Tryptophan Indole-Lyase from Proteus vulgaris: Kinetic and Spectral Properties
L. N. Zakomirdina1, V. V. Kulikova1, O. I. Gogoleva2, I. S. Dementieva1, N. G. Faleev2*, and T. V. Demidkina1*
1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, Moscow, 119991 Russia; fax: (095) 135-1405; E-mail: TVD@genome.eimb.relarn.ru2Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, Moscow, 117813 Russia; fax: (095) 135-5085; E-mail: ngfal@ineos.ac.ru
* To whom correspondence should be addressed.
Received April 17, 2002; Revision received June 6, 2002
An efficient method for purification of recombinant tryptophanase from Proteus vulgaris was developed. Catalytic properties of the enzyme in reactions with L-tryptophan and some other substrates as well as competitive inhibition by various amino acids in the reaction with S-o-nitrophenyl-L-cysteine were studied. Absorption and circular dichroism spectra of holotryptophanase and its complexes with characteristic inhibitors modeling the structure of the principal reaction intermediates were examined. Kinetic and spectral properties of two tryptophanases which markedly differ in their primary structures are compared. It was found that although the spectral properties of the holoenzymes and their complexes with amino acid inhibitors are different, the principal kinetic properties of the enzymes from Proteus vulgaris and Escherichia coli are analogous. This indicates structural similarity of their active sites.
KEY WORDS: tryptophan indole-lyase from Proteus vulgaris, pyridoxal-5´-phosphate, purification, substrates, inhibitors, spectral properties
Abbreviations: PLP) pyridoxal-5´-phosphate; PMP) pyridoxamine-5´-phosphate; DTT) dithiothreitol; LDH) lactate dehydrogenase; SOPC) S-o-nitrophenyl-L-cysteine; KIE) kinetic isotopic effect; OIA) oxindolyl-L-alanine; AS) ammonium sulfate.
Tryptophanase (tryptophan indole-lyase, EC 4.1.99.1) is a bacterial
pyridoxal-5´-phosphate-dependent enzyme present in many bacteria
[1] and catalyzing reactions of
alpha,beta-elimination (1, 2) and
beta-substitution (3) of L-tryptophan and some other natural and
synthetic amino acids:
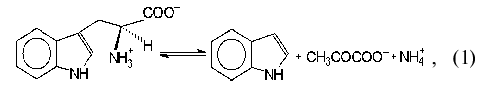
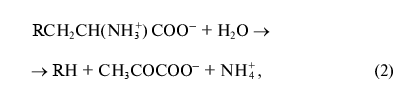
where R = OH, SH, R1S;
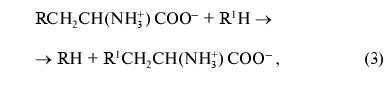
where R = OH, SH, R1S; R1 = indolyl.
This enzyme is of particular interest because of its possible use for the synthesis of L-tryptophan and its physiologically active analogs [2-4]. Recently tryptophanase was cloned from Haemophilus influenzae virulent strain and the interrelation between the ability of the strain to produce indole and cause some infectious diseases including meningitis was shown [5]. Thus, tryptophanase is convenient as a target for elaboration of efficient inhibitors which could be used for treatment of meningitis.
Newton and Snell first obtained tryptophanase from E. coli cells [6] and by now this enzyme is the best studied one [7-10]. We obtained tryptophanase crystals suitable for the X-ray structure analysis both from E. coli and Pr. vulgaris and performed preliminary X-ray structure determination [11]. Tryptophanase crystals from Pr. vulgaris appeared to be more promising for determination of the spatial structure of the enzyme as they provided higher resolution. The primary structures of the tryptophanases from these two sources markedly differ: their identity is only 52% [12]. For tyrosine phenol-lyase highly homologous to tryptophanase [13] and catalyzing chemically similar reaction, the mechanisms of action of the enzymes from various bacterial sources are known to differ significantly [14] although their structural identity is 90% [15].
The goal of the present work was to study the catalytic properties and factors controlling substrate specificity of the enzyme from Pr. vulgaris. We investigated the enzyme interaction with characteristic inhibitors and substrate analogs and found that in spite of noticeable difference in the spectral properties of holoenzymes and complexes with amino acid inhibitors, the principal catalytic properties of the enzymes from Pr. vulgaris and E. coli are analogous; this is evidently caused by structural similarity of their active sites.
MATERIALS AND METHODS
Reagents and materials. The following reagents were used in this study: lactate dehydrogenase from rabbit muscle, protein markers for SDS-PAGE, and D2O from Sigma (USA); pyridoxal-5´-phosphate (PLP), NADH, L-tryptophan, L-alanine, L-phenylalanine, S-benzyl-L-cysteine, S-methyl-L-cysteine, S-ethyl-L-cysteine, L-methionine, beta-chloro-L-alanine, beta-phenyl-DL-serine, dithiothreitol (DTT), and EDTA from Serva (USA); Lauria-Bertani medium (LB medium) from Amersham (England); tryptone and yeast extract from Difco (USA); DEAE cellulose from Whatman (England); phenyl-Sepharose from Pharmacia Biotech (Sweden). S-o-Nitrophenyl-L-cysteine (SOPC) was synthesized as described earlier [16]; oxindolyl-L-alanine (OIA) was obtained by oxidation of L-tryptophan as described in [17]. The salts used in this study were of chemically pure and extra pure grade and of Russian production.
Growth of cells. E. coli SVS 370 cells containing the tryptophanase gene from Pr. vulgaris in the pAVK2 plasmid [12] were grown with shaking (180 rpm) in LB medium for 16 h at 37°C. Then 1-methyl-DL-tryptophan was added to the medium and growth was continued for 4 h. The cells were harvested by centrifugation and stored at -80°C. On the average, from 1 liter of medium 6 g of cells was obtained.
Protein determination. During purification, the protein concentration was determined by the Lowry method [18]. The concentration of purified preparations was determined spectrophotometrically, measuring absorbance at 278 nm and taking A1cm1% = 9.19 [19].
Enzyme assay. Tryptophanase activity was determined using SOPC as the substrate by the optical density decrease at 370 nm using the molar extinction coefficient 1860 M-1*cm-1 [20]. The measurements were performed in 0.1 M potassium phosphate buffer, pH 7.8, in the presence of 0.1 mM PLP. The amount of enzyme catalyzing the conversion of 1 µmol SOPC per 1 min was taken as the activity unit of tryptophanase. The specific activity was expressed as U/mg protein.
Steady-state kinetic constants. Kinetic parameters of beta-elimination were determined at 30°C by the coupled assay with lactate dehydrogenase (LDH) by decrease in the optical density of NADH at 340 nm using the molar extinction coefficient 6220 M-1*cm-1. The reaction mixtures contained 0.1 M potassium phosphate buffer, pH 7.8, 0.1 mM PLP, 0.2 mM NADH, 8 U LDH, and varying amounts of the substrates.
Calculations were performed by the Michaelis-Menten equation using the Enzfitter program.
The inhibitory effect of amino acids on beta-elimination of L-tryptophan and SOPC was studied under the conditions described above. The data were analyzed in Dixon coordinates [21].
The reaction rate of side transamination of L-alanine was determined by decrease in peak intensity of the quinonoid intermediate (lambdamax = 501 nm), processing the data by the Michaelis-Menten equation using the Enzfitter program.
The reaction rate of side transamination of beta-chloro-L-alanine was determined by the rate of enzyme inactivation during the reaction.
Absorption spectra were recorded using a Cary 50 spectrophotometer from Varian and the circular dichroism spectra using a Jobin Ivon Mark III dichrograph (France).
Enzyme purification. The cells were suspended in five volumes (w/v) of 0.1 M potassium phosphate buffer, pH 7.0, containing 1 mM EDTA and 0.1 mM PLP and sonicated using a UZDN-A ultrasonic sonifier (Russia) at the maximal power for 8-10 min with cooling. The cell debris was removed by centrifugation (20,000g, 30 min). Nucleic acids were isolated from the supernatant by the protamine sulfate treatment (0.14 mg/mg protein), and the precipitate was removed. Ammonium sulfate (AS) was added to the supernatant to 45% saturation. The precipitate was removed by centrifugation and the supernatant was then brought to 65% saturation with AS. The pellet was dissolved in 0.05 M potassium phosphate buffer, pH 7.0, containing 1 mM EDTA, 5 mM DTT, and 0.1 mM PLP so that the saturation of protein solution with AS was 30%. Then the solution was |applied on a 50-ml phenyl-Sepharose column equilibrated with the same buffer. The enzyme was eluted with a linear gradient of AS in 0.05 M potassium phosphate buffer, AS saturation being changed from 30 to 0%. The active fractions eluted from 15 to 8% AS saturation were pooled and precipitated by AS (75% AS saturation. The precipitate was collected by centrifugation and dissolved in 0.05 M potassium phosphate buffer, pH 7.0, containing 1 mM EDTA, 5 mM DTT, and 0.1 mM PLP, dialyzed against the same buffer and applied on a 20-ml DEAE-cellulose column equilibrated with 0.05 M potassium phosphate buffer, pH 7.0, containing 1 mM EDTA, 5 mM DTT, and 0.1 mM PLP. The enzyme was eluted by a linear gradient of potassium phosphate buffer, pH 7.0 (from 0.05 to 0.4 M, total volume 120 ml). The enzyme was eluted in the range from 0.14 to 0.2 M potassium phosphate buffer. The active fractions were pooled, precipitated with AS to 75% saturation, and stored at -20°C. In both cases chromatography was performed at room temperature. The purity of the enzyme was tested by polyacrylamide gel electrophoresis according to Laemmli [22].
Apoenzyme preparation. To 1 ml of the enzyme solution (1 mg/ml) in 0.1 M potassium phosphate buffer, pH 7.0, containing 1 mM EDTA, 0.2 mM DTT, and 0.8 M AS, penicillamine was added to the final concentration 20 mM; this solution was incubated for 30 min at 25°C and then dialyzed against the same buffer at 4°C. The residual activity of apoenzyme was 1% of the holoenzyme activity.
Determination of pyridoxal-5´-phosphate concentration. PLP concentration in preparations was determined in 0.1 M NaOH according to Peterson and Sober [23] taking the molar extinction coefficient of PLP at 390 nm equal to 6600 M-1*cm-1.
Dissociation constant of pyridoxal-5´-phosphate. The PLP dissociation constant was determined by spectrophotometric titration. To the apoenzyme solution (0.7 mg/ml) in 0.1 M potassium phosphate buffer, pH 7.8, containing 1 mM DTT, aliquots of 0.65 mM coenzyme solution were added. The initial PLP concentration was 3.2 µM, the final one 80 µM. After 40 min incubation the changes in optical density in the CD spectrum at 420 nm were recorded. The results were fitted to the Scatchard type plot [24].
Identification of products of side transamination reaction of L-alanine. To determine pyridoxamine-5´-phosphate (PMP) and pyruvate, a sample containing 1.1 mg/ml holoenzyme and 300 mM L-alanine was incubated for 24 h at room temperature. Then an aliquot was taken and pyruvate was determined by reaction with LDH. Protein was precipitated with 10% TCA and isolated by centrifugation. TCA was removed from the supernatant by multiple extraction with ethyl ether, and the aqueous layer was evaporated. The pellet was dissolved in water and chromatographed on Whatman 3MM paper in the system butanol-acetic acid-water (4 : 1 : 5 v/v). A fluorescent spot with mobility equal to that of PMP was thus revealed. After staining with 0.2% ninhydrin the spot became orange, which is typical of PMP.
Isotope exchange rate. The rate of isotope exchange of alpha-proton of L-phenylalanine by the action of tryptophanase in D2O was determined as described in [25].
RESULTS AND DISCUSSION
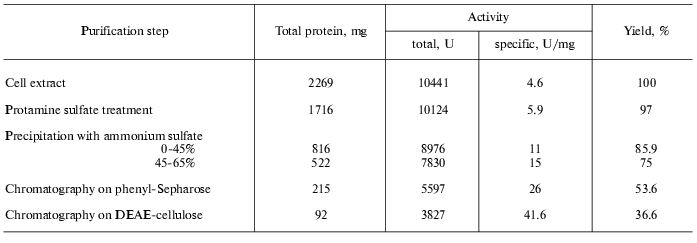
To purify tryptophanase from Pr. vulgaris, in this study we used the purification procedure presented in Table 1. It produced electrophoretically homogeneous enzyme (Fig. 1) with high yield and high specific activity (40-45 U/mg protein).
Table 1. Purification of tryptophanase from
15 g of Proteus vulgaris cell mass
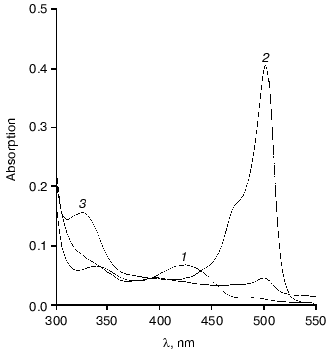
Affinity to cofactor and spectral properties of the holoenzyme. The dissociation constant of PLP determined by spectrophotometric titration of apoenzyme by coenzyme for tryptophanase from E. coli is 4.01*10-7 M [20]. However, affinity to cofactor determined from dependence of the reaction rate with SOPC on the PLP concentration is somewhat lower--2.0*10-6 M [26]. In this study we determined affinity of tryptophanase from Pr. vulgaris to coenzyme, obtaining almost equal values by both methods: 2.1*10-6 M by titration and 1.56*10-6 M by enzyme activation with PLP addition. Thus, enzymes from various sources have comparable affinities to coenzyme. This agrees with the X-ray structural data [27]; according to these data, PLP binding to the active sites occurs analogously with participation of the same functional groups. Absorption and CD spectra of tryptophanase from Pr. vulgaris and some its enzyme-inhibitor complexes are presented in Fig. 2. Analogously to tryptophanase from E. coli, the holoenzyme absorption spectrum has two characteristic bands with the maxima at 340 and 422 nm; these bands have positive dichroism and correspond to enolimine and ketoenamine tautomeric forms:Fig. 1. Electrophoresis of tryptophanase from Pr. vulgaris in 10% polyacrylamide gel according to Laemmli: 1) tryptophanase 5 µg; 2) molecular mass markers.
Fig. 2. Absorption (a) and circular dichroism (b) spectra of tryptophanase from Pr. vulgaris and its enzyme-inhibitor complexes: 1) holoenzyme (1.3 mg/ml) in 0.1 M potassium phosphate buffer, pH 7.8; 2) complex with L-alanine (280 mM); 3) complex with oxindolyl-L-alanine (0.2 mM); 4) complex with beta-phenyl-DL-serine (90 mM).
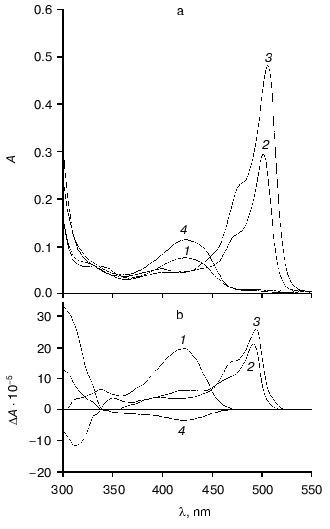
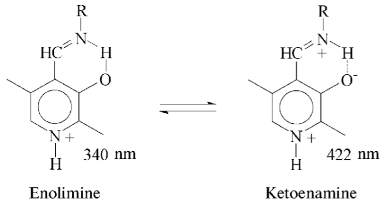
However, unlike tryptophanase from E. coli, for which enolamine is a dominant tautomer of internal aldimine at pH 7.8 [9], ketoenamine absorbing at 422 nm is a dominant tautomer in the spectrum of the enzyme from Pr. vulgaris. It should be noted that for tyrosine phenol-lyase (EC 4.1.99.2) from Citrobacter intermedius with the primary and spatial structures and the active site residues highly homologous to those of tryptophanase from Pr. vulgaris [27], ketoenamine is also a dominant tautomeric form of holoenzyme. These data suggest that for tryptophanase from Pr. vulgaris as well as for tyrosine phenol-lyase, the microenvironment of the active site in the dominant conformation is less hydrophobic [28, 29] than for tryptophanase from E. coli.
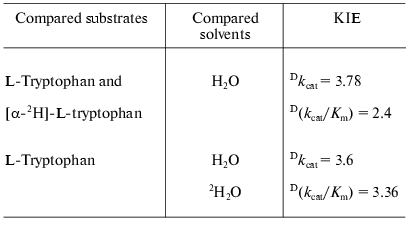
Catalytic properties. Catalytic parameters of decomposition of the natural substrate, L-tryptophan, by the enzyme from Pr. vulgaris (Table 2) are almost the same as those for the enzyme from E. coli [30]. The values of kinetic isotope effects arising from replacement of alpha-hydrogen atom of the substrate by deuterium and from transfer from H2O to D2O are presented in Table 3. These values are close to analogous isotope effects for tryptophanase from E. coli; thus, the ratios of the rates of the main elementary steps sensitive to the various isotope effects are probably very similar for the enzymes from various sources.
Table 2. Steady-state kinetic parameters of
tryptophanase from Pr. vulgaris
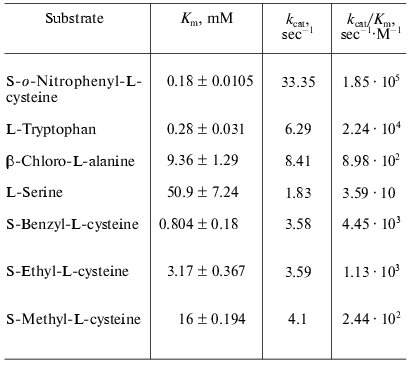
Table 3. Kinetic isotope effects on the
reaction of tryptophanase from Pr. vulgaris withthe natural
substrate
We also determined kinetic parameters in reactions of the enzyme from Pr. vulgaris with L-serine, some S-alkyl-L-cysteines, and substrates bearing a good leaving group in the beta-position: S-o-nitrophenyl-L-cysteine and beta-chloro-L-alanine (Table 2). The difference in the Km values for the enzyme from various sources in the reaction with S-alkyl-L-cysteines is notable. The affinity of tryptophanase from Pr. vulgaris to S-ethyl-L-cysteine and S-benzyl-L-cysteine is ~5-10 times lower than affinity of tryptophanase from E. coli (Km = 0.065 mM) [19], whereas affinity of tryptophanase from Pr. vulgaris to S-methyl-L-cysteine is very close to affinity of tryptophanase from E. coli (Km = 20 mM) [19]. The decomposition rates of S-alkyl-L- cysteines by tryptophanase from Pr. vulgaris appeared to be lower than the corresponding kcat values of the enzyme from E. coli.
The ability of tryptophanase from Pr. vulgaris to catalyze stereospecific exchange of the amino acid alpha-hydrogen for deuterium in D2O was studied using the reaction with L-phenylalanine as an example. The value of kcat was found to be 0.68 sec-1, this being 10% of the main reaction rate. For the enzyme from E. coli the rate of isotope exchange of L-phenylalanine is significantly higher: 60% of the main reaction rate [31].
Spectral properties of the enzyme-inhibitor complexes. Earlier we studied the absorption and CD spectra of the complexes of tryptophanase from E. coli with some amino acids [10] and found that for L-alanine and oxindolyl-L-alanine (OIA), the reaction is terminated at the stage of quinonoid intermediate and for beta-phenyl-DL-serine at the stage of external aldimine, since formation of noticeable amounts of the quinonoid structure is not observed. The absorption and CD spectra of the complexes of tryptophanase from Pr. vulgaris with L-alanine and OIA have the same absorption and circular dichroism band typical of the quinonoid intermediate with lambdamax in the region 495-502 nm, and formation of the external aldimine with beta-phenyl-DL-serine is also accompanied by the sign change of circular dichroism of the ketoenamine absorption band (Fig. 2). It should be noted that anisotropic factors of two absorption bands, the internal ketoenamine with lambdamax = 422 nm (DeltaA/A = 25.65*10-4) and quinonoid intermediate with OIA (DeltaA/A = 5.29*10-4) appeared to be approximately twice as large as the corresponding values for the enzyme from E. coli [10]. This suggests that for tryptophanases from various sources, significant difference in microenvironment of the active site takes place not only at the stage of holoenzyme but also at subsequent stages of the internal aldimine and quinonoid intermediate.
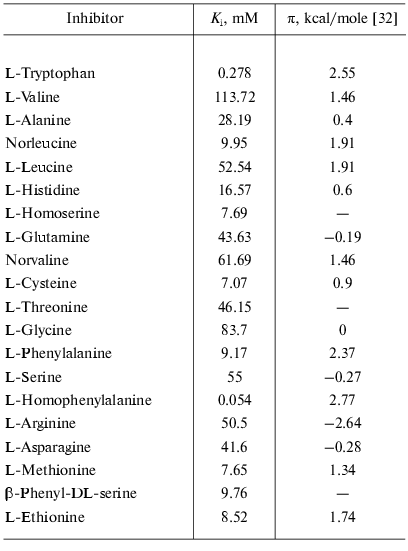
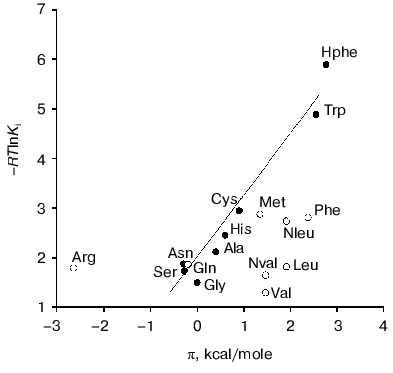
Factors governing efficient binding of inhibitors in the active site. For tryptophanase from E. coli, the relation between the structural features of amino acid inhibitors and effective inhibition constant was quantitatively analyzed using correlation methods [31]. For some amino acids considered as in optimal manner corresponding to the active site structure, a good linear correlation between -RTlnKi and hydrophobicity of the side chain (pi) was observed [32]. The negative deviations from this dependence were interpreted as being a consequence of steric interactions and positive deviations as evidence for favorable specific interactions. In this study, we applied an analogous approach for analysis of the Ki values characterizing binding of amino acid inhibitors to the enzyme from Pr. vulgaris (Table 4). The dependence of the Ki values on hydrophobicity presented in Fig. 3 is qualitatively analogous to that for the enzyme from E. coli. For some amino acids of those optimally fit to the active site of the enzyme from E. coli, -RTlnKi values correlate well (r = 0.98) with hydrophobicity of the side groups, and the slope of the line is close to unity. For amino acids with the branched side chain, negative deviations from linearity are observed and in some cases the values of these deviations differ markedly from those observed for the enzyme from E. coli; this is probably related with certain differences in the steric parameters of various tryptophanases. For both enzymes, affinity to phenylalanine is significantly lower than could be expected accounting for its hydrophobicity. This fact can be rationalized by impossibility of the “proper” orientation of the phenyl ring (nearly parallel to the cofactor plane). For arginine having a positive charge in the side chain, a positive deviation (3.1 kcal/mol) from linearity is observed; for the enzyme from E. coli, analogous deviation is 3.6 kcal/mol. This deviation indicates that there exists specific interaction with the electronegative protein moiety. According to the X-ray diffraction data [27], this moiety can be Asp133 residue, which is positioned at the distance of a hydrogen bond from the nitrogen atom of the indole ring.
Table 4. Competitive inhibition constants of
SOPC decomposition
Side transamination reaction. The side transamination reaction was observed for many PLP-dependent enzymes not being aminotransferases [33, 34]. For these enzymes, transamination results from “disruption” in the enzymatic mechanism: after removing the alpha-proton of external aldimine, protonation of the C4´ atom occurs instead of a normal catalytic act or reversion of the proton to its original position. Typical features of this reaction are inactivation of the enzyme on incubation with the substrates or inhibitors, intensity decrease in the absorption band of quinonoid intermediate with time, and appearance of an absorption band in the range 325-330 nm due to formation of the reaction products, pyridoxamine phosphate and keto acid, absorbing in this wavelength range. Addition of PLP results in restoration of the enzyme activity to the initial level.Fig. 3. Dependence of binding efficiency of L-amino acids with tryptophanase on hydrophobicity of their side chain.
On interaction of tryptophanase from Pr. vulgaris with L-alanine, the quinonoid peak with lambdamax = 501 nm was gradually decreased (Fig. 4). After 23 h incubation at 25°C the quinonoid peak intensity was ~10% of its initial value, the appearance of an absorption band at 325 nm being observed, and enzyme inactivation being 92-94%. Upon incubation of tryptophanase with L-Phe and S-ethyl-L-cysteine, we observed slower inactivation of the enzyme, by 20-30% in 24 h. The activity was restored completely on addition of PLP to the reaction mixtures. After incubation of the enzyme with L-alanine for 24 h, the reaction products, PMP and pyruvate, were identified in the reaction mixture. We determined the values of kcat for the side transamination of L-alanine by tryptophanases from Pr. vulgaris and E. coli. The reaction was monitored by the rate of disappearance of the quinonoid peak. The values obtained were 7.0*10-5 and 3.8*10-5 sec-1, respectively. Very close to these is the kcat for the side transamination of beta-chloro-L-alanine by the enzyme from E. coli (9.1*10-5 sec-1). However, for the reaction with the natural substrate, L-tryptophan, the rate of side transamination for tryptophanase from E. coli is from 2.3*10-4 to 4.0*10-4 sec-1 [34]. So, on transfer from the natural substrate to the inhibitor (L-alanine) or not natural substrate (beta-chloro-L-alanine) having a structure significantly different from that of the natural substrate, the rate of “erroneous” transamination reaction not only does not increase, but it even somewhat decreases. It is noteworthy that for tyrosine phenol-lyase (EC 4.1.99.2) from Citrobacter intermedius [35], the rate of side transamination of L-alanine is more than one order of magnitude higher (kcat = 1.53*10-3 sec-1) at the comparable values of the main reaction rates [23]. This suggests that in spite of high conservatism of the spatial structure and the active site residues of tryptophanase from Pr. vulgaris and tyrosine phenol-lyase from Citrobacter intermedius, the microenvironment of the quinonoid intermediates of L-alanine in these enzymes should differ.
This work was financially supported by the Russian Foundation for Basic Research (grants No. 01-04-48636 and 01-04-48719) and the “Leading Scientific Schools” Program (grant No. 00-15-97-844).Fig. 4. Changes in absorption spectrum of the complex of tryptophanase with L-alanine on incubation for 23 h: 1) holotryptophanase, 1 mg/ml in 0.1 M potassium phosphate buffer; 2) plus 300 mM L-alanine; 3) the same after 23 h.
REFERENCES
1.Snell, E. E. (1975) Adv. Enzymol.,
42, 287-333.
2.Nakazawa, H., Enei, H., Okumura, Sh., and Yamada,
H. (1972) Agr. Biol. Chem., 36, 2523-2528.
3.Asai, Y., Shimada, M., and Soda, K. (1982)
Manufacture of L-5-Hydroxytryptophan, Mitsui Toasu Chemicals
Inc. Jpn. Kokai Tokyo Koho JP 8283,285 (Cl. Cl 12P13/04) Appl.
80/155,883 07 Nov. 1980, 6 pp. (Chem. Abst. 097 (15)
125715w).
4.Su, Ch-Sh., Chang, Z.-J., Huang, W.-Hs., and Tsai,
H. (1996) Enzymatic Production of L-Tryptophan from Indole,
Pyruvate and Ammonium Salt, Dev. Center for Biotechnology (TW).
Ger. Offen. DE 4,427,217 (Cl. C12N11/12) Appl. 4,427,217 1 Aug. 1994,
10 pp.
5.Martin, K., Morlin, G., Smith, A., Nordyke, A.,
Eisenstark, A., and Golomb, M. (1988) J. Bacteriol., 180,
107-118.
6.Newton, W. A., and Snell, E. E. (1964) Proc.
Natl. Acad. Sci. USA, 51, 382-389.
7.Watanabe, T., and Snell, E. E. (1977) J.
Biochem., 82, 733-745.
8.Phillips, R. S., Miles, E. W., and Cohen, L. A.
(1984) Biochemistry, 23, 6228-6234.
9.Metzler, C. M., Viswanath, R., and Metzler, D. E.
(1991) J. Biol. Chem., 266, 9374-9381.
10.Zakomirdina, L. N., Sakharova, I. S., and
Torchinskii, Yu. M. (1988) Mol. Biol. (Moscow), 22,
187-194.
11.Dementieva, I. S., Zakomirdina, L. N., Sinitzina,
N. I., Antson, A. A., Wilson, K. S., Isupov, M. N., Lebedev, A. A., and
Harutyunyan, E. G. (1994) J. Mol. Biol., 235,
783-786.
12.Kamath, A. V., and Yanofsky, C. (1992) J.
Biol. Chem., 267, 19978-19985.
13.Antson, A. A., Demidkina, T. V., Gollnick, P.,
Dauter, Z., von Tersch, R. L., Long, J., Berezhnoy, S. N., Phillips, R.
S., Harutyunyan, E. G., and Wilson, K. S. (1993) Biochemistry,
32, 4195-4206.
14.Kiik, D., and Phillips, R. (1988)
Biochemistry, 27, 7333-7338.
15.Iwamori, S., Oikawa, T., Ishiwata, Ken-Ichi, and
Makiguchi, N. (1992) Biotechnol. Appl. Biochem., 16,
77-85.
16.Boyland, E., Manson, D., and Nery, R. (1962)
J. Chem. Soc.,2, 606-612.
17.Savige, W. E., and Fontana, A. (1980) Int. J.
Peptide Protein Res., 15, 285-297.
18.Lowry, O. H., Rosobrough, N. J., Farr, A. L., and
Randall, R. J. (1951) J. Biol. Chem., 193, 265-275.
19.Phillips, R. S., and Gollnick, P. D. (1989) J.
Biol. Chem., 264, 10627-10632.
20.Suelter, C. H., Wang, J., and Snell, E. E. (1976)
Analyt. Biochem., 76, 221-232.
21.Dixon, M. (1953) Biochem. J., 55,
170-171.
22.Laemmli, U. K. (1970) Nature, 227,
680-685.
23.Peterson, E. A., and Sober, H. A. (1954) J.
Am. Chem. Soc., 76, 169-175.
24.Klotz, I. M., and Hunston, D. L. (1971)
Biochemistry, 10, 3065-3069.
25.Faleev, N. G., Ruvinov, S. B., and Demidkina, T.
V. (1988) Eur. J. Biochem., 177, 395-401.
26.Newton, W. A., Morino, Y., and Snell, E. E.
(1965) J. Biol. Chem., 240, 1211-1218.
27.Isupov, M. N., Antson, A. A., Dodson, E. J.,
Dodson, G. G., Dementieva, I. S., Zakomirdina, L. N., Wilson, K. S.,
Dauter, Z., Lebedev, A. A., and Harutyunyan, E. H. (1998) J. Mol.
Biol., 276, 603-623.
28.Llor, J., and Cortijo, M. (1977) J. Chem. Soc.
Perkin Trans., 9, 1111-1113.
29.June, D. S., Suelter, S. H., and Dye, J. L.
(1981) Biochemistry, 20, 2714-2719.
30.Phillips, R. S. (1991) Biochemistry,
30, 5927-5934.
31.Faleev, N. G., Ruvinov, S. B., Zakomirdina, L.
N., Sakharova, I. S., Torchinskii, Yu. M., and Belikov, V. M. (1991)
Mol. Biol. (Moscow), 25, 752-760.
32.Frommel, C. (1984) J. Theor. Biol.,
111, 247-260.
33.Braunstein, A. E. (1973) in The Enzyme
(Boyer, P. D., ed.) 3rd ed., Vol. 9, Academic Press, New York, pp.
379-481.
34.Miles, E. W. (1985) in Transaminases
(Christen, Ph., and Metzler, D., eds.) Wiley, New York, pp.
470-481.
35.Demidkina, T. V., Myagkikh, I. V., and Azhayev,
A. V. (1987) Eur. J. Biochem., 170, 311-316.