. Site-directed mutagenesis of these residues demonstrated the critical importance of Asp812 residue for the catalysis; the substitution of His811 residue proved to be slightly less crucial. For instance, the His811Gln mutant retained 25% of enzymatic activity, although other substitutions at the same position resulted in almost complete inactivation [52]. It is believed that Asp812 and Asp537 are directly involved in the catalysis by forming a complex with Mg2+, while the role of His811 is to increase the nucleophilicity of the adjacent Asp812 [10, 11].
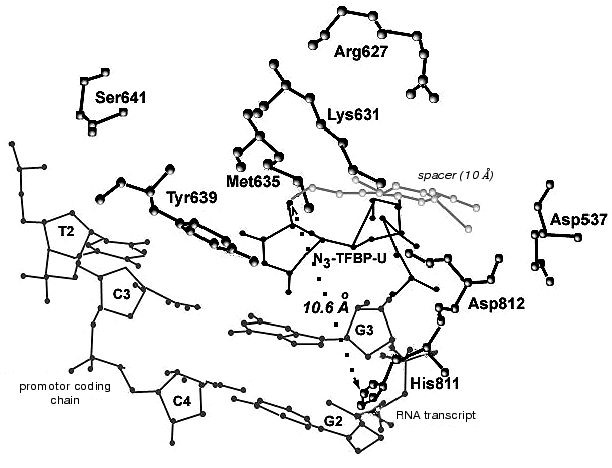
During the UV-irradiation of T7 RNAP complex with reaction product GGGGGGGAU* (9-mer oligoribonucleotide), where U* is 5´-[5-(E)-(4-azido-2,3,5,6-tetrafluorobenzamido)propenyl-1]-UTP residue (N3-TFBP-UTP) (3), which is a photoaffinity reagent and potential protein modifier [53], a covalent RNA-protein complex was obtained. Treatment of the modified enzyme by guanyl-specific T1 nuclease, complete trypsinolysis, and following separation of the peptides identified the modified peptide Tyr802-Lys826, which contains the most possible “candidates” for the binding with photoaffinity nucleotide, residues His811 and Asp812. Even though the modified amino acid was not unequivocally determined, the analysis of the spectral changes in the complex compared to the original N3-TFBP-UTP (significant shift in absorption maximum to longer wavelength also observed during the interaction of perfluoroazide, the structural fragment of N3-TFBP-UTP with the free histidine in model system) strongly suggested that His811 is modified [54]. Hence, it was experimentally illustrated that during the enzymatic reaction His811 is located sufficiently close to the last nucleotide inclusion site into the RNA-chain of the product. Computer modeling based on X-ray data also confirmed that this particular range is situated 10 Å from the 3´-end of the RNA-product, the distance exactly matching the length of the spacer between the nucleotide and photoreactive moiety of the modifier molecule (Fig. 5).
Thus, the comprehensive study of the properties of artificially obtained T7 RNAP forms that were affinity modified or contained point amino acid substitutions in different regions of the enzyme molecule revealed a number of previously unknown regularities in T7 RNAP functioning. In particular, it was shown that the role of the N-terminal domain not only confined to binding of the RNA-product, but is also essential for binding to the template. The functionally important and directly participating in enzyme-promoter binding fragment 563-571 was determined. Motif B of T7 RNAP was thoroughly examined and the roles of particular amino acid residues were postulated, also showing that this region participates in the formation of the enzyme active site, where certain amino acids organize three-dimensional structure and support its stability, while the others (and especially Tyr639) are directly involved in the enzymatic act. There is also new information available regarding the spatial deposition of His811 residue inside motif C. It was revealed that this residue is located near the elongating end of the RNA-chain during the functioning of the enzyme.Fig. 5. Binding model of RNA transcript containing N3-TFBP-UTP residue in the T7 RNAP active site. Modeling was performed using WebLab ViewerPro 3.7 software (Molecular Simulations Inc.). Data from [9] were used (PDB 1QLN).
REFERENCES
1.McAllister, W. T., and Raskin, C. A. (1993) Mol.
Microbiol., 10, 1-6.
2.Sousa, R. (1996) Trends. Biochem. Sci.,
21, 186-190.
3.Cermakian, N., Ikeda, T. M., Cedergren, R., and
Gray, M. W. (1996) Nucleic Acids Res., 24, 648-654.
4.Chamberlin, M., McGrath, J., and Waskell, L. (1970)
Nature, 228, 227-231.
5.Moffatt, B. A., Dunn, J. J., and Studier, F. W.
(1984) J. Mol. Biol., 173, 265-269.
6.Sousa, R., Chung, Y. T., Rose, J. P., and Wang,
B.-C. (1993) Nature, 364, 593-599.
7.Jeruzalmi, D., and Steitz, T. A. (1998) EMBO
J., 17, 4101-4113.
8.Cheetham, G. M. T., Jeruzalmi, D., and Steitz, T.
A. (1999) Nature, 399, 80-83.
9.Cheetham, G. M. T., and Steitz, T. A. (1999)
Science, 286, 2305-2309.
10.Cheetham, G. M. T., and Steitz, T. A.
(2000) Curr. Opin. Struct. Biol., 10,
117-123.
11.Steitz, T. A. (1993) Curr. Opin. Struct.
Biol., 3,31-38.
12.Joyce, C. M., and Steitz, T. A. (1994) Annu.
Rev. Biochem., 63, 777-822.
13.Delarue, M., Poch, O., Tordo, N., Moras, D., and
Argos, P. (1990) Protein Eng., 3, 461-467.
14.Tunitskaya, V.L., Luchin, S.V., Memelova, L.V.,
Lyakhov, D.L., Rechinsky, V.O., and Kochetkov, S.N. (1989) Mol.
Biol. (Moscow), 23, 1273-1278.
15.Tunitskaya, V. L., Mishin, A. A., Tyurkin, V. V.,
Lyakhov, D. L., Rechinsky, V. O., and Kochetkov, S. N. (1988) Mol.
Biol. (Moscow), 22, 1642-1649.
16.Rusakova, E. E., Tunitskaya, V. L., and
Kochetkov, S. N. (1999) Mol. Biol. (Moscow), 33,
353-367.
17.Pozhitkov, A. E., Lavrik, I. N., Sergeev, M. M.,
and Kochetkov, S. N. (1998) Mol. Biol. (Moscow), 32,
93-97.
18.Lyakhov, D. L., Ilgenfritz, H., Chernov, B. K.,
Dragan, S. M., Rechinsky, V. O., Pokholok, D. K., Tunitskaya, V. L.,
and Kochetkov, S. N. (1992) Mol. Biol. (Moscow), 26,
1022-1035.
19.Protasevich, I. I., Memelova, L. V., Kochetkov,
S. N., and Makarov, A. A. (1994) FEBS Lett., 349,
429-432.
20.Ikeda, R. A., and Richardson, C. C. (1987) J.
Biol. Chem., 262, 3790-3799.
21.Martin, C. T., Muller, D. K., and Coleman, J. E.
(1988) Biochemistry, 27, 3966-3974.
22.Muller, D. K., Martin, C. T., and Coleman, J. E.
(1988) Biochemistry, 27, 5763-5771.
23.Tunitskaya, V. L., Akbarov, A. Kh., Luchin, S.
V., Memelova, L. V., Rechinsky, V. O., and Kochetkov, S. N. (1990)
Eur. J. Biochem., 191, 99-103.
24.Akbarov, A. Kh., Tunitskaya, V. L., and
Kochetkov, S. N. (1990) Biochem. Int., 20, 1033-1040.
25.Lyakhov, D. L., Ilgenfritz, H., Rechinsky, V. O.,
Tunitskaya, V. L., Chernov, B. K., and Kochetkov, S. N. (1991) Dokl.
Akad. Nauk SSSR, 317, 1005-1008.
26.Schenborn, E. T., and Mierendorf, R. C., Jr.
(1985) Nucleic Acids Res., 13, 6223-6236.
27.Filippova, S. E., Ivanovskaya, M. G., Romanova,
E. A., Tunitskaya, V. L., and Kochetkov, S. N. (2002) Mol. Biol.
(Moscow), in press.
28.Rechinsky, V. O., Kostyuk, D. A., Lyakhov, D. L.,
and Kochetkov, S. N. (1991) Mol. Biol. (Moscow), 25,
1588-1593.
29.Rechinsky, V. O., Kostyuk, D. A., Lyakhov, D. L.,
Chernov, B. K., and Kochetkov, S. N. (1993) Mol. Gen. Genet.,
238, 455-458.
30.Rechinsky, V. O., Tunitskaya, V. L., Dragan, S.
M., Kostyuk, D. A., and Kochetkov, S. N. (1993) FEBS Lett.,
320, 9-12.
31.Rechinsky, V. O., Chernov, B. K., Dragan, S. M.,
Kostyuk, D. A., Tunitskaya, V. L., and Kochetkov, S. N. (1995) Mol.
Gen. Genet., 247, 110-113.
32.Tabor, S., and Richardson, C. C. (1995) Proc.
Natl. Acad. Sci. USA, 92, 6339-6343.
33.Rusakova, E. E., Yankin, A. P., Memelova, L. V.,
Tunitskaya, V. L., a nd Kochetkov, S. N. (1999) Mol. Biol.
(Moscow), 33, 598-602.
34.Grachev, M. A., Zaychikov, E. E., Lukhtanov, E.
A., Maksimova, T. G., and Mustayev, A. A. (1987) Bioorg. Khim.,
13, 568-570.
35.Maksimova, T. G., Mustayev, A. A., Zaychikov, E.
F., Lyakhov, D. L., Tunitskaya, V. L., Akbarov, A. Kh., Luchin, S. V.,
Rechinsky, V. O., Chernov, B. K., and Kochetkov, S. N. (1991) Eur.
J. Biochem., 195, 841-847.
36.Tunitskaya, V. L., Bazhulina, N. P., Dragan,
S. M., Kuznetsova, N. V., Lyakhov, D. L., Rechinsky, V. O.,
Morozov, Yu. V., and Kochetkov, S. N. (1993) Mol. Biol.
(Moscow), 27, 51-57.
37.Tunitskaya, V. L., Dragan, S. M., Kostuyk, D. A.,
Lyakhov, D. L., Memelova, L. V., Rechinsky, V. O., and Kochetkov, S. N.
(1994) Biochemistry (Moscow), 59, 355-362.
38.Bonner, G., Patra, D., Lafer, E. M., and Sousa,
R. (1992) EMBO J., 11, 3767-3775.
39.Osumi-Davis, P. A., de Aguilera, M. C., Woody, R.
W., and Woody, A.-Y. M. (1992) J. Mol. Biol., 226,
37-45.
40.Kochetkov, S. N., Rechinsky, V. O., Tunitskaya,
V. L., Kostyuk, D. A., Lyakhov, D. L., and Memelova, L. V. (1994) in
Modern Enzymology: Problems and Trends, Nova Science Publ. Inc.,
New York, pp. 277-292.
41.Kochetkov, S. N., Kostyuk, D. A., Lyakhov, D. L.,
Memelova, L. V., Rechinsky, V. O., and Tuniyskaya, V. L. (1996) in
Chemical Modification of Enzymes (Kurganov, B. I., Nagradova, N.
K., and Lavrik, O. I., eds.) Nova Science Publ. Inc., New York, pp.
347-387.
42.Tunitskaya, V. L., Rusakova, E. E., Memelova, L.
V, Kochetkov, S. N., van Aershot, A., Herdewijn, P., Efimtseva, E. V.,
Ermolinsky, B. S., and Mikhailov, S. N. (1999) FEBS Lett.,
442, 20-24.
43.Rusakova, E. E., Tunitskaya, V. L., Memelova, L.
V., Ermolinsky, B. S., Kochetkov, S. N., and Mikhailov, S. N. (1999)
Nucleosides and Nucleotides, 18, 1359-1360.
44.Sousa, R., and Padilla, R. (1995) EMBO
J.,14, 4609-4621.
45.Tunitskaya, V. L., Memelova, L. V., Kostuyk, D.
A., Gudima, S. O., and Kochetkov, S. N. (1997) Mol. Biol.
(Moscow), 31, 365-370.
46.Kostuyk, D. A., Dragan, S. M., Rechinsky, V. O.,
Tunitskaya, V. L., Chernov, B. K., and Kochetkov, S. N. (1995) Dokl.
Akad. Nauk, 346, 824-827.
47.Kostyuk, D. A., Dragan, S. M., Lyakhov, D. L.,
Rechinsky, V. O., Tunitskaya, V. L., Chernov, B. K., and Kochetkov, S.
N. (1995) FEBS Lett., 369, 165-168.
48.Gudima, S. O., Kostyuk, D. A., Grishchenko, O.
I., Tunitskaya, V. L., Memelova, L. V., and Kochetkov, S. N. (1998)
FEBS Lett., 439, 302-306.
49.Gudima, S. O., Kazantseva, E. G., Kostyuk, D. A.,
Shchaveleva, I. L., Grishchenko, O. I., Memelova, L. V., and Kochetkov,
S. N. (1997) Nucleic Acids Res., 25, 4614-4618.
50.Aphasizhev, R. A., Theobald-Dietrich, A.,
Kostyuk, D. A., Kochetkov, S. N., Kisselev, L. L., Giege, R., and
Fasiolo, F. (1997) RNA, 3, 893-904.
51.Rusakova, E. E., Tunitskaya, V. L., Memelova, L.
V., Kochetkova, S. V., Kostyuk, D. A., and Kochetkov, S. N. (1998)
FEBS Lett., 423, 189-192.
52.Osumi-Davis, P. A., Sreerama, N., Volkin, D. B.,
Middaugh, R. C., Woody, R. W., and Woody, A.-Y. M. (1994) J. Mol.
Biol., 237, 5-19.
53.Tunitskaya, V. L., Kochetkova, S. V., Godovikova,
T. S., and Kochetkov, S. N. (2000) Mol. Biol. (Moscow),
34, 60-66.
54.Tunitskaya, V. L., Memelova, L.V., Skoblov, Yu.S.
and Kochetkov, S. N. (2003) Mol. Biol. (Moscow), in press.