REVIEW: Antibody Proteases: Induction of Catalytic Response
A. G. Gabibov1,2*, A. Friboulet3, D. Thomas3, A. V. Demin4, N. A. Ponomarenko1, I. I. Vorobiev1, D. Pillet3, M. Paon3, E. S. Alexandrova1, G. B. Telegin5, A. V. Reshetnyak2, O. V. Grigorieva2, N. V. Gnuchev4, K. A. Malishkin6, and D. D. Genkin6
1Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, Moscow, 117198 Russia; fax: (095) 310-7007; E-mail: gabibov@ibch.ru2School of Chemistry, Lomonosov Moscow State University, Moscow, 119899 Russia
3Compiegne Technological University, Compiegne, France
4Institute of Gene Biology, Russian Academy of Sciences, ul. Vavilova 34/5, Moscow, 117984 Russia
5Branch of Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, Pushchino, Moscow Region, 142290 Russia; fax: (27) 790-527
6ASGL-Scientific Laboratories, Charity, P.O. Box 32, St. Petersburg, Russia
* To whom correspondence should be addressed.
Received May 27, 2002; Revision received July 12, 2002
Most of the data accumulated throughout the years on investigation of catalytic antibodies indicate that their production increases on the background of autoimmune abnormalities. The different approaches to induction of catalytic response toward recombinant gp120 HIV-1 surface protein in mice with various autoimmune pathologies are described. The peptidylphosphonate conjugate containing structural part of gp120 molecule is used for reactive immunization of NZB/NZW F1, MRL, and SJL mice. The specific modification of heavy and light chains of mouse autoantibodies with Val-Ala-Glu-Glu-Glu-Val-PO(OPh)2 reactive peptide was demonstrated. Increased proteolytic activity of polyclonal antibodies in SJL mice encouraged us to investigate the production of antigen-specific catalytic antibodies on the background of induced experimental autoimmune encephalomyelitis (EAE). The immunization of autoimmune-prone mice with the engineered fusions containing the fragments of gp120 and encephalitogenic epitope of myelin basic protein (MBP89-104) was made. The proteolytic activity of polyclonal antibodies isolated from the sera of autoimmune mice immunized by the described antigen was shown. Specific immune response of SJL mice to these antigens was characterized. Polyclonal antibodies purified from sera of the immunized animals revealed proteolytic activity. The antiidiotypic approach to raise the specific proteolytic antibody as an “internal image” of protease is described. The “second order” monoclonal antibodies toward subtilisin Carlsberg revealed pronounced proteolytic activity.
KEY WORDS: proteolysis, catalytic antibodies, abzymes, antiidiotypic antibodies, reactive immunization, peptidylphosphonates, HIV-1, gp120, autoimmune inbred lines MRL, NZB/NZW F1, and SJL
The generation of a protease which would be able to recognize and hydrolyze a definite protein epitope in vivo is one of the fundamental problems of biochemistry, molecular biology, and molecular medicine. The successful realization of the approaches for directed destruction of extracellular proteins in blood of patients has considerable interest for modern pharmacology. In this sense biocatalysts that belong to the immunoglobulin superfamily (abzymes) have a considerable advantage over traditional proteases. Among the potential targets of “catalytic vaccines” are envelope virion proteins, cytokines, and different pathogenic proteins. Now the principal difficulties in the design of proteolytic antibodies based on the theory of transition state prevent considerable results [1, 2]. However, a series of recent studies in abzymology demonstrated that high probability of spontaneous catalytic antibody formation in a course of autoimmune process as well as the possibility of induction high catalytic response against various antigens in model animals with autoimmune pathologies give reasons for definite optimism in the field of the design of new biocatalysts which are abzymes [3-6].
In the present paper reviews some approaches for obtaining proteolytic abzymes. These approaches include: 1) the induction of epitope-specific catalytic response on a background of development of autoimmune process using covalently modified peptide antigens; 2) the induction of catalytic response by means of fusion proteins containing the antigen which provokes autoimmune process development; 3) the obtaining of antiidiotypic antibodies as antibodies of “internal image” of enzymes catalyzing proteolytic cleavage.
THE BASIC PRINCIPLES OF CATALYTIC RESPONSE INDUCTION
The description of proteolytic abzyme target. Coat protein of human immunodeficiency virus (HIV-1) gp120 was suggested as a main target for proteolytic abzymes. The selection of this object was determined by unsuccessful attempts to neutralize HIV-1 by antibodies which were obtained by direct animal immunization by surface antigens and their fragments. This phenomenon is the consequence of the hypervariability of gp120 immune dominant epitopes. At the same time, many antibodies to these protein constant regions having no neutralizing effect were obtained. HIV infection can be considered as the intervention of exposed variable and cryptic constant antigenic determinants. Cleavage of gp120 in the patient's body will overcome the viral immunological mimicry, destroy the antigen surface package, expose its constant epitopes and, consequently, provide the possibility to eliminate the HIV infection by the immune system of the patient [7]. There are experimental data concerning the disruption of the structure and properties of gp120 in terms of CD4 binding by the action of proteases [8]. At the same time, many antibodies recognizing both virion-bound and soluble gp120 are known to exist [9]. However, these antibodies are not neutralizing because of low binding constant or the lack of interference with infection mechanism. This limitation, which occurs for binding antibodies, does not occur in the case of abzymes as far as the antigen cleavage is discussed earlier to result in multiple consequences concerned with immune response activation and conformational reorganization of the protein. Nevertheless, the most direct way to obtain specific abzyme protease against exposed conservative regions of gp120 is to obtain antibody that would have specificity exactly to these parts of the protein. Moreover, selected parts of the protein have to be sensitive to proteolysis.
The analysis of published data has shown that two sites of gp120 satisfied to requirements discussed above: the regions of Lys432 and Glu269 [8]. The first region was defined by the inhibition of gp120 ability to bind CD4 after trypsin treatment, which cleaves after Lys or Arg residues. This region is disposed near the CD4 binding site and represents the loop from Met426 to Lys432. Cleavage of this loop leads to the loss of protein conformation essential for interaction with CD4. The second conservative region was determined due to repression of CD4 binding by gp120 after protease V8 treatment, which cleaves after Asp and Glu residues. This site in contrast to the first region is located on the opposite side of the gp120 molecule and most distant from the CD4 binding site. The region is a loop as well but more extended containing residues from Gly23 and Val270. It is obvious to conclude from all of the data presented above that, in spite of the compact gp120 structure and the presence of oligosaccharides and disulfide bonds, this molecule possesses a very metastable conformation which tends to relax readily due to protease cleavage. The distant localization of the regions described above from structure modifying parts of the gp120 causes their accessibility to proteolysis. Besides, these regions do not belong to the gp120 trimeric interface discovered by recent crystallographic studies of the protein [10, 11]. The choice of potential antigens for immunization was based on the reasons presented above. The conformational and consequently functional load, which is carried by these regions in the absence of stabilizing factors such as disulfide bonds and intersubunit contacts consisting the trimer and gp41 complex, causes both the sequence conservatism (the peptide NTEGSDTITLPCRIKQIINMWQEVGK is the most conservative in the protein, for example) and the protein structure relaxation upon the proteolysis. Thus, the arguments described above determined the choice of the potential targets for gp120 cleavage.
Induction of epitope-specific catalytic response by “reactive immunization” method. Three mouse strains were used for obtaining the epitope specific abzymes. Inbred mouse strains with autoimmune diseases represent a perfect model for investigation of different aspects of autoimmunity. MRL mice during aging acquire a number of autoimmune disorders generally resembling those observed in SLE and rheumatoid arthritis [12]. The New Zealand hybrid mouse strain (NZB/NZW F1) spontaneously develop SLE-like nephritis [13]. In contrast, SJL mouse strain is characterized by multiple inducible autoimmune abnormalities including experimental allergic encephalomyelitis, which is considered to be a model for human multiple sclerosis [14].
It is proposed to use “reactive immunization” methods for induction of catalytic response in model mice with autoimmune disorder [15]. This method based on the induction of antibody that forms a covalent bond with hapten molecule in the contrast to “traditional” antibodies. This binding results from chemical reactions between antibody and chemically active hapten. Selection of the active hapten determines the kind of reaction occurring between it and antibody, and therefore the type of the reaction catalyzed by the antibody. The possibility of such a mechanism of immune response was shown by obtaining abzyme with aldolase activity [16]. The activity of produced antibodies was comparable with activity of natural enzymes [17].
Chemically active haptens used for reaction immunization are synthesized on the basis of mechanism-based enzyme inhibitors [18, 19]. These inhibitors posses the ability to interact with the enzyme active site due to the catalytic function encoded by the enzyme, but do not pass through the complete path of catalyzed conversion. The main property of mechanism-based inhibitors is the mimicry of some stable (particularly due to a formation of covalent bond between inhibitor and enzyme) analog of the reaction transition state. This mimicry may be either initially caused by the structure of the inhibitor or formed during the reaction. This property points out the mechanism-based inhibitors from the class of suicidal inhibitors in general. The latter are able to inhibit covalently the enzyme activity in different ways that are not related with the catalytic mechanism, but mechanism-based inhibitors form a covalent bond with just the reactive catalytic residue in the active site of the enzyme while mimicking the mentioned above transition state of the reaction. Among all the known proteases only serine and cysteine proteases have a stage of formation of covalent enzyme-substrate complex during the catalytic reaction. After nucleophilic Ser (or Cys) attacks to carbonyl atom the C-terminal part of the cleaved protein leaves the active site, and as a result acyl protein containing the ester bond between Ser (or Cys) and N-terminus of the cleaved protein forms. At this stage of acyl protein formation the mechanism-based covalent proteinase inhibitors function. There are many such inhibitors of cysteine and particularly of serine proteinases that can take part in the first stage of the enzyme-substrate reaction and form a stable covalent complex with enzyme. The oldest and best known inhibitors are those based on phosphonates. Another type of inhibitors consists of different peptidyl aldehydes including some natural ones. The mechanism-based inhibitors might appear to be very effective potential haptens for proteolytic antibody obtained by the reactive immunization technique.
During induction of catalytic antibodies with known mechanism of action by reactive immunization, the most important point for hapten selection will be the presence of covalent interaction between enzyme and substrate at the stage of transformation as just this stage will be imitated in hapten binding. There is a principal possibility of covalent catalysis by abzyme and there are many examples of such catalysis.
First, covalent intermediate formation was shown for esterolytic antibody 17E8 [20, 21]. Second, our own investigation of antiidiotypic esterase antibody 9A8 has unambiguously demonstrated the formation of such complex with inhibitor. The analysis of antibody active center has shown that the mechanism of charge transfer is the same as the catalytic mechanism of serine hydrolases which class both proteinases and esterases belong to. Therefore, different phosphonate inhibitors of serine proteinases covalently modify the Ser residue of the 9A8 active site. At the same time, the modification of abzyme by mechanism-dependent inhibitor was demonstrated in the latter case by a direct method, mass-spectrometric analysis of the modified part of the antibody. Moreover, the second amino acid of the abzyme active center (His35 of heavy chain) presents as a catalytic residue in many abzymes [22]. These facts show that covalent catalytic apparatus of serine hydrolases can be reproduced in the case of abzymes and, furthermore, it can be induced in different ways: both by classic approach in the frame of analogs of transition states and by antiidiotypic methods as in the case of 9A8. Therefore, the possibility of induction of serine or homologous cysteine proteinase using the reactive immunization method is rather high. However, the significant restriction is that proteolysis in contrast to esterolysis is an energetically unfavorable reaction. The active center of serine proteases is much more complicated compared to the esterases, and an immediate interaction with the cleaved bond and the multiple point interaction with N-terminal part of the protein are required [23]. This is necessary to delocalize the energy of the cleaved bond along the maximum part of the enzyme-substrate complex and, therefore, for reducing the energy barrier of the reaction. Besides, the N-terminal part determines the protease specificity, and the latter can be changed by mutating the region that binds this part [24].
Thus one can conclude that to obtain the specific proteolytic abzyme by the reactive immunization method it is necessary that the hapten has a structure consisting of two parts: the “head” is a mechanism-based proteinase inhibitor, and the “tail” is the peptide chain corresponding to the length of the region interacting in the active site of the protein [23, 25]. It can be noted that highly efficient natural covalent inhibitors like leupeptin and its analogs have such a structure [26].
On the basis of these theses it is obvious that the hapten structure for gp120 cleavage in a specific and accessible for proteolysis region must be comprised of the sequence of the corresponding peptide with C-terminal amino acid modified by the mechanism-based inhibitor group. This hapten can be immobilized by means of the N-terminal amino acid on a carrier protein in many hundreds of copies for immunization. An important property of the selected hapten is that it has relatively poor unspecific reactivity. Otherwise the hapten will be toxic for the organism, making it useless for immunization. Finally, the hapten has to be stable in aqueous solutions.
Epitope-specific catalytic response during the development of inducible autoimmune disorder. As mentioned above, the inbred mouse strain SJL is characterized by the development of inducible autoimmune abnormality, namely experimental autoimmune encephalomyelitis during immune system activation by fragments of myelin basic protein (MBP). The experimental autoimmune encephalomyelitis (EAE) is a CD4+-mediated autoimmune disease [27]. This disorder is characterized by perivascular CD4+ T-lymphocyte and monocyte activation followed by primary demyelinization of axonal filaments of the central nervous system resulting in the progressive paralysis of extremities. As for mouse strain SJL (H-2S haplotype), it was shown that the disease is chronic with cycling periods of aggravation and remission. This fact permits the use of the described model to obtain the immune response during the extended and stable autoimmune disease. EAE can be induced by the immunization of myelin proteolipid protein (PLP), myelin basic protein, and peptides corresponding to immunodominant epitopes of these proteins (MBP84-104, PLP139-151, PLP178-191) in the SJL mice. It is reasonable to suggest that the autoimmune process can be induced by fusion proteins containing disease inducing peptides and epitopes for catalytic response. The method of immunization by fusion antigens is developed on a novel basis and leads to the creation of antibodies specific to some earlier unknown epitopes of the selected antigen. As far as gp120 is concerned the spectrum of catalytic epitopes revealed as the result of “autoimmunization” is supposed to be wider and definitely different in terms of composition from the usual spectrum of immunodominant epitopes in human and mouse. For our study the 89-104 peptide of myelin basic protein [28, 29] was selected and expressed as a part of different fusion proteins in E. coli.
Antibodies of “internal image” as a reflection of the active site of proteinases. An alternative method for obtaining catalytic antibody is based on the ability of the immune system to create the antibody of an internal image and on the theory of idiotypic network. This theory was formulated by N. Jerne [30] and supposed for the immune system to be a network of idiotype-antiidiotype interaction. According to this conception, for each antibody Ab1 (idiotype) raised against the antigen and idiotope carrier (i1) there is its own antibody (Ab2) directed against the Ab1 in a complementary manner and carrying idiotope (i2). This scheme is repeated multiply.
Therefore, the binding center of antiidiotypic antibody (Ab2) can represent the “internal image” of the original antigen. This hypothesis resulted to the idea concerning obtaining catalytic antibody by means of reflection of the enzyme active sites. In this case the antibody obtained against the enzyme active site functions as an antigen at the second stage of immunization. In the course of the mentioned immunization obtaining the antiidiotypic antibody with features reflecting the first antigen (enzyme) is induced. This suggestion was confirmed experimentally by obtaining catalytic antiidiotypic antibodies 9A8 against acetylcholinesterase [31] and 9G4H9 against beta-lactamase [32].
The probability and precision of the reproduction of enzyme structure by antibody are not known so far. In both cases of esterolytic (9A8) and amidase (9G4H9) antibodies no similarity among amino acid sequences of the antibodies and enzymes were found. However, the antibody active sites contain catalytic residues identical to those of the corresponding enzymes [33]. The substrate specificity and interaction with inhibitors of abzymes and initial enzymes are different as well. So, the substrate specificity of esterase mAb 9A8 looks like the specificity of the corresponding enzyme--acetylcholinesterase [31]. In contrast, amidase mAb 9G4H9 exhibits a selective specificity for penicillin substrates while beta-lactamase catalyzes efficiently the hydrolysis of both penicillin and cephalosporin substrates [32].
The results discussed above provide evidence for the fact that indeed catalytic antibody thus obtained can reflect the structure of the initial antigens--enzymes--but their substrate specificity can differ.
As a model enzyme for a catalytically active image of proteinase active site subtilisin Carlsberg was selected in the present work. This subtilisin is a well characterized serine proteinase from Bacillus licheniformis that hydrolyses a wide range of the substrates with high specificity.
MATERIALS AND METHODS
Materials, chemicals, and animals. The following set of chemicals and enzymes was used in the present work: Tris(hydroxymethyl)aminomethane (Tris), ammonium persulfate, sodium borate, EDTA, reagent BCA for protein determination, hydro- and dihydrosodium phosphate, sodium chloride, sodium acetate, magnesium chloride, o-phenylenediamine, 4-chloro-1-naphthol, uridyl-2´-3´-cyclophosphate, bovine serum albumin fraction V (BSA) (Sigma, USA); acrylamide, N´,N´-methylene-bis-acrylamide, SDS, urea sequence grade, kit for plasmid DNA sequencing, membrane Hybond C extra (Amersham, USA); agar, tryptone, yeast extract (Difco, United Kingdom); restriction endonucleases, isopropyl-beta-D-thiogalactopyranoside .IPTG) (Fermentas, Lithuania); RNase A, protein ladder LMW, Protein G Sepharose Fast Flow (Pharmacia, Sweden); metallochelator sorbent Talon (Clontech, USA); T4 DNA-ligase, Taq DNA-polymerase, alkaline phosphatase, DNA-polymerase I Klenow' fragment (USB, USA); plasmids pET32b(+), pET28a(+), E. coli strains BL21(DE3) and TOPP10 (Novagen, USA); bis(sulfosuccinimidyl)suberate (BS3), Keyhole Limpet Hemocyanin (KLH) (Pierce, USA). The other chemicals were produced in Russia. SPF (specific pathogen free) inbred mice, strains MRL, SJL, and F1 hybrid mice ..B×NZW were from the vivarium of the Pushchino Branch (Moscow Region) of the Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, and originated from Harlan (UK).
Diphenyl 4-aminoalkylphosphonate synthesis. The mixture of triphenylphosphite, isobutanal, and benzylcarbamate, 0.1 mol each in 15 ml glacial acetic acid, was incubated while mixing until heat was dissipated. Then the material was incubated at 80°C for 1 h. At the end of the reaction the mixture was concentrated using a rotary evaporator. The oil-like residue was dissolved in 180 ml of methanol and crystallized at -20°C for 3 h. Then the precipitate of diphenyl 1-(N-benzyloxylcarbonyl)-aminoalkylphosphonate was collected by filtration and recrystallized by dissolution in a minimal volume of chloroform followed by addition of four volumes of methanol. The yield of the reaction was 30%. The product was dissolved in 33% solution of HBr in acetic acid and incubated for 1 h at room temperature. After rotary evaporation, hydrobromide 1-(N-benzyloxycarbonyl)-aminoalkylphosphonate was crystallized by addition of anhydrous diethyl ether. The free phosphonate was produced by passing ammonium gas through the ether suspension of hydrobromide phosphonate until a solid precipitate was formed and the suspension became transparent. The obtained ammonium bromide was isolated by filtering and diethyl ether was evaporated using a water bath under atmospheric pressure. The yield of the product was 90%. Chemical formula and the structure of the compounds synthesized were confirmed by PMR spectrometry and elemental microanalysis.
Synthesis of peptidylphosphonate LAEEEV-Phos. The blocked peptide Boc-Val-Ala-(t-Bu)Glu-(t-Bu)Glu-(t-Bu)Glu was produced by standard solid phase peptide synthesis methods. To obtain the hapten Leu-Ala-Glu-Glu-Glu-Val-Phos (LAEEEV-Phos), where Phos means the substitution of alpha-carboxylic group to diphenylphosphonate, 7 µmol of the peptide was dissolved in 0.5 ml of anhydrous acetonitrile, then 7 µmol of dry Val-Phos and 7.8 µmol of dicyclohexylcarbodiimide were added, and the mixture was incubated for 1 h. Four volumes of 20 mM potassium phosphate buffer, pH 7.0, were added to the reaction mixture, and products of the reaction were separated by reverse phase HPLC using a Waters C18 NovaPak column 150 × 3.9 mm in a gradient of acetonitrile concentrations from 20 to 80%. Aliquots of collected fractions were analyzed by MALDI-TOF, and fractions that contains the compounds with masses 1145 ([M +H]+), 1167 ([M +Na]+), or 1183 ([M +K]+) were combined and evaporated in a Speedvac vacuum centrifuge. The precipitate was dissolved in 0.1 ml 100% trifluoroacetic acid (TFA) and incubated for 1 h at room temperature to release the blocking groups. Anhydrous diethyl ether (1 ml) was added to the deblocked peptidyl phosphonate, and the precipitate was centrifuged for 10 min at 12,500 rpm, decanted, and air dried. The final yield was 20%. Molecular mass and purity of obtained peptidylphosphonate were analyzed by MALDI-TOF and RP-HPLC.
Conjugate of peptidylphosphonate with carrier protein. PBS buffer (0.3 ml) containing 3 mg of the carrier protein KLH was added by drops to 0.1 ml PBS buffer containing 1 mg bis(sulfosuccinimidyl)suberate (BS3) with intensive mixing, and the solution obtained was incubated for 1 h at 37°C. Unreacted BS3 was removed by ultrafiltration with 10 volumes of PBS using a Microcon 100. PBS (100 µl) containing 1.5 mg peptidylphosphonate was added to the obtained solution and the final mixture was incubated for 1 h at 37°C. Free succinimide groups were inactivated by adding 2 µl of 2-ethanolamine. Following removal of low molecular weight components, the product was sterilized by filtration, the protein concentration was determined by means of modified Lowry method using bichinoic acid, and the final product was used for reactive immunization.
Synthesis of peptidylphosphonate biotinylated analog. The solution of 0.3 mg of peptidylphosphonate in 50 µl of acetonitrile was mixed with 0.2 mg of dry N-hydroxysuccinimide ester of biotin and incubated for 1 h. Then 1 µl of ethanolamine was added to the reaction mixture, which was then incubated additionally for 30 min.
Construction of expression plasmids. The DNA fragment encoding MBP 89-104 peptide was synthesized by polymerase chain reaction (PCR) using two overlapping oligonucleotides. The DNA product was cloned into pET32b plasmid using NotI and XhoI restriction sites. To identify the recombinant proteins at all stages of expression, isolation and purification, pET32CHmbp plasmid containing the sequence encoding immune dominant epitope of human protein p62 (c-myc) [34] was constructed. The DNA fragments encoding different parts of gp120 (I, II, and III) were amplified by PCR using synthetic oligonucleotides followed by fusion in-frame that was performed by “splicing by overlap extension” technique. The sequence of gene HXB2-env served as initial template (clone was obtained from AIDS Research and Reference Reagent Program, Division of AIDS, NIH) [35]. The resulting I-III fragment was cloned into pBlueScript plasmid followed by transfer to the expression vectors pET32b and pET32CHmbp utilizing NcoI and BamHI restrictases. The fragments NcoI-XhoI from these plasmids were transferred to pET28a utilizing the corresponding restriction sites. All procedures were carried out using standard protocols [36].
Production of recombinant proteins. Expression of recombinant proteins was performed according to the following general scheme: competent E. coli cell strain BL21 (DE3) transformed by a plasmid DNA were seeded onto Petri dishes containing 30 µg/ml of kanamycin or 50 µg/ml of ampicillin and 2% of glucose, and incubated for 12-14 h at 30°C. 2× YT medium (1 liter) containing 30 µg/ml of kanamycin or 100 µg/ml of ampicillin and 0.1% of glucose was inoculated with colonies. The cell culture was grown at 37°C with good aeration to OD600 ~ 0.6-1.0 (not more than for 3 h), and the expression was induced with 1 mM of IPTG for 12-14 h at 30°C. The fraction of insoluble recombinant proteins (inclusion bodies) was isolated using standard protocol [37], solubilized in chromatographic buffer A (50 mM NaH2PO4/Na2HPO4, 300 mM NaCl, pH 8.0) containing 7 M urea, incubated on ice for 1 h, and centrifuged for 20 min at 20,000g. Recombinant protein was purified by metal chelate chromatography with Talon resin according to the instructions of the resin manufacturer [38]. Proteins were eluted by buffer A containing 7 M urea and 50 mM EDTA-Na and dialyzed against water at room temperature. The precipitate was solubilized with 0.02% SDS, stabilized by 5 mM solution of dithiothreitol (DTT), and stored at 4°C.
Immunization of mice. Female SJL mice, 6-8 week-old, were immunized twice with a week interval by the following doses of the recombinant protein gp120 I-III-mbp: 150 and 300 µg per mouse in complete Freund's adjuvant containing 2 mg/ml of M. tuberculosis. Emulsion was injected subcutaneously in the shaved backs of the mice distributing it over three sites in first case of immunization and in the paw pad in the second case. Additionally 400 ng of pertussis toxin was injected intraperitoneally a day before as well as after two days following the first immunization by the antigen. After 17 days a boost in PBS buffer containing 50 µg of immunogen per mouse was performed intraperitoneally. At the same time, test bleeding from eye sinus for mice from each group was carried out to monitor immune response development. The presence of the antigen-specific antibodies in the serum was analyzed by ELISA. The reactive immunization of MRL, SJL, and NZB/NZW F1 mice by peptidylphosphonate conjugated with carrier was done using a “short” scheme published earlier [5]. The test bleeding from eye sinus for three mice from each group and for control non-immunized mice of each group, as well as the analysis of immune response were performed in the same way as was described for immunization by gp120. After 21 days from the beginning of the experiment, the mice were sacrificed and their blood sera were used for further experiments.
Analysis of antigen specificity of polyclonal antibodies. Immunoenzyme analysis was performed using a standard protocol [39] with some modifications. To detect the antibody to the recombinant gp120 mouse blood serum diluted 1 : 50 and 1 : 100 was incubated in an immunological strip with preliminarily immobilized antigen. The antigen-antibody complex was revealed by means of goat antibodies against Fc fragment of mouse IgG conjugated with horseradish peroxidase (HRP). In the case of the reactive immunization mouse blood serum diluted 1 : 12 and 1 : 48 was incubated in an immunological strip with preliminarily immobilized goat antibodies against mouse IgG, then biotinylated antigen was added and the antigen-antibody complex was revealed by means of neutravidin conjugated with HRP. Biotinylated peptidylphosphonate and Val-phosphonate as well as nitrophenyl-methyl-p-biotinylphenylmethylphosphonate were applied as antigens.
Detection of proteolytic activity of antibody sample by enzymatic test. First, a calibrating curve was constructed in the range of RNase A concentrations 0.01-1 ng/µl dissolved in buffer PBS/TA. The reaction of polyC hydrolysis was performed for 5 min at 37°C, then it was stopped by adding 50% aqueous solution of trichloroacetic acid (TCA) to the final concentration 10%, and frozen in liquid nitrogen. Samples were centrifuged, diluted 10-fold with water, and the optical density of solutions was measured at 274 nm. The value of optical density of a control sample that did not contain ribonuclease was subtracted from the values obtained in the previous step.
For these experiments we used such quantities of RNase A for which the optical density of the reaction products was 60-70% on the linear part of the optical density-ribonuclease concentration dependence curve. For proteolytic activity to be measured the reaction mixture containing 0.15-1.05 µM of antibodies and necessary amount of RNase A in the same buffer was incubated at 37°C for 14 h.
Isolation of antibodies from blood serum and ascites. The isolation of antibodies from serum and ascitic fluid was performed by triple re-precipitation by an equal volume of ammonium sulfate saturated solution followed by affinity chromatography using Protein G Sepharose Fast Flow according to a standard protocol [39].
Specific labeling of polyclonal antibodies by biotinylated peptidylphosphonate. The polyclonal antibodies (10 µg), BSA (20 µg) as a negative control, and trypsin (10 µg) as a positive control were incubated for 3 h at 37°C in solution containing 0.1 µM of biotinylated peptidylphosphonate. The samples were mixed with 2× Laemmli loading buffer, heated on the water bath, and separated by polyacrylamide gel electrophoresis.
Laemmli gel electrophoresis. Electrophoretic separation of proteins under denaturing conditions was performed according to the Laemmli protocol [39]. The concentrating and separating gels contained 6 M urea.
Immunoblotting. Immunoblotting was performed as reported elsewhere [39]. BSA was added to the hybridization buffer to 0.5%. The membrane was hybridized with neutravidin conjugated with HRP. The 4-chloro-1-naphthol was used as peroxidase substrate.
Isolation of the substrates to detect antibody proteolytic activities with fluorescence test. The fluorescent substrates BSA-FITC and gp120-FITC were synthesized according to [40]. The synthesis of gp120-FITC was performed using weight ratio of protein/FITC 1 : 1.
Detection of proteolytic activity of antibody samples. To determine the proteolytic activity in antibody samples, 3 µg of BSA-FITC or gp120-FITC was incubated with 15 µg of polyclonal antibodies or 1 µg of mAb 6B8E12 in 20 mM Tris-HCl buffer, pH 7.5, containing 50 mM NaCl at 37°C during 24 and 48 h. Three parallel measurements of fluorescence intensity were made after 0, 24, and 48 h at the wavelength of emission 530 nm and excitation 480 nm. Proteolytic activity (A) was calculated as a ratio of difference of median fluorescence intensity at the time point t (Ft) and median fluorescence intensity in the initial time point (F0) to fluorescence intensity at initial point (F0) in percent:
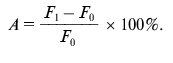
Production of antiidiotypic antibody 6B8E12. The antibodies against subtilisin were obtained by means of immunization of BALB/c mice with subtilisin Carlsberg according to a described protocol [41]. Antiidiotypic mAb 6B8E12 was isolated after immunization of BALB/c mice with antibody against subtilisin as reported previously [31].
Analysis of proteolytic activity of antibodies which are idiotype-specific. To determine the proteolytic activity of monoclonal antibodies, the chromogenic substrates succinyl-Ala-Ala-Pro-p-nitroanilide (AAPFpNa) and glutaryl-Gly-Gly-Leu-p-nitroanilide (GGLpNa) were used. The analysis was performed in immunological strips with preliminarily immobilized anti mouse IgG goat antibodies as much as 0.5 µg per sample. After the following incubation with purified antibodies (0.1 µg per sample) the chromogenic substrates AAPFpNa and GGKpNa were added to 50 and 100 µg in 0.1 M Tris-HCl, pH 8.6. The enzymatic reaction was monitored by the increase in optical density at 410 nm.
Due to the experiments described, the possibility in principal of producing catalytic monoclonal antibody with proteolytic activity was demonstrated. Undoubtedly, investigation of the catalytic mechanism and structural and functional features of the new biocatalysts is promising.
RESULTS AND DISCUSSION
Induction of catalytic response by reactive immunization. On the basis of data presented above concerning the synthesis of phosphonate derivative of a peptide followed by mouse reactive immunization, the sequence Val-Ala-Glu-Glu-Glu-Val was selected. This peptide corresponds to amino acid residues 265-270 of gp120 of HIV-1 because the proteolytic cleavage of the protein gp120 in this region leads to disrupting of interaction with CD4.
At the first stage the diphenyl-1-(N-benzyloxycarbonyl)-aminoalkylphosphonate with blocked free amino group was synthesized by reaction of condensation of triphenylphosphite, isobutanal, and benzylcarbamate. The purified intermediate was studied by elemental analysis and PMR spectroscopy to confirm the theoretically predicted elemental composition and structure. Then the blocked group was removed, and isolated diphenyl-4-aminoalkylphosphonate was analyzed and used for further study. The obtained diphenylvalylphosphonate was demonstrated by elemental analysis and PMR spectroscopy to be an exact analog of the corresponding amino acid with replaced carboxylic group to diphenylphosphonate.
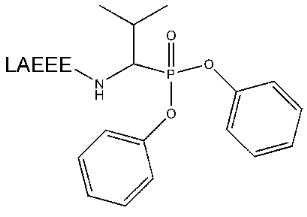
The synthesis of the peptide Boc-Val-Ala-(t-Bu)Glu-(t-Bu)Glu-(t-Bu)Glu with blocked N-terminal and side amino groups was carried out using the standard method for Fmoc-derivatives of amino acid. Blocking groups were chosen in a way for deblocking conditions not to lead to degradation of the phosphonate group. Such blocking of all of the above mentioned groups caused the phosphonate derivative to join to the C-terminus of the peptide. The further condensation of this peptide with Val phosphonate derivative was performed by activation of the free carboxylic group by dicyclohexylcarbodiimide. The products of the reaction were separated by RF-HPLC in increasing acetonitrile gradient and analyzed by MALDI-TOF. The fraction containing the substance with molecular mass 1145 [M + H]+, 1167 [M + Na]+, or 1183 [M + K]+ daltons was lyophilized and deblocked. The preliminary analysis performed demonstrated that the optimal conditions for deblocking is the use of 100% TFA. This allowed deblocking quantitatively protected groups in the absence of changes in phosphonate group detected by mass spectrometry. The peptidylphosphonate (Fig. 1) obtained in this way did not require further purification and after precipitation was used in the next stages of the work.
It is well known that small molecules are poor immunogens. For this reason, to perform an effective immunization, the synthesized reactive peptides were conjugated with KLH. The method with preliminary activation of a carrier followed by hapten attachment was chosen for N-terminal amino acid of a peptide to join to available amino groups of KLH.Fig. 1. Structure of peptidyl-Val-phosphonate.
Immunization of mouse strains MRL, SJL, and NZB/NZW F1 was performed using an earlier published scheme for obtaining catalytic antibodies to TSA (transient state analogs) of enzymatic reaction [5]. Mice demonstrating the maximal antigen-specific response in the immunological tests were selected for bleeding test after 21 days from experiment start.
The antigen specificity and catalytic activity of the polyclonal antibodies isolated from mouse blood sera were tested.
For primary comparative monitoring the specific antigen immune response in the case of all three mouse strains, some different modified ELISA technique was applied. The following compounds were used as antigens: the original biotin-labeled peptidylphosphonate, biotinylated Val-phosphonate, and nitrophenylmethyl-p-biotinyl-phenylmethylphosphonate. In the latter case a specific covalent modification of abzyme active center was reported previously. The result of comparative analysis (Table 1) was that the antibodies of mice under investigation have a high specificity to the modified antigen peptide fragment, do not react with free Val-phosphonate under the conditions applied, and bind covalently to more active and less specific modifying agent. At the same time, it is necessary to note that New Zealand hybrids produce some more antigen specific antibodies in comparison with the two other autoimmune mouse strains, but as far as the level of covalent modification is concerned antibodies from MRL mouse strain was the most effective.
Table 1. Immunoenzyme analysis of serum
mouse of SJL, MRL, and NZB/NZW F1 strains immunized with
peptidylphosphonate
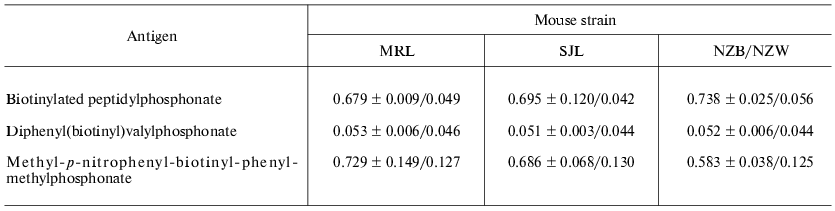
Note: The mean value of optical density and confidence interval
(p = 0.05) were shown for three immunized mice and one control
(non-immunized) mouse of corresponding strain.
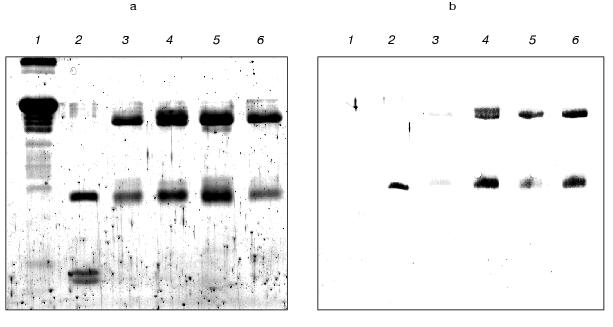
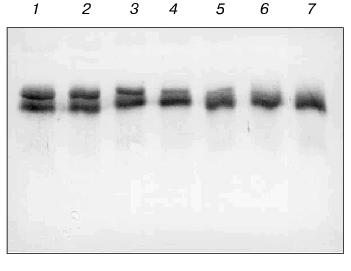
The further detailed investigation of a kind of interaction between reacting peptide and antibodies was carried out using preliminarily purified IgG preparations. The covalent membrane immobilized complex of antigen-antibody was detected by immunoblotting. The results of this experiment (Fig. 2) provide evidence for the presence of both light and heavy immunoglobulin chains, which are able to be covalently modified by the peptide Val-Ala-Glu-Glu-Glu-Val-PO(OPh)2, in the polyclonal antibodies prepared from autoimmune mice immunized by the peptide. The immunoenzyme analysis of the data concerning the reactive immunization of autoimmune animals by peptidylphosphonate showed the obvious specificity of induced antibodies to the peptide component. This fact we think demonstrates good prospects for this approach for epitope specific abzyme production.
The isolation of proteolytic antibodies against gp120 during inducible autoimmune disorder. On the basis of the literature data, the peptide VVHFFKNIVTPRTPPPS corresponding to the region 89-104 of myelin basic protein was selected to induce experimental autoimmune encephalomyelitis in SJL mouse strain [28, 29]. The DNA sequence encoding this peptide was synthesized by PCR from two overlapping oligonucleotides containing additionally stop-codon and restriction sites, and cloned into a vector for bacterial expression.Fig. 2. Laemmli gel image (a) and Western blot (b) of polyclonal antibodies modified covalently by the antigen (1 µg) and isolated from immunized SJL (4), MRL (5), and NZB/NZW F1 (6) mice. Lanes 1-3 correspond to 10 µg of BSA, 1 µg of trypsin, and 1 µg of IgG isolated from a BALB/c mouse.
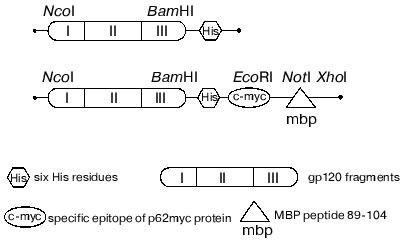
The data concerning the structure and immunogenic and functional activity of the coat glycoprotein gp120 published earlier [42-44] revealed the most perspective in terms of immunization region of the protein with relatively constant sequence. The chimeric polypeptide consisting of three gp120 fragments (named I, II, III) with deleted first, second, and third hypervariable regions was selected for further research. These fragments of gp120 were produced by PCR while gene HXB2-env served as a template. The final product of the reaction was cloned into the expression constructions produced earlier and encoded for the following fusion proteins (Fig. 3): gp120 I-III-mbp (1) that was used for immunization of SJL mice, and gp120 I-III (2) that is a substrate to investigate the specific proteolytic abzyme activities.
The procedure of expression in the bacterial system, isolation, and further purification of recombinant protein preparations by metal chelation affinity chromatography were worked out. The major part of the recombinant protein quantity was in inclusion bodies; therefore, the isolation was carried under the denaturing conditions followed by solubilization in the presence of denaturing detergent. The protein preparations were characterized by mass-spectrometry of the trypsin digest as well as by N-terminal sequencing. The determined molecular masses coincided with theoretical values within the errors of the method (~2.5%). This fact together with the results of N-terminal sequencing provided evidence that the produced protein corresponded to its theoretical sequence.Fig. 3. Schemes of recombinant protein genes expressed which contain pET28a vector.
Immunization of SJL mouse strain by the recombinant protein gp120 I-III-mbp was carried out using earlier worked out scheme of immune response induction to a foreign antigen during EAE development. After 21 days from the beginning of the experiment those mice demonstrating the maximal antigen specific response were used for blood test.
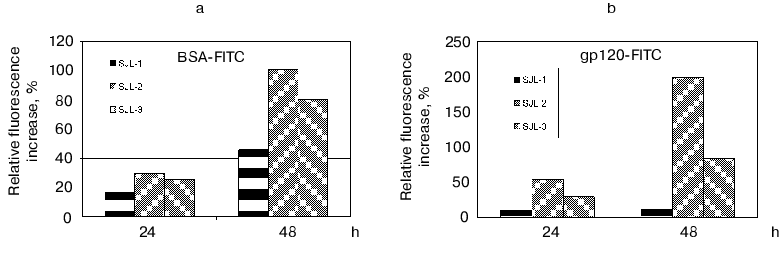
To determine the proteolytic activity, the polyclonal antibodies from mouse blood sera were purified by ammonium sulfate precipitation and subsequent affinity chromatography on a G-Sepharose column. The activity was detected using the fluorescent test [45]. The principle of the method is based on the reported phenomenon of fluorescence quenching in the protein abundantly labeled by fluorophores (this phenomenon is caused by an interaction of aromatic rings of different fluorophore molecules) and the fluorescence increases greatly after polypeptide chain breaks occur. The BSA and recombinant trx-gp120 I-III abundantly labeled by FITC (further referred as BSA-FITC and gp120-FITC, respectively) were used as the proteolysis substrates in this test. If the reaction occurred, the fluorescence increase was detected in time-dependent manner in comparison with a control. The results obtained (Fig. 4) demonstrated for both substrates, first, the increasing proteolytic activity in the samples prepared from mice immunized by gp120 I-III-mbp in comparison with control group of SJL mouse strain; second, the presumable increasing of antigen-specific proteolytic activity with regard to the general proteolytic activity: the signal increase for gp120-FITC was as much as 10-20 times more than for non-immunized mice, whereas this value for BSA-FITC was only two times more; and third, the negative correlation of proteolytic activity with immunogen dose applied for immunization. Double increase of immunogen dose resulted to insignificant decrease in unspecific (BSA-FITC) activity and significant decrease of specific (gp120-FITC) activity.
The observed proteolytic activity is well inhibited by AEBSF (100% for control mouse group and 90% for immunized mice), while phenylalanine chloromethyl ketone completely inhibits the antibody activity only in the case of samples isolated from control group of mice, and 30% of decrease is observed for immunized mice. The date show the presence of abzymes with different structure of active site in the polyclonal antibodies isolated from immunized mice.Fig. 4. Determination of proteolytic activity of antibody samples isolated from SJL mouse serum, immunized with fusion protein gp120 I-III-mbp in the different doses. SJL-1, control mice; SJL-2, mice immunized with 150 µg of antigen per mouse; SJL-3, mice immunized with 300 µg/mouse. BSA-FITC (a) and gp120-FITC (b) were used as substrates.
The preincubation of isolated antibody samples with antispecies antibodies inhibits the proteolytic activity completely, giving evidence for the abzymatic nature of this activity.
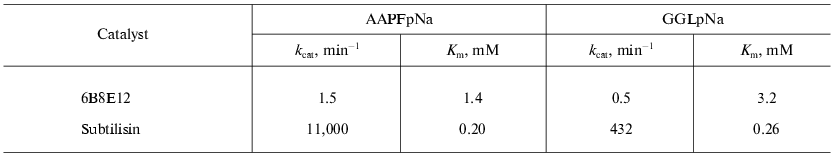
Isolation of antiidiotypic antibody with proteolytic activity. The antiidiotypic mAb 6B8E12 was revealed after immunization of mouse BALB/c strain by idiotypic mAb 5H3H4. MAb 5H3H4 obtained by immunization of mice by subtilisin was able to bind the enzyme active site and specifically inhibited its proteolytic activity [46]. In the first stage of the investigation 40 mAbs recognizing variable part of 5H3H4 idiotype were selected using the ELISA technique. At the next stage the hybridoma clones producing the antibody specific to 5H3H4 idiotype were screened to reveal the proteolytic activity. The ability to hydrolyze the chromogenic subtilisin substrate: succinyl-alanyl-alanyl-prolyl-p-nitroanilide (AAPFpNa) and glutaryl-glycyl-glycyl-leucyl-p-nitroanilide (GGLpNa) of purified antibodies isolated from the corresponding clones was tested. Among twelve positive clones the mAb 6B8E12 was selected for further experiments. The values of Michaelis constants for the abzyme and enzyme are comparable in spite of significant difference of catalytic constants (Table 2).
Table 2. The catalytic properties of
antiidiotypic mAb 6B8E12 and subtilisin
The presence of proteolytic activity of mAb 6B8E12 was confirmed by means of fluorescent tests examining the hydrolysis of the protein substrate, BSA-FITC (Fig. 5). Detected activity was inhibited quantitatively by idiotype 5H3H4, whereas the idiotypic mAb against active site of acetylcholinesterase AE-2 selected as a negative control did not affect the reaction.
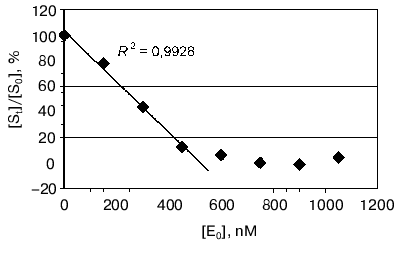
Moreover the proteolytic activity of mAb 6B8E12 was characterized by enzymatic test reported earlier [45], where ribonuclease A was used as a proteolytic substrate. MAb 6B8E12 is able to inactivate RNase A activity at molar ratio of enzyme/substrate corresponding to 50 : 1 (Fig. 6). The western blot analysis of RNase A hydrolytic products (Fig. 7) demonstrated the site-specificity of polypeptide substrate cleavage by mAb 6B8E12.Fig. 5. Determination of proteolytic activity of antibody 6B8E12 by means of fluorescence test. The concentration of mAb 6B8E12 and of antibodies from mouse strain BALB/c was 22 nM. BSA-FITC was used as substrate.
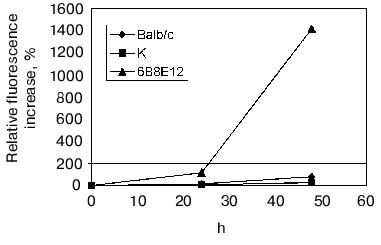
Fig. 6. Determination of proteolytic activity of antibody 6B8E12 by means of enzymatic test. The abscissa is concentration of antibody in the reaction mixture, the ordinate presents the ratio of ribonucleolytic activity of hydrolyzed RNase A to the activity of RNase A incubated in the solution containing no antibody 6B8E12. The reaction was performed for 14 h at room temperature.
This work was supported by the Russian Foundation for Basic Research (grant 99-04-49163), Copernicus grant IC15-CT96-0909, and ASGL Scientific Research Laboratories.Fig. 7. Western blot analysis of antibody 6B8E12 proteolysis of RNase A. Immunoblot of biotinylated RNase A (3.33 pmol in each lane) incubated with antibody 6B8E12 at concentrations of 2.4, 1.9, 1.4, 0.9, and 0.5 µM (lanes 1-5, respectively). Lane 6 corresponds to the biotinylated RNase A sample incubated in the same conditions in the absence of antibody 6B8E12, lane 7 to the untreated sample of biotinylated RNase A. The concentration of biotinylated RNase A was 0.7 µM in all reaction mixtures, and the hydrolysis was carried out for 15 h at room temperature.
REFERENCES
1.Tramontano, A., Janda, K. D., and Lerner, R. A.
(1986) Science, 234, 1566-1570.
2.Pollack, S. J., Jacobs, J. W., and Schultz, P. G.
(1986) Science, 234, 1570-1573.
3.Paul, S. (1994) Appl. Biochem.
Biotechnol.,47, 241-253.
4.Shuster, A. M., Gololobov, G. V., Kvashuk, O. A.,
Bogomolova, A. E., Smirnov, I. V., and Gabibov, A. G. (1992)
Science, 256, 665-667.
5.Tawfik, D. S., Chap, R., Green, B. S., Sela, M.,
and Eshhar, Z. (1995) Proc. Natl. Acad. Sci. USA, 92,
2145-2149.
6.Li, L., Paul, S., Tyutyulkova, S., Kazatchkine, M.
D., and Kaveri, S. (1995) J. Immunol., 154,
3328-3332.
7.Poignard, P., Saphire, E. O., Parren, P. W. H. I.,
and Burton, D. R. (2001) Annu. Rev. Immunol., 19,
253-274.
8.Pollard, S. R., Meier, W., Chow, P., Rosa, J. J.,
and Wiley, D. C. (1991) Proc. Natl. Acad. Sci. USA,
88, 11320-1134.
9.Ditzel, H. J., Binley, J. M., Moore, J. P.,
Sodroski, J., Sullivan, N., Sawyer, L. S., Hendry, R. M., Yang, W. P.,
Barbas III, C. F., and Burton, D. R. (1995) J.
Immunol.,154, 893-906.
10.Wyatt, R., Kwong, P. D., Desjardins, E., Sweet,
R. W., Robinson, J., Hendrickson, W. A., and Sodroski, J. G. (1998)
Nature,393, 705-711.
11.Kwong, P. D., Wyatt, R., Robinson, J., Sweet, R.
W., Sodroski, J., and Hendrickson, W. A. (1998) Nature,
393, 648-659.
12.Theofiloulos, A. N., and Dixon, F. J. (1985)
Adv. Immunol.,37, 269-390.
13.Knight, J. G., Adams, D. D., and Purves, H. D.
(1977) Clin. Exp. Immunol., 28, 352-358.
14.Bernard, C. C., and Carnegie, P. (1975) J.
Immunol.,114, 1537-1540.
15.Wirsching, P., Ashley, J. A., Lo, C. H., Janda,
K. D., and Lerner, R. A. (1995) Science, 270,
1775-1782.
16.Wagner, J., Lerner, R. A., and Barbas III, C. F.
(1995) Science, 270, 1797-1800.
17.Barbas III, C. F., Heine, A., Zhong, G.,
Hoffmann, T., Gramatikova, S., Bjornestedt, R., List, B., Anderson, J.,
Stura, E. A., Wilson, I. A., and Lerner, R. A. (1997)
Science, 278, 2085-2092.
18.Krishnan, R., Zhang, E., Hakansson, K., Arni, R.
K., Tulinsky, A., Lim-Wilby, M. S., Levy, O. E., Semple, J. E., and
Brunck, T. K. (1998) Biochemistry, 37,
12094-12103.
19.Bone, R., Sampson, N. S., Bartlett, P. A., and
Agard, D. A. (1991) Biochemistry, 30, 2263-2272.
20.Zhou, G. W., Guo, J., Huang, W., Fletterick, R.
J., and Scanlan, T. S. (1994) Science, 265,
1059-1064.
21.Guo, J., Huang, W., Zhou, G. W., Fletterick, R.
J., and Scanlan, T. S. (1995) Proc. Natl. Acad. Sci. USA,
92, 1694-1698.
22.Charbonnier, J. B., Golinelli-Pimpaneau, B.,
Gigant, B., Tawfik, D. S., Chap, R., Schindler, D. G., Kim, S. H.,
Green, B. S., Eshhar, Z., and Knossow, M. (1997) Science,
275, 1140-1142.
23.Perona, J. J., and Craik, C. S. (1997) J.
Biol. Chem., 272, 29987-29990.
24.Harris, J. L., and Craik, C. S. (1998) Curr.
Opin. Chem. Biol., 2, 127-132.
25.Stewart, J. D., and Benkovic, S. J. (1995)
Nature, 315, 388-391.
26.Schultz, R. M., Varma-Nelson, P., Ortiz, R.,
Kozlowski, K. A., Orawski, A. T., Pagast, P., and Frankfater, A. (1989)
J. Biol. Chem.,264, 1497-1507.
27.Pettinelli, C. B., and McFarlin, D. E. (1981)
J. Immunol., 127, 1420-1423.
28.Sakai, K., Zamvil, S. S., Mitchell, D. J., Lim,
M., Rothbard, J. B., and Steinman, L. (1988) J. Neuroimmunol.,
19, 21-32.
29.Tan, L. J., Kennedy, M. K., and Miller, S. D.
(1992) J. Immunol., 148, 2748-2755.
30.Jerne, N. K. (1974) Ann. Immun.,
125C, 373-378.
31.Izadyar, L., Friboulet, A., Remy, M. H., Roseto,
A., and Thomas, D. (1993) Proc. Natl. Acad. Sci. USA, 90,
8876-8880.
32.Avalle, B., Thomas, D., and Friboulet, A. (1998)
FASEB J., 12, 1055-1060.
33.Kolesnikov, A. V., Kozyr, A. V., Alexandrova, E.
S., Koralewski, F., Demin, A. V., Titov, M. I., Avalle, B., Tramontano,
A., Paul, S., Thomas, D., Gabibov, A. G., and Friboulet, A. (2000)
Proc. Natl. Acad. Sci. USA, 97, 13526-13531.
34.Evan, G. I., Lewis, G. K., Ramsay, G., and
Bishop, J. M. (1985) Mol. Cell. Biol., 5, 3610-3616.
35.Page, K. A., Landau, N. R., and Littman, D. R.
(1990) J. Virol., 64, 5270-5276.
36.Sambrook, J., Fritsch, E. F., and Maniatis, T.
(1989) Molecular Cloning. Laboratory Manual, 2nd Ed., Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, New York.
37.pET System Manual (1997) 7th Ed., Novagen
Inc.
38.TALON Metal Affinity Resin User Manual
(PT1320-1) (1997) Clontech Laboratories Inc.
39.Harlow, E., and Lane, D. (1988) Antibodies: a
Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, New York.
40.Voss, E. W., Workman, C. J., Jr., and Mummert, M.
E. (1996) Biotechniques,20, 286-291.
41.Pillet, D., Paon, M., Vorobiev, I. I., Gabibov,
A. G., Thomas, D., and Friboulet, A. (2002) J. Immunol. Meth.,
in press.
42.Hansen, J. E., Lund, O., Nielsen, J. O., Brunak,
S., and Hansen, J.-E. S. (1996) Proteins, 25, 1-11.
43.Shioda, T., Oka, S., Xin, X., Liu, H., Harukuni,
R., Kurotani, A., Fukushima, M., Hasan, M. K., Shiino, T., Takebe, Y.,
Iwamoto, A., and Nagai, Y. (1997) J. Virol., 71,
4871-4881.
44.Sullivan, N., Sun, Y., Sattentau, Q., Thali, M.,
Wu, D., Denisova, G., Gershoni, J., Robinson, J., Moore, J., and
Sodroski, J. (1998) J. Virol., 72, 4694-4703.
45.Ponomarenko, N. A., Alexandrova, E. S., Vorobiev,
I. I., Durova, O. M., Kozyr, A. V., Kolesnikov, A. V., Telegin, G. B.,
Kalinina, A. R., Suchkov, S. V., and Gabibov, A. G. (2000) Dokl.
Akad. Nauk, 375, 256-259.
46.Avalle, B., Friboulet, A., and Thomas, D. (1998)
Ann. N. Y. Acad. Sci., 864, 118-130.