The Regulatory alpha Subunit of Phosphorylase Kinase May Directly Participate in the Binding of Glycogen Phosphorylase
I. E. Andreeva1,2*, N. A. Rice3,4, and G. M. Carlson3
1Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, Moscow, 117071 Russia; fax: (095) 954-2732; E-mail: i_andreeva@yahoo.com2Current address: Department of Molecular Physiology and Biophysics, Baylor College of Medicine, Houston, TX 77030 USA; fax: (713) 798-3475; E-mail: andreeva@bcm.tmc.edu
3Division of Molecular Biology and Biochemistry, School of Biological Sciences, University of Missouri-Kansas City, Kansas City, Missouri 64110-2499 USA; fax: (816) 235-5595; E-mail: carlsonGM@umkc.edu
4Current address: Department of Molecular, Cellular, and Developmental Biology, University of Colorado at Boulder, 80309 USA; E-mail: ayersn@stripe.colorado.edu
* To whom correspondence should be addressed.
Received April 17, 2002; Revision received June 5, 2002
The yeast two-hybrid screen has been used to identify potential regions of interaction of the largest regulatory subunit, alpha, of phosphorylase kinase (PhK) with two fragments of its protein substrate, glycogen phosphorylase b (Phb). One fragment, corresponding to residues 17-484 (PhbN´), contained the regulatory domain of the protein, but in missing the first 16 residues was devoid of the sole phosphorylation site of Phb, Ser14; the second fragment corresponded to residues 485-843 (PhbC) and contained the catalytic domain of Phb. Truncation fragments of the alpha subunit were screened for interactions against these two substrate fragments. PhbC was not found to interact with any alpha constructs; however, PhbN´ interacted with a region of alpha (residues 864-1014) that is near the phosphorylatable region of that subunit. PhbN´ was also screened for interactions against a variety of fragments of the catalytic gamma subunit of PhK; however, no interactions were detected, even with full-length gamma. Our results support the idea that amino acid residues proximal to the convertible serine of Phb are important for its specific interaction with the catalytic subunit of PhK, but that regions distinct from the convertible serine residue of Phb and from the catalytic domain of PhK may also be involved in the interaction of these two proteins.
KEY WORDS: glycogen phosphorylase b, phosphorylase kinase, yeast two-hybrid system, protein-protein interaction
Glycogen phosphorylase b (Phb) (EC 2.4.1.1) and phosphorylase kinase (PhK) (EC 2.7.1.38) are key regulatory enzymes of glycogen catabolism. In skeletal muscle, the Ca2+-dependent phosphorylation of Phb by PhK converts it to the active Pha form, thus coupling contraction to increased glycogenolysis [1-3]. Structurally, muscle Phb is a homodimer, with each monomer having two separate domains, an N-terminal regulatory domain (residues 1-484) and a C-terminal catalytic domain (residues 485-842) [4]. PhK is much larger and more complex than its protein substrate: it is a hexadecamer of four copies of four different subunits, (alphabetagammadelta)4, with a total mass of 1.33*106 daltons [5]. The masses of the individual alpha, beta, gamma, and delta subunits are 138.4 [6], 125.2 [7], 44.7 [8], and 16.7 kD [9], respectively. The activity of the catalytic gamma subunit of PhK is allosterically controlled by its regulatory alpha, beta, and delta subunits, the last being an intrinsic molecule of calmodulin [5]. PhK is the only kinase known that can convert Phb to Pha, which it does by phosphorylating a single seryl residue (Ser14) in the N-terminal regulatory region of Phb [1].
It is not yet fully understood how PhK, Phb, and glycogen interact to initiate glycogen catabolism. Various studies have shown that the alpha subunit of PhK is important for its interaction with glycogen. Studies that examined the chemical modification of PhK by iodoacetate demonstrated that glycogen protected only the alpha subunit of the holoenzyme from carboxymethylation [10]. Glycogen was also reported to stimulate the activity of an alphagammadelta, but not gammadelta, complex of PhK [11], further suggesting a role for the alpha subunit in the stimulation of PhK by glycogen. In studies investigating the structural features contributing to PhK-Phb complex formation, Xu and Carlson [12] suggested that elements outside of the catalytic domain of PhK and outside the phosphorylatable region of Phb may both be involved in forming the PhK-Phb complex. Thus, it is possible that a regulatory subunit(s) of PhK may be involved in its interactions with both glycogen and Phb.
Previous electron and immunoelectron microscopy studies of an alphagammadelta complex and the (alphabetagammadelta)4 holoenzyme, respectively, suggested that the alpha subunit of PhK is exposed and peripherally located in the holoenzyme [13-15], consistent with its high susceptibility to proteolysis by a wide variety of proteases [16]. Based upon the probable exposed location of the alpha subunit, it is feasible to hypothesize that in the PhK-Phb enzyme-substrate complex, presumably formed on the surface of the glycogen particle, Phb is oriented in such a manner as to interact not just with the catalytic gamma subunit of PhK, but also with its much larger alpha and/or beta subunits [13-15, 17]. In this study, we have investigated potential interactions between Phb and the alpha and gamma subunits of PhK by using the yeast two-hybrid system. To identify regions of Phb outside of its regulatory phosphorylation domain that may be involved in its interaction with PhK, we have used a truncated form of Phb (Phb´) lacking its 16 N-terminal amino acids, including the single seryl residue phosphorylated by PhK, Ser14.
Eukaryotic transcription factors contain two functional domains, a DNA binding domain (BD), which binds to an upstream promoter sequence, and an activation domain (AD) that directs transcription of downstream genes. The yeast two-hybrid technology is based on the fact that a functional transcription factor can be reconstituted through noncovalent interaction of two independent hybrid proteins containing a DNA BD and AD [18]. For two proteins that associate, their coexpression in yeast as BD and AD chimeric proteins results in the creation of a novel transcription activator that directs the activation of a phenotypically detectable downstream reporter gene.
In this work, we have identified an interaction between the N-terminal half of Phb, missing its phosphorylation site (PhbN´, residues 17-484) and the regulatory alpha subunit of PhK (residues 864-1014). This is the first study to designate a specific region of Phb distinct from its phosphorylation site to associate with a region of PhK distinct from its catalytic site on the gamma subunit.
MATERIALS AND METHODS
Bacteria and yeast strains. Escherichia coli strain DH5alpha was used for all plasmid manipulations according to standard procedures [19, 20]. Yeast strain Saccharomyces cerevisiae EGY 48 (MATalpha, his3, trp1, ura3, LexAop-LEU2) (CLONTECH) was used for two-hybrid analyses [21].
DNA constructs. All constructs of PhK alpha and gamma were engineered as previously described [22, 23]. Rabbit skeletal muscle Phb cDNA was kindly provided by Dr. Donald J. Graves (Iowa State University, USA) and used as a template for the preparation of a construct expressing the regulatory domain of Phb, residues 17-484 (PhbN´), and the catalytic domain construct, residues 485-843 (PhbC). Constructs were generated by PCR using the following primers (Operon Technologies, Inc.) to yield cDNAs flanked with 5´ EcoRI and 3´ XhoI restriction sites: PhbN´ sense strand oligonucleotide (5´-TGAATTCCGCGGCCTGGCCGGC-3´) and antisense strand oligonucleotide (5´-GCTCTCGAGTTAGGTCTTGTTCTGGAACT-3´); PhbC sense strand oligonucleotide (5´-TGAATTCAACGGCATCACCCCTCG-3´) and antisense strand oligonucleotide (5´-GCTCTCGAGCTAGGGTATCTTCTCGTCG-3´). PhbN´ and PhbC constructs were cloned into both pLexA and pB42 AD two-hybrid vectors using the EcoRI and XhoI restriction sites. The correct sequences of LexA and pB42 Phb fusion proteins were verified by dideoxy sequencing and restriction mapping.
Two-hybrid screening. To test for interactions between Phb and either PhK alpha or gamma, series of C-terminal deletion mutants of PhK alpha and gamma were assayed against PhbN´ (alpha and gamma) and PhbC (alpha only) in all possible binary combinations. Briefly, yeast strain EGY48, possessing the pSH18-34 lacZ reporter plasmid, was cotransformed with both fusion plasmids by a modified lithium acetate procedure [24]. Cotransformants were grown at 30°C for 3 days on synthetic complete medium containing 2% glucose, but lacking histidine, tryptophan, and uracil (SD-His-Trp-Ura). Positive interactions between Phb and PhK alpha or gamma constructs were identified by the transcriptional activation of the LEU2 gene by growth on defined media lacking leucine (-Leu) and by activation of the lacZ reporter gene. Primary single colonies were restreaked onto plates with synthetic complete medium containing 2% galactose, 1% raffinose, but lacking histidine, tryptophan, uracil, and leucine (SD Gal/Raf-His-Trp-Ura-Leu), in the presence of 80 µg/ml 5-bromo-4-chloro-3-indolyl-beta-D-galactopyranoside (X-gal) to screen for galactose-dependent lacZ expression. beta-Galactosidase activity of all positive interactions, as determined by the aforementioned in vivo plate assays, was subsequently quantified by liquid o-nitrophenyl-beta-D-galactopyranoside (ONPG) spectroscopic assays [25]. Activity is expressed as Miller units according the following formula: A = OD420·1000/V(ml)·tau(sec)·OD600 [22].
RESULTS
PhbN´ does not interact with PhK gamma in the two-hybrid system. To identify potential contacts between the gamma subunit of PhK and regions outside the phosphorylation site of Phb, we tested a series of full-length and truncated gamma constructs (Fig. 1) against the N-terminus of Phb lacking the first 16 residues (PhbN´) in the yeast two-hybrid system as both DNA BD and AD fusion proteins (Table 1). All truncated mutants were designed based upon the known crystal structure of the gamma subunit in an attempt to avoid possible disruption of secondary structural elements [26]. Full length alpha and gamma constructs of PhK (alphaFL and gammaFL, respectively) were used in the assays as positive interaction controls because they have previously been shown to strongly interact in the two-hybrid system [23]. When all constructs were tested in every possible binary combination, no positive interactions were observed between any gamma construct and PhbN´ regardless of the transcriptional domain (Table 1), suggesting that the N-terminal phosphorylatable region of Phb, containing Ser14,is absolutely essential in the interaction of the regulatory domain of Phb with the gamma subunit of PhK.
Table 1. Domain mapping of phosphorylase b N´ domain interactions with phosphorylase kinase catalytic gamma subunitFig. 1. Deletion mutants of phosphorylase kinase catalytic gamma subunit. Constructs were engineered as both transcriptional DNA BD LexA or AD pB42 fusion proteins as described under “Materials and Methods”. Amino acid numbers are given above the first construction. C-Terminal constructs are named as subunit followed by the last C-terminal residue expressed beginning from residue 1, e.g., gamma65C is a fusion protein of amino acids 1-65 of the gamma subunit. The last two N-terminal constructions are named by the first N-terminal residue expressed ending with the final residue of gamma (position 386). Sequence domains are represented by the various patterns as described in [23]: checkered, unique N-terminal region; gray, hinge region; hatched, C-terminal regulatory domain; and black, calmodulin binding domains.
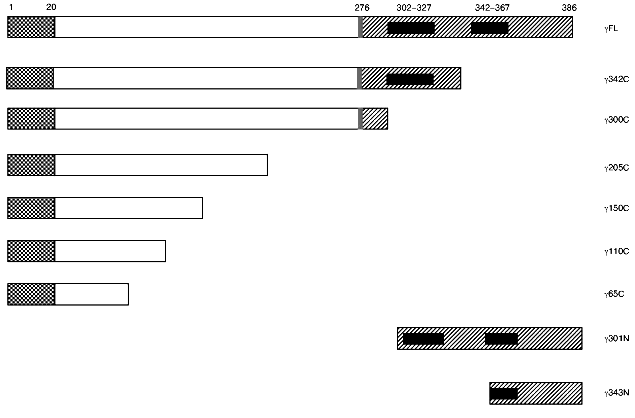
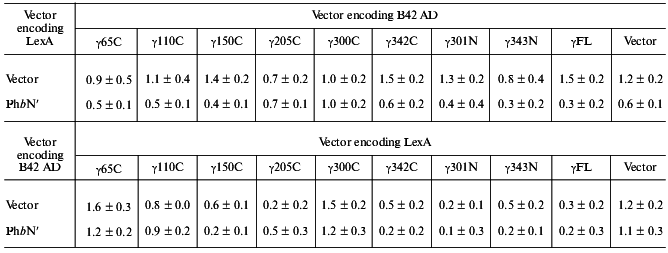
Note: beta-Galactosidase activity from yeast lysates was determined using ONPG as substrate. Activity was expressed as Miller units according to the following formula: A = OD420*1000/V(ml)*tau(sec)*OD600 [22]. Data represent the mean ± S. E. of three assays performed in duplicate. A positive interaction is defined as >2.0 Miller units. Interactions between PhK alphaFL and gammaFL, as described [23], were used as positive controls in the assay, and were 78.0 ± 10.0 for LexA alphaFL versus B42 AD gammaFL and 23.4 ± 5.0 for B42 AD alphaFL versus LexA gammaFL.
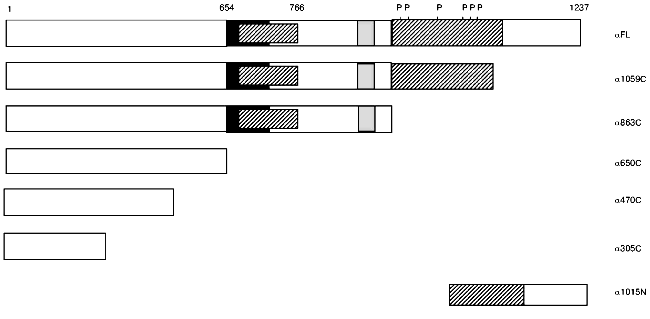
The regulatory domain of Phb interacts with the regulatory alpha subunit of PhK in the two-hybrid system. To define regions of potential interaction between Phb and the alpha subunit of PhK, we used, in addition to full length PhK alpha (residues 1-1237), six additional constructs of alpha encoding various deletion mutants fused to both the LexA DNA BD and B42 AD sequences [22, 23]. Interactions between PhK alphaFL and gammaFL, as described above, were used as positive controls in the assay, and the various alpha constructs were tested against both the regulatory (PhbN´, residues 17-484) and catalytic (PhbC, residues 485-842) domains of Phb. Of the five C-terminal and one N-terminal deletion mutants (Fig. 2) assayed, no significant positive interactions were observed between any alpha construct and PhbC (data not shown). However, PhbN´ induced beta-galactosidase activity above baseline levels when screened against both alpha1059C and alphaFL DNA BD fusion proteins, 2.6 and 3.3 Miller units, respectively. When PhbN´ was coexpressed with alpha1059C and alphaFL as the reciprocal AD chimeric proteins, no interactions were observed. While the expression of all of the alpha and gamma fusion proteins has been confirmed by immunoblot analysis [22, 23], the observed difference in transcriptional activation between the same pair of constructs, PbN´-alpha1059C and PhbN´-alphaFL, expressed in opposite domains is probably due to dissimilar levels of nuclear transport or protein expression [21]. Such “domain effects” have been observed by others and are a common limitation of two-hybrid analyses [21]. Our results suggest that the domain of alpha that interacts with PhbN´ lies between residues 864-1237. To further define the interacting domain of alpha, an N-terminal deletion mutant containing only its extreme C-terminus, alpha1015N (Fig. 2), was assayed for its interaction with PhbN´. No detectable interaction was observed (Table 2), thus implying that it is a portion of PhKalpha between residues 864-1014 that associates with Phb.
Table 2. Domain mapping of phosphorylase b N´ domain interactions with phosphorylase kinase regulatory alpha subunitFig. 2. Deletion mutants of phosphorylase kinase regulatory alpha subunit. Constructs were engineered as both transcriptional DNA BD LexA or AD pB42 fusion proteins as described under “Materials and Methods”. Amino acid numbers are given above the first construct. C-Terminal constructs are named as subunit, followed by the last C-terminal residue expressed beginning from residue 1, e.g., alpha305C is a fusion protein of amino acids 1-305 of the alpha subunit. The last N-terminal construction is named by the first N-terminal residue expressed ending with the final residue of alpha (position 1237). Functionally significant domains are represented by various patterns as described in [22]: black, region missing in alpha subunit of phosphorylase kinase; hatched, regions missing in beta subunit; and gray, leucine zipper.
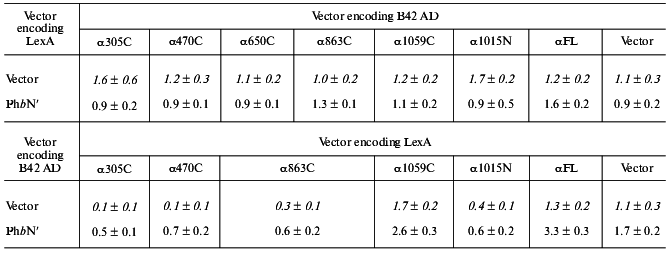
Note: beta-Galactosidase activity from yeast lysates was determined using ONPG as substrate. Activity was expressed as Miller units according to the following formula: A = OD420*1000/V(ml)*tau (sec)*OD600 [22]. Data represent the mean ± S. E. of three assays performed in duplicate. Control interactions between empty vectors and different alpha constructs (shown in italic) were taken from [27]. A positive interaction is defined as >2.0 Miller units. Interactions between PhK alphaFL and gammaFL, as described [23], were used as positive controls in the assay, and were 61 ± 10.0 for LexA alphaFL versus B42 AD gammaFL and 14.4 ± 2.0 for B42 AD alphaFL versus LexA gammaFL.
DISCUSSION
Because PhK is the only known kinase that can phosphorylate Phb, and conversely, Phb is the only known physiological substrate for PhK, it is of great interest to understand the apparently high specificity of their interaction. Kinetic studies on the phosphorylation by the PhK holoenzyme [28], or by its truncated catalytic gamma subunit [28], of relatively short synthetic peptide analogs corresponding to the region surrounding the convertible Ser14 of Phb have indicated that several residues near Ser14 greatly influence its phosphorylation. These results were largely confirmed in the context of full-length Phb by kinetic studies on the phosphorylation of site-directed mutants within its N-terminal region using the truncated gamma subunit of PhK [29]. Evidence that the N-terminal region of Phb may also significantly contribute to complex formation with the gamma subunit came from binding studies using an immunochemical approach, which showed that elimination of the N-terminal 16 residues of Phb dramatically inhibited its ability to form a complex with the truncated catalytic gamma subunit [12]. Our current results using the two-hybrid approach further support the importance of the N-terminal region of phosphorylase b for complex formation with the catalytic subunit of PhK, because not a single gamma construct, including full-length, was found to interact with PhbN´, which is missing the first 16 residues. The results from all of these different approaches incontrovertibly show that amino terminal residues of Phb, particularly among its first 16, are key contributors to its specific interaction with PhK.
Additional regions of Phb away from its amino terminus containing the convertible serine have also been implicated in contributing to its specific interaction with PhK, particularly with the holoenzyme form of the kinase. For example, the kinetic parameters (using the PhK holoenzyme) and kinase specificity for phosphorylation of a fragment of Phb containing its first 91 residues much more closely resemble those for full-length Phb than for a model peptide corresponding to residues 5-18 [30]. Moreover, elimination of the first 16 residues of Phb resulted in only a relatively small decrease (58% increase in apparent Kd) of complex formation with the PhK holoenzyme, in contrast to a decrease too large to measure with the truncated gamma subunit [12]. The sum of the results cited in this and the preceding paragraph indicate that residues proximal to the convertible Ser14 of Phb are critical in its interactions with the catalytic domain of PhK, but also suggest that regions far from that convertible serine may interact with regions of PhK far from its catalytic domain. This latter idea was the basis of the current study, namely, to determine if a regulatory subunit of PhK could interact with a form of Phb lacking its amino terminal region.
Both PhK and Phb bind directly to glycogen [31], with the binding of PhK apparently enhanced by Phb [17, 32]. Given that it is the alpha subunit of PhK that has been implicated in the binding of glycogen [10, 11], we asked whether that same large regulatory subunit might also interact with Phb. The region of the alpha subunit identified in this study as potentially interacting with Phb´ occurs in the stretch of residues from 863-1059, within the C-terminal half of alpha. The only potential contacts between alpha and Phb´ were with the N-terminal half of the latter (PhbN´). That no interactions between alpha and PhbC were observed is in agreement with the results of a previous report that the Fab fragment of an antibody against residues 727-748 of Phb, which is within the PhbC domain, did not affect the interaction between Phb and the PhK holoenzyme [12]. An additional layer of complexity in the interactions among phosphorylase b, PhK, and glycogen is introduced by the protein PTG (protein targeting to glycogen). This protein, which binds glycogen and thus targets protein phosphatase 1 to the glycogen particle, also binds Pha and PhK [33]. Although the subunit of PhK that interacts with PTG has yet to be determined, the alpha subunit is an obvious candidate to screen for this function.
This work was supported by the Russian Foundation for Basic Research (grants 00-15-97787 and 02-04-49099) and National Institutes of Health, USA (grant DK32953).
REFERENCES
1.Pickett-Gies, C. A., and Walsh, D. A. (1986) in
The Enzymes (Boyer, P. D., and Krebs, E. G., eds.) 3rd ed., Vol.
17, Academic Press, San Diego, CA, pp. 395-459.
2.Madsen, N. B. (1986) in The Enzymes (Boyer,
P. D., and Krebs, E. G., eds.) 3rd ed., Vol. 17, Academic Press, San
Diego, CA, pp. 365-394.
3.Livanova, N. B. (1993) Biochemistry
(Moscow), 58, 1234-1239.
4.Barford, D., and Johnson, L. N. (1989)
Nature, 340, 609-616.
5.Cohen, P., Burchell, A., Foulkes, J. G., Cohen, P.
T. W., Vanaman, T. C., and Nairn, A. C. (1978) FEBS Lett.,
92, 287-293.
6.Zander, N. F., Meyer, H. E., Hoffmann-Pozorske, E.,
Crabb, J. W., Heilmeyer, L. M. G., Jr., and Kilimann, M. W. (1988)
Proc. Natl. Acad. Sci. USA, 85, 2929-2933.
7.Kilimann, M. W., Zander, N. F., Kuhn, C. C., Crabb,
J. W., Meyer, H. E., and Heilmeyer, L. M. G., Jr. (1988) Proc. Natl.
Acad. Sci. USA, 85, 9381-9385.
8.Reimann, E. M., Titani, K., Ericsson, L. H., Wade,
R. D., Fisher, E. H., and Walsh, K. A. (1984) Biochemistry,
23, 4185-4192.
9.Grand, R. J. A., Shenolikar, S., and Cohen, P.
(1981) Eur. J. Biochem., 113, 359-367.
10.Sanchez, V. E. (1993) Ph. D. Thesis,
University of Tennessee, Memphis.
11.Chan, K. F., and Graves, D. J. (1982) J. Mol.
Biol., 257, 5939-5947.
12.Xu, Y.-H., and Carlson, G. M. (1999)
Biochemistry, 38, 9562-9569.
13.Hinrichsen, A., Bockl, H. J., and Schramm, H. J.
(1989) Biol. Chem. Hoppe-Seyler, 370, 125-133.
14.Wilkinson, D. A., Marion, T. N., Tillman, D. M.,
Norcum, M. T., Hainfeld, J. M., Seyer, J. M., and Carlson, G. M. (1994)
J. Mol. Biol., 235, 974-982.
15.Nadeau, O. W., Carlson, G. M., and Gogol, E. P.
(2002) Structure, 10, 23-32.
16.Trempe, M. R., and Carlson, G. M. (1987) J.
Biol. Chem., 262, 4333-4340.
17.Andreeva, I. E., Makeeva, V. F., Kurganov, B. I.,
Chebotareva, N. A., and Livanova, N. B. (1999) FEBS Lett.,
445, 173-176.
18.Fields, S., and Sternglanz, R. (1994) Trends
Genet., 10, 286-292.
19.Ausembel, F. M., Brent, R., Kingston, R. E.,
Moore, D. D., Seidman, J. G., Smith, J. A., and Struhl, K. (1996)
Current Protocols in Molecular Biology,John Wiley and Sons, Inc.,
N.Y.
20.Sambrook, J., Fritsch, E. F., and Maniatis, T.
(1989) Molecular Cloning: A Laboratory Manual,2nd ed., Cold
Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
21.Estojak, J., Brent, R., and Golemis, E. A. (1995)
Mol. Cell. Biol., 15, 5820-5829.
22.Ayers, N. A., Wilkinson, D. A., Fitzgerald, T.
J., and Carlson, G. M. (1999) J. Biol. Chem.,274,
35583-35590.
23.Rice, N. A., Nadeau, O. W., Yang, Q., and
Carlson, G. M. (2002) J. Biol. Chem.,277,
14681-14687.
24.Geitz, D., St. Jean, A., Woods, R. A., and
Shiestl, R. H. (1992) Nucleic Acids Res.,20, 1425.
25.Miller, J. H. (1972) Experiments in Molecular
Genetics, Cold Spring Harbor Laboratory Press, Cold Spring Harbor,
N.Y.
26.Owen, D. J., Noble, M. E., Garmar, E. F.,
Papageorgiou, A. C., and Johnson, L. N. (1995) Structure,
3, 467-482.
27.Ayers, N. A. (1999) Ph. D. Thesis,
University of Tennessee, Memphis.
28.Graves, D., Bartlesson, C., Biorn, A., and Pete,
M. (1999) Pharmacol. Ther., 82, 143-155.
29.Biorn, A. C., Bartleson, C., and Graves, D. J.
(2000) Biochemistry, 39, 15887-15894.
30.Tabatabai, L. B., and Graves, D. J. (1978) J.
Biol. Chem., 253, 2196-2202.
31.Cohen, P. (1978) Curr. Top. Cell. Regul.,
14, 117-196.
32.Andreeva, I. E., Makeeva, V. F., Kurganov, B. I.,
Chebotareva, N. A., and Livanova, N. B. (1999) Biochemistry
(Moscow), 64, 159-168.
33.Printen, J. A., Brady, M. J., and Saltiel, A. R.
(1997) Science, 275, 1475-1478.