REVIEW: Effect of Interactions Between Amino Acid Residues 43 and 61 on Thermal Stability of Bacterial Formate Dehydrogenases
V. V. Fedorchuk1, A. G. Galkin1, I. E. Yasny1, L. B. Kulakova1, A. M. Rojkova1, A. A. Filippova2, and V. I. Tishkov1*
1Department of Chemical Enzymology, School of Chemistry, Lomonosov Moscow State University, Moscow, 119992 Russia; fax: (095) 939-2742; E-mail: vit@enz.chem.msu.ru2Department of General Chemistry, Sechenov Moscow Medicine Academy, Moscow, Russia
* To whom correspondence should be addressed.
Received April 26, 2002
NAD+-dependent formate dehydrogenases (EC 1.2.1.2, FDH) of methylotrophic bacteria Pseudomonas sp. 101 (PseFDH) and Mycobacterium vaccae N10 (MycFDH) exhibit high homology. They differ in two amino acid residues only among a total of 400, i.e., Ile35 and Glu61 in MycFDH substitute for Thr35 and Lys61 as in PseFDH. However, the rate constant for MycFDH thermal inactivation in the temperature range of 54-65°C is 4-6-times higher than the corresponding rate constant for the enzyme from Pseudomonas sp. 101. To clarify the role of these residues in FDH stability the dependence of the apparent rate constant for enzyme inactivation on phosphate concentration was studied. Kinetic and thermodynamic parameters for thermal inactivation were obtained for both recombinant wild-type and mutant forms, i.e., MycFDH Glu61Gln, Glu61Pro, Glu61Lys and PseFDH Lys61Arg. It has been shown that the lower stability of MycFDH compared to that of PseFDH is caused mainly by electrostatic repulsion between Asp43 and Glu61 residues. Replacement of Lys61 with an Arg residue in the PseFDH molecule does not result in an increase in stability.
KEY WORDS: formate dehydrogenase, thermal inactivation, thermal stability, directed mutagenesis, Pseudomonas sp. 101, Mycobacterium vaccae N10
NAD+-dependent formate dehydrogenases (EC 1.2.1.2, FDH), being composed of two identical subunits and not containing metal ions or prosthetic groups in the active site, present a unique group among the enzymes catalyzing formate-ion oxidation to CO2 in the cell. Formate dehydrogenases of this group are widespread in nature (for details see [1, 2]) and belong to the superfamily of D-specific NAD(P)+-dependent dehydrogenases of 2-hydroxy acids [3]. FDHs of this type have been found in all strains of methanol-utilizing yeast of Candida, Hansenula, and Pichia species [1]. In contrast, this type of FDH is rather rare among methylotrophic bacteria, and currently only five strains synthesizing this type of FDH are known, i.e., Pseudomonas sp. 101 [4], Moraxella C-1 [5], Paracoccus sp. 12A [6], Mycobacterium vaccae N10 [7], and Hyphomicrobium sp. JC17 [8]. The genes of all these enzymes have been cloned and expressed in E. coli cells [7-10]. In addition, the genes of bacterial FDHs were found in the genome of uncultured proteobacterium EBAC31A08 (EMBL Accession AF279106) and in pSymA megaplasmid of symbiotic nitrogen-fixing bacterium Sinorhizobium meliloti [11]. The complete gene sequences for FDHs of methylotrophic yeast Pichia angusta (previously named Hansenula polymorpha) [12], Candida methylica [13], Candida boidinii [14, 15], baker's yeast Saccharomyces cerevisiae (EMBL Accession Z75296), fungi Aspergillus nidulans [16], Neurospora crassa [17], potato mitochondria [18], and barley [19] have been recently established.
All FDHs are characterized by similar values of Km for formate and NAD+ [2]. However, the specific activity of bacterial FDHs is about 1.5-2-times higher than that of eucaryotic FDHs. The second feature of bacterial FDHs distinguishing them from analogous enzymes of other sources is their higher thermal stability. Pseudomonas sp. 101 FDH can be stored at 4°C in phosphate buffer, pH 7.0, for a couple of years without changing its activity. At the same time, the commercially available lyophilized preparations of FDH from C. boidinii yeast (Boehringer Mannheim, Germany, purity <50%), containing special stabilizing additives, loses 50% of the initial activity in two weeks being dissolved in phosphate buffer and stored at 4°C. The period of half-inactivation of highly purified preparations of the above enzyme is even lower, no more than 2-3 days [15].
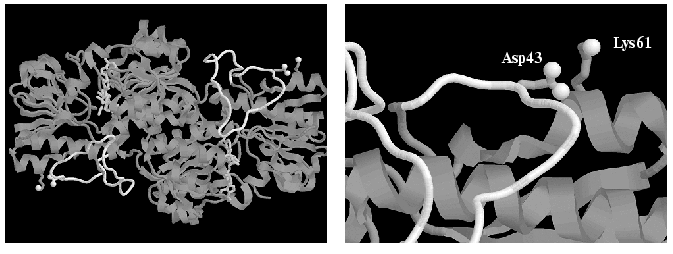
Comparison of amino acid sequences of FDHs from different sources (Fig. 1) shows that the bacterial enzymes contain a 29-residue longer N-terminal region. In accordance with the data of X-ray analysis [20], this region represents a long non-structured loop (Fig. 2). One may assume that the interaction of amino acid residues of this loop with the amino acid residues of the rest of the protein globule is one of the reasons for higher stability of bacterial FDHs. FDH from M. vaccae N10 (MycFDH), whose gene was cloned earlier, is the optimal one to test the above hypothesis [7]. This enzyme differs from FDH of Pseudomonas sp. 101 (PseFDH) by two substitutions only, i.e., Ile35 for Thr and Glu61 for Lys, but the rate of thermal inactivation of MycFDH is 4-6-times higher than that of PseFDH. The first of the above residues is located directly in the loop (Fig. 1), and the second one is neighboring Asp43 (Fig. 2) located in the loop, which is missing in non-bacterial FDHs. The mutant forms of MycFDH with the Glu61Lys, Glu61Gln, and Glu61Pro replacements were obtained earlier [7]. The preliminary studies on the thermal inactivation of these mutants at 60.5°C demonstrated their higher stability [7]. However, the comparison of rate constants for thermal inactivation at one fixed temperature cannot be used as a characteristic of the stabilization effect because the temperature dependence of these rate constants for the wild-type and mutant forms may be completely different. To exemplify the above statement we can point to the data on the thermal stability of the PseFDH Arg284Gln mutant: to achieve a 10-fold increase in the inactivation rate constant for the wild-type enzyme one needs to go 4°C up (see “Results”), while for the noted mutant to get the same enhancement one has to increase the temperature 25°C up [21]. This results in a similar stability of the wild-type and Arg284Gln PseFDH mutant at 54°C, and in a 20-fold increase in stability of the mutant compared to the wild-type enzyme at 68°C. In addition, the temperature dependence of the inactivation rate constant gives information on the component, i.e., entropy or enthalpy, predominantly determining the enzyme stability.
Fig. 1. Amino acid sequences of N-terminal region of formate dehydrogenases from different sources: PseFDH, Pseudomonas sp. 101; MycFDH, Mycobacterium vaccae N10; MorFDH, Moraxella sp. C-1; ParFDH, Paracoccus sp. 12A; HypFDH, Hyphomicrobium sp. JC17; SmeFDH, Sinorhizobium meliloti; UncFDH, uncultured proteobacterium EBAC31A08; StuFDH, potato; BarFDH, barley; SceFDH, baker's yeast; CmeFDH, Candida methylica; HanFDH, Pichia angusta; NeuFDH, Neurospora crassa; AspFDH, Aspergillus nidulans. Potato and barley FDH sequences are shown without signal peptides. Residues 35, 43, and 61 are shown in bold and seven Pro residues in the sequence 11-46 of bacterial FDHs are shown in bold italic.

The goal of the present work was to quantitatively estimate the effect of amino residues in positions 35 and 61 on thermal stability of bacterial FDH. To clarify the role of these residues in FDH stability, the dependence of the apparent rate constant for enzyme inactivation on phosphate concentration was studied. Kinetic and thermodynamic parameters for thermal inactivation were obtained for both recombinant wild-type and mutant forms, i.e., MycFDH Glu61Gln, Glu61Pro, Glu61Lys and PseFDH Lys61Arg.Fig. 2. A general view of the holo-form of Pseudomonas sp. 101 formate dehydrogenase (PDB2NAD.ENT [17]) and the enlarged fragment with Asp43 and Lys61 residues. The “non-structured loop” in the sequence 11-46 is shown in white. Oxygen atoms of carboxy-group of Asp43 and nitrogen atom of Lys61 are shown in white balls. The picture was made using RasMol 2.6b program.
MATERIALS AND METHODS
The following enzymes were used: phage T4 DNA-polymerase (10 U/µl), DNA-ligase (400 U/µl), and polynucleotide kinase (16 U/µl), all from New England Biolabs (USA). All reagents used for genetic engineering manipulations were of “molecular biology grade” (Sigma, USA).
Site-directed mutagenesis was performed in accordance with the Kunkel method as described in [7]. To confirm the unique mutation sites the gene-containing plasmid regions were sequenced in both directions using an Applied Biosystems model 370A automated DNA sequencer (USA) and an ABI PRISM DNA Sequencing Kit based on fluorescent labeled terminators produced by the same company. The mutagenesis efficiency was about 60-100%. For the thermal stability studies each mutant enzyme was produced using the plasmids isolated from two individual clones.
Production and purification of MycFDH, PseFDH, and their mutant forms. Biomass of E. coli TG1 cells with the corresponding plasmid was produced by cultivation of a single colony in 200 ml of 2YT medium (16 g/liter bactotryptone, 10 g/liter yeast extract (both from Difco, USA), and 5 g/liter NaCl, pH 7.0) containing 150 µg/ml ampicillin for 12-15 h at 37°C. The inducer of FDH biosynthesis, beta-isopropyl-D-thiogalactoside (IPTG), was added in the beginning of cultivation up to 0.5 mM. The cells were collected by centrifugation at 8000g on a Beckman J-21 centrifuge (USA) for 10 min. The further purification of MycFDH and PseFDH and their mutant forms was performed using the standard protocol developed for the recombinant FDH of Pseudomonas sp. 101 expressed in E. coli [22]. The protocol included cell disruption in a ultrasonic disintegrator, ammonium sulfate fractionation (40% saturation) and FPLC hydrophobic chromatography (Pharmacia Biotech, Sweden) on a 1 × 10 cm column packed with highly substituted Phenyl Sepharose Fast Flow (Pharmacia Biotech). The enzyme preparations obtained were at least 90-95% purity as judged from SDS-PAGE. For the purposes of thermal stability studies, all enzyme preparations were transferred into a K-phosphate buffer of the needed concentration, pH 7.0, by gel filtration through Sephadex G-25 (Pharmacia Biotech).
For each individual enzyme four independent preparations from four biomass samples were obtained.
Formate dehydrogenase activity was assayed with an automated Reaction Rate Analyzer, model 2086 Mark 2 (LKB, Sweden), following NADH formation at 340 nm. Protein concentration was determined by absorbance at 280 nm using the value of A1%1cm,280 = 16 [23] and the molecular mass of recombinant FDH equal to 88 kD [7, 9].
The dependence of inactivation rate on phosphate buffer concentration was studied at 61°C in the concentration range of 0.01-1.26 M. The temperature dependence of the inactivation rate constant was studied in the temperature range 54-65°C in 0.1 M phosphate buffer. Enzyme solution (50 µl) dissolved in 0.1 M K-phosphate buffer, pH 7.0, preliminary heated to 45°C, was added to 550 µl of the same buffer, preliminary heated to the needed temperature (with the accuracy of ±0.1°C), to the final enzyme concentration of 0.05-0.1 mg/ml, vigorously shaken, and incubated at the designated temperature. To measure the enzyme residual activity 50 µl aliquots were taken at fixed intervals. The thermal inactivation rate constant kin was determined from the time course of enzyme inactivation treated as first-order kinetics by nonlinear regression using the Origin 4.1 program. Each value of kin for the native and mutant FDH forms represents an average from at least four independent measurements. The activation parameter DeltaH. of thermal inactivation process was determined as a slope of a ln(kin/T) versus 1/T plot using linear regression with the statistic weights of (kin/T)2/s2, where T is the absolute temperature, and s is the error of kin determination calculated from primary plots. The value of DeltaS. was determined from the slope of the DeltaG. versus T plot.
Modeling and structural optimization of FDH mutants was performed with the Insight II program on a Silicon Graphics workstation. To visualize and analyze protein three-dimensional structures RasMol 2.6b and WebLab Viewer Pro 3.7 (MSI) programs were used.
RESULTS
To clarify the role of amino acid residues in positions 35 and 61 in the sequence of bacterial FDH in the enzyme thermal stability the physicochemical characteristics of the inactivation process were studied for the wild-type MycFDH and PseFDH and their single point mutants: MycFDH Glu61Gln, Glu61Pro, Glu61Lys and PseFDH Lys61Arg. The first mutation, Glu61Gln, removes the negative charge, and Glu61Pro mutation provides both the removal of the negative charge and the increase in the rigidity of the polypeptide chain. Glu61Lys mutant of MycFDH and wild-type PseFDH differ in only one amino acid residue in position 35, the former has Ile, and the latter has Thr. The comparison of inactivation rates of these enzymes allows one to determine the contribution of the Thr35Ile replacement to the enzyme stability. The construction of PseFDH Lys61Arg mutant was aimed to further improve the thermal stability of the enzyme. The improvement of stability could originate from the facts that: 1) a guanidinium group of arginine residue is a much stronger base than epsilon-amino group of the lysine residue, and thus, the ionic pair Asp43-Arg61 will be stronger than Asp43-Lys61; and 2) comparative statistical analysis of structures of proteins from mesophylic and thermophylic microorganisms [24] demonstrates the presence of Arg residue instead of Lys in alpha-helix regions of enzymes with high thermal stability.
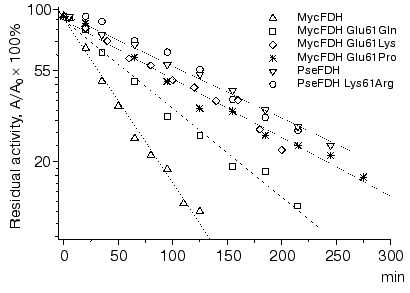
The time course of wild-type and mutant FDHs inactivation at 60°C is presented in Fig. 3. As one can see from these data, the time dependence of enzyme residual activity is linear in semi-logarithmic plots (lnA versus t). The value of the slope of the straight lines, i.e., the apparent inactivation rate constant kin, was independent of enzyme concentration in the range of 0.03-0.25 mg/ml. The linear character of dependences in Fig. 3 and the independence of the apparent rate constants on the initial enzyme concentration indicate that FDH thermal inactivation proceeds in accordance with first order kinetics. In other words, this indicated the unfolding of the protein globule is not preceded by enzyme dissociation into individual active subunits. It must be noted that PseFDH is known for strong interaction between its subunits. For instance, the application of 8 M urea is not sufficient to dissociate the enzyme into individual subunits for the purpose of specific modification of Cys145 residue located in the region of the inter-subunit contact. The data presented in Fig. 3 prove the important role of the residue in position 61 in the enzyme stability. The lowest stability is observed for the wild-type MycFDH. The removal of the negative charge (Glu61Gln mutation) improves the thermal stability decreasing the inactivation rate constant by 2.5-fold. Moreover, introduction of a positive charge of Lys residue in this position, Glu61Lys mutant of MycFDH, yields an enzyme whose stability is close to that of the wild-type PseFDH. A comparable stabilization effect is achieved by introducing a proline residue in position 61 (Fig. 3). The replacement Lys61Arg in the PseFDH molecule did not lead to stability improvement.
The efficiency of electrostatic interaction significantly drops with an increase in the ionic strength. To discriminate between the contribution of residues in positions 35 and 61 into the FDH stability the dependence of the apparent rate constant for the enzyme inactivation on the concentration of phosphate buffer was studied. The choice of phosphate for the purpose of ionic strength studies was based on the big radius of its anion. The latter prevents phosphate from penetrating inside the protein globule, and thus, phosphate can affect only ion pairs located at the surface of the enzyme molecule. The dependence of the apparent rate constants for the studied enzymes on phosphate concentration in the range 0.01-1.26 M is presented in Fig. 4. The obtained bell-shaped curve is typical for most proteins. The initial increase in ionic strength causes enzyme destabilization and an increase in inactivation rate constant because of the decreased efficiency of electrostatic interactions. At some point, all ion pairs on the protein surface are destroyed and electrostatic interaction makes no more contribution to the enzyme stability. This leads to the maximum destabilizing effect. The further increase in the ionic strength enhances hydrophobic interactions, and this stabilizes FDH. We note here the three most important and interesting results.Fig. 3. Time course of thermal inactivation of wild-type and mutant FDHs from M. vaccae N10 and Pseudomonas sp. 101 at 60°C. Since the pairs of enzymes MycFDH Glu61Lys-MycFDH Glu61Pro and wild-type PseFDH-PseFDH Lys61Arg show very close stability, their inactivation curve is shown as one straight line. Enzyme concentration 0.05-0.1 mg/ml, 0.1 M K-phosphate buffer, pH 7.0.
1. Maximum destabilization effect for the wild-type MycFDH is observed at phosphate concentration of 0.1 M, while for all mutant forms of this enzyme and for PseFDH the maximum decrease in stability occurs at 0.2-0.3 M concentrations.Fig. 4. Dependence of rate constants of thermal inactivation of the wild-type and mutant forms of M. vaccae N10 and Pseudomonas sp. 101 FDHs on phosphate concentration. Enzyme concentration 0.05-0.1 mg/ml, K-phosphate buffer, pH 7.0, 61°C.
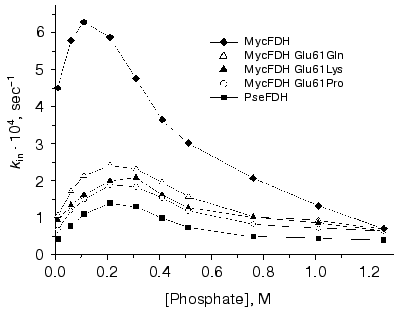
2. Maximum 3-fold decrease of enzyme stability (expressed as the ratio of inactivation rate constants at 0.01 and 0.21 M phosphate) is observed for PseFDH and MycFDH E61K, which contain the ion pair Asp43-Lys61. In the case when the above mutation is absent, for instance in MycFDH Glu61Pro and Glu61Gln mutants, the destabilization effect is smaller and is equal to 2. The minimum decrease in stability (1.4-fold) is observed for the wild-type MycFDH because an increase in the ionic strength diminishes the repulsion of carboxy-groups of Asp43 and Glu61.
3. At high phosphate concentrations destroying all surface ion pairs, the rate constants for the wild-type MycFDH and all its mutant forms are equal and only 1.5-fold higher than kin for PseFDH. This means that, under the above conditions, the difference in stability of PseFDH and all MycFDH forms is based only on the nature of the amino acid residue in position 35.
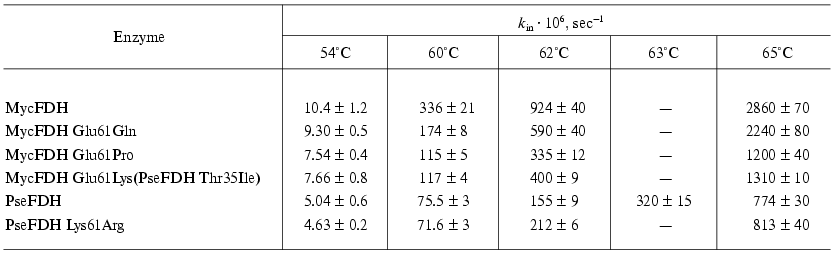
To get the complete information on the mechanism of wild-type and mutant FDH inactivation the temperature dependence of the apparent rate constants for enzyme inactivation was studied in the 54-65°C range (Table 1). To conduct these experiments we have chosen the concentration of phosphate buffer equal to 0.1 M since it is the concentration which provides the maximum difference in stability of wild-type and mutant MycFDHs and PseFDH. The data presented in Table 1 indicate the different effect of temperature on the inactivation rates of the studied enzymes. We note that the increase in rigidity of the polypeptide chain caused by the replacement of Glu61 with Pro provides the enzyme with a higher stability at 62°C than the restoration of the ionic pair Asp43-Lys61 (Table 1). Different temperature dependence is also observed for the wild-type PseFDH (ion pair Asp43-Lys61) and its K61R mutant (ion pair Asp43-Arg61). At 54 and 60°C, a slight but reliable and repeatedly reproducible increase in stability of PseFDH K61R mutant compared to the wild-type PseFDH is observed, while at 62°C and above the wild-type enzyme is more stable (Table 1).
Table 1. Rate constants of thermal
inactivation of the wild-type and mutant forms of M. vaccae N10
and Pseudomonas sp. 101 FDHs
The secondary plots of ln(kin/T) dependence on 1/T were linear. The linear character of the secondary plots proves that thermal inactivation of wild-type and mutant FDH forms can be described by the theory of an activated complex. In accordance with the latter [25], the temperature dependence of the apparent rate constant for thermal inactivation, kin, is presented by the following equation:
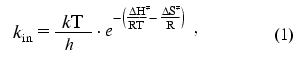
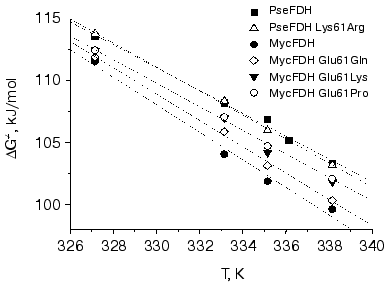
where k and h are the Boltzmann and Plank constants, respectively, R is the universal gas constant, and DeltaH. and DeltaS. are the activation parameters of thermal inactivation. The values of DeltaH. and DeltaS. can be calculated from slopes and intercepts of the secondary plots ln(kin/T) versus 1/T, respectively. However, the extrapolation of the linear dependence to the ordinate axis to determine the value of DeltaS. gives a high error in its determination. The more precise value for this parameter can be obtained from the dependence of DeltaG. on T (Fig. 5). The calculated values of DeltaH. and DeltaS. are shown in Table 2. We note that the conversion from PseFDH to MycFDH is accompanied by changes in both DeltaH. and DeltaS., and moreover, the change in DeltaH. value is ~60 kJ/mol or 15%.
Table 2. Activation parameters for thermal
inactivation of the wild-type and mutant forms of M. vaccae N10
and Pseudomonas sp. 101 FDHs
Fig. 5. Dependence of free activation energy of thermal denaturation of the wild-type and mutant forms of M. vaccae N10 and Pseudomonas sp. 101 FDHs on absolute temperature.
DISCUSSION
The data obtained in this study confirm the important role of the non-structured loop in the region of 15-43 residues in providing high thermal stability of bacterial FDHs. The replacement of Lys61 residue with Glu residue in the MycFDH molecule results in a significant drop in the enzyme stability. The data on thermal stability of Glu61Gln and Glu61Lys mutants of MycFDH indicate the importance of electrostatic interaction between the residues in positions 43 and 61. The removal of the negative charge in position 61 in the first mutant results in slight stabilization of the enzyme, while the restoration of the ionic pair Asp43-Lys61 yields the MycFDH mutant, whose rate constant of thermal inactivation is only 1.5-fold higher that the corresponding rate constant for PseFDH (Table 1). The destabilization of MycFDH compared to PseFDH is partially due to the mutation of residue 35 (Ile35Thr). The side group of residue 35 is exposed to the solution, and the replacement of the hydrophilic side group of Thr residue with a bulky hydrophobic side group of Ile can worsen the entropy. As shown in our earlier works on Ser-Ala mutations in alpha-helix regions of PseFDH [26], the entropy change originating from the replacement of a polar group with a nonpolar one results in a 9-40% change in the enzyme stability. Nevertheless, the contribution of this mutation is much smaller compared to introduction of glutamic acid to replace lysine at position 61.
The observed changes in thermal stability in our experiments are rather small compared to the effects that could be predicted for optimized electrostatic interactions [20]. The data on X-ray structural analysis for both apo- and holo-forms of PseFDH confirm the total exposure of the Asp43 carboxy-group and the Lys61 amino group into solution (Fig. 2). Thus, the efficient interaction between two oppositely charged groups is strongly weakened because of the high dielectric constant of water. This may explain the absence of additional stabilization in the case of Lys61Arg replacement in PseFDH.
As noted earlier, bacterial FDHs possess an additional 29-residue sequence in the N-terminal region (Fig. 1), which forms a non-structured loop (Fig. 2, shown in gray color) absent in the eukaryotic enzymes. However, the term “non-structured” was used only to emphasize the formal absence of typical elements of secondary structure. In reality, the loop must be of high rigidity, because it has seven Pro residues among total 35 (11-46 in sequence). Thus, this region of the amino acid sequence of bacterial FDH is divided into small blocks of 4-7 amino acid residues by proline residues. The analysis of thermal stability changes induced by Glu61Pro mutation (Table 1) leads to the conclusion that electrostatic interactions between 43 and 61 residues may be necessary for the fixation of the polypeptide chain in the region of residue 61 but not for the stabilization of the already rigid “non-structured” loop formed by residues 11-46. The Lys61 residue is the second one in the alpha1-helix (Figs. 1 and 2), and based on general considerations, its mutation to Pro (Lys61Pro) had to change the helix conformation. However, the results of computer modeling for this mutant structure demonstrate no distortion of the alpha1-helix with the introduced mutation. At first sight this may seem unexpected; however, a similar situation is observed for PseFDH in the case of Pro105 residue located at the second position in the alpha3-helix [20] (not shown in Fig. 1). A similar stability of Glu61Pro and Glu61Gln mutants of MycFDH (Fig. 3, Table 1) is a strong argument supporting the above assumption.
In conclusion, the experiments presented in this work have shown that the “non-structured” loop in the region of residues 11-46, which is present in all bacterial formate dehydrogenases and absent in analogous eukaryotic enzymes, is one of the reasons for the higher stability of bacterial enzymes. The data obtained demonstrate that one of the “weak” points in the structure of bacterial FDHs is the region of the polypeptide chain including residue 61. This region can be stabilized both by electrostatic interaction with Asp43, located in the “non-structured” loop and by increasing the rigidity of the polypeptide chain and introducing Pro residue in position 61. We are currently working on the increase in thermal stability of Pseudomonas sp. 101 FDH, which is the most stable enzyme among the NAD+-dependent formate dehydrogenase family. A number of site-directed mutations have yield the enzymes with the thermal stability prevailing over that of the wild-type enzyme by a factor of 10-20. These experiments will be reported in detail in our further publications.
This work was supported by grants of the Russian Foundation for Basic Research 99-04-49156 and 02-04-49415.
REFERENCES
1.Rodionov, Yu. V. (1981) Uspekhi Mikrobiol.,
16, 104-138.
2.Popov, V. O., and Lamzin, V. S. (1994) Biochem.
J., 301, 625-643.
3.Vinals, C., Depiereux, E., and Feytmans, E. (1993)
Biochem. Biophys. Res. Commun., 192, 182-188.
4.Egorov, A. M., Avilova, T. V., Dikov, M. M., Popov,
V. O., Rodionov, Yu. V., and Berezin, I. V. (1979) Eur. J.
Biochem., 99, 569-576.
5.Asano, Y., Sekigawa, T., Inukai, H., and Nakazawa,
A. (1988) J. Bacteriol., 170, 3189-3193.
6.Iida, M., Kitamura-Kimura, K., Maeda, H., and
Mineki, S. (1992) Biosci. Biotech. Biochem., 56,
1966-1970.
7.Galkin, A., Kulakova, L., Tishkov, V., Esaki, N.,
and Soda, K. (1995) Appl. Microbiol. Biotechnol., 44,
479-483.
8.Mitsunaga, T., Tanaka, Y., Yoshida, T., and
Watanabe, K. (2000) Japan Patent JP245471A2.
9.Tishkov, V. I., Galkin, A. G., Marchenko, G. N.,
Tsygankov, Y. D., and Egorov, A. M. (1993) Biotechnol. Appl.
Biochem., 18, 201-207.
10.Shinoda, T., Satoh, T., Mineki, S., Iida, M., and
Taguchi, H. (2002) Biosci. Biotech. Biochem., 66,
271-276.
11.Barnett, M. J., Fisher, R. F., Jones, T., Komp,
C., Abola, A. P., Barloy-Hubler, F., Bowser, L., Capela, D., Galibert,
F., Gouzy, J., Gurjal, M., Hong, A., Huizar, L., Hyman, R. W., Kahn,
D., Kahn, M. L., Kalman, S., Keating, D. H., Palm, C., Peck, M. C.,
Surzycki, R., Wells, D. H., Yeh, K.-C., Davis, R. W., Federspiel, N.
A., and Long, S. R. (2001) Proc. Natl. Acad. Sci. USA,
98, 9883.
12.Hollenberg, C. P., and Janowicz, Z. (1989)
European Patent EP1987000110417, Bulletin 89/03.
13.Allen, S. J., and Holbrook, J. J. (1995)
Gene, 162, 99-104.
14.Sakai, Y., Murdanoto, A. P., Konishi, T.,
Iwamatsu, A., and Kato, N. (1997) J. Bacteriol., 179,
4480-4485.
15.Slusarczyk, H., Felber, S., Kula, M. R., and
Pohl, M. (2000) Eur. J. Biochem., 267, 1280-1289.
16.Saleeba, J. A., Cobbett, C. S., and Hynes, M. J.
(1992) Mol. Gen. Genet., 235, 349-358.
17.Chow, C. M., and RajBhandary, U. L. (1993) J.
Bacteriol., 175, 3703-3709.
18.Colas des Francs-Small, C., Ambard-Bretteville,
F., Small, I. D., and Remy, R. (1993) Plant Physiol.,
102, 1171-1177.
19.Suzuki, K., Itai, R., Suzuki, K., Nakanishi, H.,
Nishizawa, N. K., Yoshimura, E., and Mori, S. (1998) Plant
Physiol., 116, 725-732.
20.Lamzin, V. S., Dauter, Z., Popov, V. O.,
Harutyunyan, E. H., and Wilson, K. S. (1994) J. Mol.
Biol.,236, 759-785.
21.Galkin, A. G., Kutsenko, A. S., Bajulina, N. P.,
Esipova, N. G., Lamzin, V. S., Mezentzev, A. V., Shelukho, D. V.,
Tikhonova, T. V., Tishkov, V. I., Ustinnikova, T. B., and Popov, V. O.
(2002) Biochim. Biophys. Acta, 1594, 136-149.
22.Tishkov, V. I., Galkin, A. G., Gladyshev, V. N.,
Karzanov, V. V., and Egorov, A. M. (1992) Biotekhnologiya, No.
5, 52-59.
23.Rodionova, Yu. V., Avilova, T. V., and Popov, V.
O. (1977) Biokhimiya, 43, 2020-2026.
24.Menendez-Arias, L., and Argos, P. (1989) J.
Mol. Biol., 206, 397-406.
25.Cornish-Bowden, A. (1976) Principles of Enzyme
Kinetics, Chap. 1, Butterworth & Co, London-Boston.
26.Rojkova, A. M., Galkin, A. G., Kulakova, L. B.,
Serov, A. E., Savitsky, P. A., Fedorchuk, V. V., and Tishkov, V. I.
(1999) FEBS Lett., 445, 183-188.