
|
REVIEW: Role of Nitric Oxide in the Progression of PneumoconiosisV. CastranovaHealth Effects Laboratory Division, National Institute for Occupational Safety and Health, 1095 Willowdale Road, Morgantown, West Virginia 26505, USA; fax: 304-285-5938; E-mail: vic1@cdc.gov
|
Received April 30, 2003
Conflicting evidence has been reported as to whether nitric oxide (NO) possesses anti-inflammatory or inflammatory properties. Data are presented indicating that in vitro or in vivo exposure to selected occupational dusts, i.e., crystalline silica, organic dust contaminated with endotoxin, or asbestos, results in upregulation of inducible nitric oxide synthase (iNOS) and the production of NO by alveolar macrophages and pulmonary epithelial cells. Nitric oxide production is associated temporally and anatomically with pulmonary damage, inflammation, and disease progression in response to occupational dusts. Blockage of inducible nitric oxide synthase by administration of NOS inhibitors or in iNOS knockout mice decreases the magnitude of injury and inflammation following in vivo exposure to silica, endotoxin, or asbestos. Therefore, NO may play an important role in the initiation and progression of pneumoconiosis.
KEY WORDS: silicosis, byssinosis, asbestosis, iNOS, nitric oxide, LPS, endotoxin, cotton dust, crystalline silica, asbestos, pneumoconiosis
Abbreviations: iNOS) inducible nitric oxide synthase; NO) nitric oxide; ROS) reactive oxygen species; NF-kappaB) nuclear factor kappaB; AP-1) activator protein 1; LPS) lipopolysaccharide; O2-*) superoxide anion; BAL) bronchoalveolar lavage; NOx) nitrate and nitrite; TNF-alpha) tumor necrosis factor alpha; IL-1) interleukin-1; PGE2) prostaglandin E2; TiO2) titanium dioxide; CL) chemiluminescence; IkappaBalpha) inhibitor protein to nuclear factor kappaB.
Pneumoconiosis is a pulmonary disease associated with the inhalation of
particles. This review will discuss the role of nitric oxide (NO) in
the initiation and progression of pulmonary disease due to exposure to
crystalline silica, organic dusts contaminated with bacterial
endotoxin, and asbestos fibers.
Occupational exposure to crystalline silica is associated with pulmonary damage, inflammation, and fibrosis [1]. The rate of progression of silicosis, i.e., acute, accelerated, or chronic, is associated with the intensity of dust exposure. Occupations, such as sand blasting and rock drilling, are often characterized by exposures to high levels of respirable crystalline silica and rapid onset of disease. Exposure to crystalline silica is also associated with an increased risk of lung cancer [2]. Both silica-induced fibrosis and cancer have been related to the oxidant damage due to the production of reactive oxygen species (ROS) [3, 4]. Data indicate that ROS can be generated from the surface of silica as well as from pulmonary phagocytes exposed to silica particles [5]. This silica-induced generation of ROS has been reported to signal activation of transcription factors, such as nuclear factor kappaB (NF-kappaB) and activator protein-1 (AP-1), which induce the production of mRNAs for chemokines, inflammatory cytokines, fibrogenic mediators, and growth factors [6-8].
Acute exposure to organic dusts, such as dust generated from compost, grain, hay, silage, etc., can lead to fever, malaise, chest tightness, and pulmonary infiltration of neutrophils into the alveolar space [9]. This disease, organic dust toxic syndrome, is associated with inhalation of plant material contaminated with bacterial endotoxin, the active component of which is lipopolysaccharide (LPS). Chronic exposure to an organic dust can lead to obstructive lung disease, such as byssinosis due to cotton dust exposure [10]. Elevated production of ROS, resulting from the infiltration and activation of phagocytes in the pulmonary air spaces, has been a characteristic of pneumoconiosis due to organic dust exposure [11].
Exposure to asbestos fibers can lead to pulmonary fibrosis (asbestosis), mesothelioma, or lung cancer [12]. ROS can be generated by asbestos fibers as well as by phagocytic cells exposed to asbestos [13]. ROS generated by asbestos have been shown to be important signals for induction of transcription and the production of inflammatory and fibrotic cytokines, oncogenes and factors causing disregulation of growth control [14, 15].
As described above, ROS have been shown to play an important role in the initiation and progression of pneumoconiosis. To date the association between NO production and pulmonary disease is less clear. NO can react with superoxide anion (O2-*) to form a reactive species, peroxynitrite [16]. Peroxynitrite has been proposed to cause lipid peroxidation, mitochondrial and DNA damage, inhibition of enzyme and protein function, inhibition of surfactant production, and inactivation of surfactant apoproteins [16-20]. However, both anti-inflammatory and pro-inflammation activity for NO has been reported. Therefore, the role of NO in the initiation and progression is unresolved.
This manuscript will summarize the effects of silica, LPS containing dust, and asbestos on NO production by lung cells. It will present data supporting both an anti-inflammatory and pro-inflammatory role for NO. Lastly, it will present recent evidence supporting an important role for NO in the initiation and progression of particle-induced lung disease.
INDUCTION OF NITRIC OXIDE PRODUCTION FOLLOWING EXPOSURE TO
OCCUPATIONAL DUSTS
In vitro exposure of RAW 264.7 cells, a mouse peritoneal macrophage cell line, to silica has been reported to induce the production of NO [21]. However, in vitro exposure to crystalline silica failed to induce NO production from primary rat alveolar macrophages or primary rat alveolar type II epithelial cells [22, 23]. It appears that primary lung cells require intercellular mediators to upregulate NO production in response to silica [22]. Indeed, intratracheal instillation of rats with silica significantly enhanced mRNA levels for iNOS and NO production by bronchoalveolar lavage (BAL) cells as well as NO-dependent chemiluminescence (CL) from alveolar macrophages [22, 24]. These data are shown in Table 1. Intratracheal instillation of rats with silica also increased NO production from isolated alveolar type II epithelial cells as shown in Table 2 [25]. Upregulation of NO production has also been reported in a silica inhalation study with rats. In this study, NO levels in the BAL fluid increased significantly after 16 days of exposure, were maintained at this increased level through 40 days of exposure, and increased dramatically at 79 and 116 days of exposure [26]. In situ immunohistochemistry noted increased iNOS levels in alveolar macrophages and type II cells at 79 and 116 days of silica inhalation [26]. In summary, data are convincing that exposure to crystalline silica increases mRNA and protein levels of the inducible form of nitric oxide synthase and activates iNOS to generate NO. Alveolar macrophages and alveolar type II epithelial cells appear to be the major sites of this induction.
Table 1. Response of bronchoalveolar lavage
cells to intratracheal instillation of crystalline silica to rats*
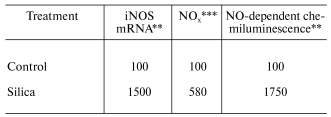
*Values are percent of control measured with bronchoalveolar
lavage cells harvested 24 h post-exposure (10 mg silica/100 g body
weight).
**Data calculated from [24]; mean values
of three experiments; silica significantly greater than control
(p < 0.05).
***Data calculated from [22]; mean values
of three experiments; silica significantly greater than control (p
< 0.05).
Table 2. Response of alveolar type II cells
to intratracheal instillation of crystalline silica to rats*
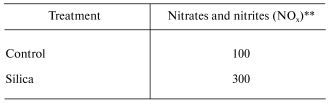
*Values are percent of control measured from alveolar type II
epithelial cells isolated by enzymatic digestion 24 h post-exposure
(10 mg silica/100 g body weight) and purified by centrifugal
elutriation.
**Data calculated from [25]; mean values of
three experiments; silica significantly greater than control (p
< 0.05).
Organic dusts are often contaminated by the gram-negative bacterial product, lipopolysaccharide (LPS). In vitro exposure of fetal lung epithelial cells, primary rat alveolar macrophages, primary rat alveolar type II epithelial cells, and a rat type II cell line (RLE-6TN) to LPS has been shown to stimulate significant production of NO [23, 27]. Inhalation of LPS results in enhancement of mRNA levels for iNOS in BAL cells, elevated nitrate and nitrite (NOx) levels in BAL fluid, and an increase in NO-dependent chemiluminescence from alveolar macrophages from exposed rats as shown in Table 3 [28]. Huffman et al. [28] showed that inhalation of cotton dust contaminated with LPS caused a similar upregulation of NO message in BAL cells, NOx levels in BAL fluid, and NO production from rat phagocytes (Table 4). In conclusion, LPS appears to be an important etiologic agent in the induction of NO production in response to organic dusts. Alveolar type II epithelial cells and alveolar macrophages appear to be important targets for LPS-stimulated induction of NO.
Table 3. Response of bronchoalveolar lavage
cells to inhalation exposure of rats to LPS*
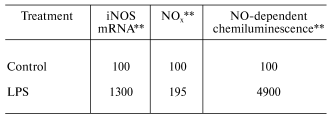
*Values are percent of control measured with bronchoalveolar
lavage cells harvested 18 h post-exposure (2.2*104 EU
LPS/m3 for 3 h).
**Data calculated from [28]; mean values
of 3-6 experiments; LPS significantly greater than control (p
< 0.05).
Table 4. Response of bronchoalveolar lavage
cells to inhalation exposure of rats to cotton dust*
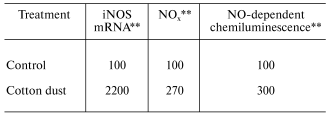
*Values are percent of control measured with bronchoalveolar
lavage cells harvested 18 h post-exposure (40 mg cotton
dust/m3 for 3 h).
**Data calculated from [28]; mean values
of 3-6 experiments; cotton dust significantly greater than control
(p < 0.05).
In vitro exposure of alveolar macrophages or lung epithelial cells to asbestos has been shown to increase NO production [29-31]. Intratracheal instillation of rats with asbestos results in enhanced iNOS expression in lung cells measured by in situ immunohistochemistry [32]. Immunohistochemistry also indicates the inhalation of asbestos increased nitrotyrosine residues in lung tissue [33]. Thus, asbestos appears to be a potent activator of significant NO production in alveolar macrophages and lung epithelial cells.
THE YING AND YANG OF NITRIC OXIDE
The data presented above clearly indicate that exposure of the lung to crystalline silica, organic dusts containing LPS, or asbestos induces iNOS activity and the production of NO. However, there is uncertainty concerning the role of NO in the progression of pneumoconiosis, because both anti-inflammatory and pro-inflammatory potential have been proposed for NO.
Anti-inflammatory potential of nitric oxide. A significant body of literature exists suggesting that NO can be protective against oxidant injury and inflammation. Having an unpaired electron, NO can react with lipid radicals to dissipate the reactivity of these intermediates, terminating the chain of lipid peroxidation and mitigating oxidant damage. NO has also been shown to inactivate the ubiquitous transcription factor, nuclear factor kappaB (NF-kappaB) [8]. Matthews et al. [34] have reported that NO can nitrosate cysteine 62 on the p50 subunit of NF-kappaB, decreasing its ability to bind to gene promoter regions on DNA. NO has also been reported to increase IkappaBalpha levels, resulting in depression of NF-kappaB activity [35]. Since NF-kappaB is critical for the production of mRNAs controlling a number of inflammatory cytokines, these inhibitory effects of NO on NF-kappaB activity have been proposed to down-regulate the response to inflammatory stimuli. For example, NO generation systems have been reported to decrease LPS-induced activation of NF-kappaB in alveolar macrophages [36]. Such inhibition has been associated with inhibition of LPS induction of TNF-alpha, IL-1, and macrophage inflammatory protein 2 (MIP-2) in alveolar macrophages [37]. Furthermore, NOS inhibitors have been reported to enhance silica- and LPS-induced activation of NF-kappaB in a peritoneal macrophage cell line (RAW 264.7 cells) [21]. An anti-inflammatory action of NO has also been reported in vivo. Kang et al. [38] reported that inhalation of NO (10 ppm) significantly decreased pulmonary damage and inflammation in response to intravenous exposure of rabbits to LPS (5 mg LPS/kg body weight) as measured by suppression of NF-kappaB activation, lung damage (BAL levels of protein and lactate dehydrogenase activity), inflammation (neutrophil influx), and the activity of alveolar macrophages (chemiluminescence).
Pro-inflammatory potential of nitric oxide. In contrast to the evidence above, data also exist indicating that NO can form peroxynitrite, which has been associated with lipid peroxidation, mitochondrial and DNA damage, inhibition of enzyme and protein function, inhibition of surfactant production, and inactivation of surfactant apoproteins [16-20]. NO generating systems have also been shown to activate NF-kappaB in RAW macrophages [39]. Furthermore, NOS inhibitors can decrease both silica- and LPS-induced NF-kappaB activation in macrophage cells [39]. Lastly, NO generating systems stimulate the production of the inflammatory mediator, prostaglandin E2 (PGE2), from rat alveolar macrophages and rat lung fibroblasts in vitro (Table 5).
Table 5. NO stimulated production of
prostaglandin E2*

*Values are percent of control PGE2 production following
a 6-h exposure of primary rat alveolar macrophages (AM) or lung
fibroblasts to a NO generation system. Data from Blake and Castranova
(unpublished); mean values of four experiments; SIN-1 significantly
greater than control (p < 0.05).
DOES NITRIC OXIDE PLAY A ROLE IN PNEUMOCONIOSIS?
Despite the conflicting information concerning the inflammatory potential of NO, there is an increasing body of evidence implicating NO in the initiation and progression of pneumoconiosis. Blackford et al. [40] exposed rats by intratracheal instillation to four types of particles of different pathogenicity, i.e., crystalline silica, coal mine dust, carbonyl iron or titanium dioxide (TiO2). They reported an association between the potency of these particles to induce mRNA for iNOS in BAL cells or increase NO-dependent chemiluminescence from alveolar macrophages and the resultant degree of pulmonary damage (BAL protein) and inflammation (BAL neutrophils), with a potency sequence of silica > coal > carbonyl iron > TiO2 (Table 6). Castranova et al. [41] reported a relationship between progression of pneumoconiosis and induction of NO. That is, NO dependent chemiluminescence from alveolar macrophages harvested by BAL was greater in a silica-exposed coal miner with an abnormal chest radiograph than an asymptomatic coal miner exposed to silica dust. Both exposed miners exhibited greater NO production than alveolar macrophages from a healthy, non-smoking control (Table 7). Therefore, these studies show a correlation between progression of pulmonary disease and NO production. In addition, Porter et al. [26] reported a temporal correlation between NO production in the lung (measured by the levels of NOx in BAL fluid) and markers of pulmonary damage and inflammation following inhalation exposure of rats to silica. A dramatic increase in NO production was associated with initiation of pulmonary fibrosis in this model. This study also reported a spatial relationship between pulmonary regions with high immunohistochemical staining for iNOS and nitrotyrosine and regions of high silica deposition, elevated inflammation and lipidosis, and development of granulomatous lesions. Alveolar macrophages and alveolar type II epithelial cells were demonstrated to be the source of this silica-induced NO production [26]. Strong evidence that NO plays an important role in the development of silica-induced disease was presented by Srivastava et al. [42]. They compared the pulmonary response of wild type and iNOS knockout mice to silica exposure (250 mg/m3, 5 h/day, for 10 days) by inhalation. In wild type mice, lesion number and size progressed from 1-12 weeks post-exposure. In contrast, lesion number and size were 80% lower in iNOS knockout mice one week post-exposure with lack of progression through 12 weeks post-exposure.
Table 6. Comparison of pulmonary damage and
inflammation resulting from intratracheal instillation of dusts with
different pathogenic potentials to rats* (data calculated from [40])
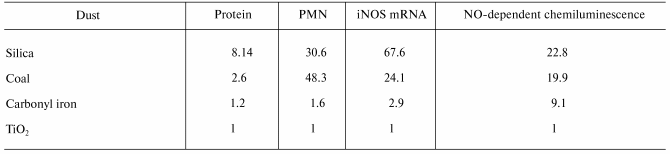
*Relative stimulatory potency is normalized for exposure to
an equal number of particles. PMN, polymorphonuclear neutrophils.
Table 7. Relationship between NO production
and pneumoconiosis in miners*

*Values are percent of control. Alveolar macrophages were
obtained by bronchoalveolar lavage.
**Data calculated from [41]; values are
from a single volunteer in each group.
NO production has also been associated with initiation of pulmonary inflammation in response to endotoxin exposure. Mikawa et al. [43] have shown that exposure to endotoxin caused an increase in neutrophil infiltration into the alveolar space as well as induction of iNOS by lung cells. Treatment with aminoguanidine, a NOS inhibitor, significantly decreased the endotoxin-induced upregulation of iNOS and the level of pulmonary inflammation. Similarly, Kristof et al. [44] reported significantly less pulmonary inflammation in iNOS knockout mice than wild type mice in response to intravenous exposure to endotoxin.
Recent data also describe the association between NO production and the adverse effects of asbestos. An association between the magnitude of NO production by lung epithelial cells and DNA oxidation has been reported in vitro [31]. In addition, a temporal correlation has been demonstrated between NO production by alveolar macrophages and neutrophil infiltration into the lung following inhalation of asbestos [45]. Lastly, Dorger et al. [46] demonstrated that intratracheal instillation of asbestos increased iNOS protein levels in lung cells and immunohistochemical staining for iNOS and nitrotyrosine in lung tissue in rats one day after treatment. In contrast, asbestos failed to increase iNOS levels in hamsters. This difference in NO response correlated with a striking difference in pulmonary damage and inflammation in these two species after exposure to asbestos, with the rat exhibiting neutrophil infiltration, damage of the alveolar air/blood barrier and edema, while asbestos failed to significantly alter these parameters in the hamster.
In summary, although several studies have reported that NO can exhibit anti-inflammatory potential, data exist indicating a synergistic effect of NO on silica- or LPS-induced activation of the transcription factor, NF-kappaB. In addition, treatment of lung cells with exogenous NO increases the production of PGE2, a potent inflammatory mediator. A growing body of evidence indicates that in vivo exposure of rats to silica, LPS, cotton dust, or asbestos increases iNOS levels and the production of NO by alveolar macrophages and possibly alveolar type II epithelial cells. The induction of NO production by lung cells has been associated with the pathogenic potential of selected dusts in a rat model, with disease progression in silica-exposed miners, and with the initiation and progression of pulmonary damage and inflammation in rats exposed to silica, LPS, or asbestos. Therefore, data support the hypothesis that exposure to occupational particles can induce pulmonary levels of iNOS and increase NO production by lung cells, such induction of NO may play an important role in the initiation of pulmonary damage and inflammation leading to the progression of pneumoconiosis.
REFERENCES
1.Driscoll, K. E., and Guthrie, G. D. (1997) in
Comprehensive Toxicology. Vol. 8. Toxicology of the
Respiratory System (Roth, R. A., ed.) Elsevier Science, Inc., New
York, pp. 373-391.
2.IARC (1997) IARC Monograph Evaluating
Carcinogenic Risks in Humans, 68, 41-242.
3.Castranova, V., and Vallyathan, V. (2000)
Environ. Health Perspect., 108 (Suppl. 4), 675-684.
4.Shi, X., Castranova, V., Halliwell, B., and
Vallyathan, V. (1998) J. Toxicol. Environ. Health (Pt. B),
1, 181-197.
5.Castranova, V. (1994) Environ. Health
Perspect., 102 (Suppl. 10), 65-68.
6.Rojanasakul, Y., Ye, J., Chen, F., Wang, L., Cheng,
N., Castranova, V., Vallyathan, V., and Shi, X. (1999) Mol. Cell.
Biochem., 200, 119-125.
7.Ding, M., Shi, X., Lu, Y., Huang, C., Leonard, S.,
Roberts, J., Antonini, J., Castranova, V., and Vallyathan, V. (2001)
J. Biol. Chem., 276, 9108-9114.
8.Chen, F., Castranova, V., Shi, X., and Demers, L.
M. (1999) Clin. Chem., 45, 7-17.
9.Rylander, R. (1997) in Comprehensive
Toxicology. Vol. 8. Toxicology of the Respiratory
System (Roth, R. A., ed.) Elsevier Science, Inc., New York, pp.
415-424.
10.Merchant, J. A. Halprin, G. M., Hudson, A. R.,
Kilburn, K. H., McKenzie, W. N., Hurst, D. J., and Bermazohn, P. (1975)
Arch. Environ. Health, 30, 222-229.
11.Castranova, V., Robinson, V. A., and Frazer, D.
G. (1996) Environ. Health Perspect., 104 (Suppl. 1),
41-53.
12.Dement, J. M., Merchant, J. A., and Green, F. H.
Y. (1987) in Occupational Respiratory Diseases (Merchant, J. A.,
Boehlecke, B. A., Taylor, G., and Pickett-Harner, M., eds.) DHHS
(NIOSH) Publication No. 86-102, pp. 287-327.
13.Castranova, V. (1998) Appl. Occup. Environ.
Hyg., 13, 613-616.
14.Brody, A. R. (1997) in Comprehensive
Toxicology. Vol. 8. Toxicology of the Respiratory System
(Roth, R. A., ed.) Elsevier Science, Inc., New York, pp. 393-413.
15.Ding, M., Dong, Z., Chen, F., Pack, D., Ma,
W.-Y., Ye, J., Shi, X., Castranova, V., and Vallyathan, V. (1999)
Cancer Res., 59, 1884-1889.
16.Van der Vliet, A., Eiserich, J. P., and
Halliwell, B. (1997) J. Biol. Chem., 272, 7617-7625.
17.Haddad, I. Y. (1996) Am. J. Physiol.,
270, L898-L906.
18.Haddad, I. Y. (1996) Am. J. Physiol.,
270, L281-L288.
19.Hogg, N., and Kalyanaraman, B. (1999) Biochim.
Biophys. Acta, 1411, 378-384.
20.Szabo, C., and Ohshima, H. (1997) Nitric
Oxide, 1, 373-385.
21.Chen, F., Kuhn, D. C., Sun, S. C., Gaydos, L. J.,
and Demers, L. M. (1995) Biochem. Biophys. Res. Commun.,
214, 839-846.
22.Huffman, L. J., Judy, D. J., and Castranova, V.
(1998) J. Toxicol. Environ. Health (Pt. A), 53,
29-46.
23.Kanj, R., Kang, J., and Castranova, V. (2002)
FASEB J., 16, A861.3.
24.Blackford, J. A., Antonini, J. M., Castranova,
V., and Day, R. D. (1994) Am. J. Respir. Cell Mol. Biol.,
11, 426-431.
25.Castranova, V., Pailes, W. H., Judy, D. J., and
Huffman, L. J. (1994) FASEB J., 8, A854.
26.Porter, D. W., Millecchia, L., Robinson, V. A.,
Hubbs, A., Willard, P., Pack, D., Ramsey, D., McLaurin, J., Khan, A.,
Landsittel, D., Teass, A., and Castranova, V. (2002) Am. J. Physiol:
Lung Cell Mol. Physiol., 283, L485-L493.
27.Gutierrez, H. H., Pitt, B. R., Schwarz, M.,
Watkins, S. C., Lowenstein, C., Caniggia, I., Chumley, P., and Freeman,
B. A. (1995) Am. J. Physiol.: Lung Cell Mol. Physiol.,
268, L501-L508.
28.Huffman, L. J., Judy, D. J., Robinson, V. A., and
Castranova, V. (1997) Inhal. Toxicol., 9, 567-579.
29.Thomas, G., Ando, T., Verma, K., and Kagan, E.
(1994) Am. J. Respir. Cell Mol. Biol., 11, 707-715.
30.Mongan, L. C., Jones, T., and Patrick, G. (2000)
Cytokine, 12, 1243-1247.
31.Chao, C. C., Park, S. H., and Aust, A. E. (1996)
Arch. Biochem. Biophys., 326, 152-157.
32.Iguchi, H., Kojo, S., and Ikeda, M. (1996) J.
Appl. Toxicol., 16, 309-315.
33.Tanaka, S., Choe, N., Hemenway, D. R., Zhu, S.,
Matalon, S., and Kagan, E. (1998) J. Clin. Invest., 102,
445-454.
34.Matthews, J. R., Botting, C. H., Panico, M.,
Morris, H. R., and Hay, R. T. (1996) Nucleic Acids Res.,
24, 2236-2242.
35.Peng, H. B., Libby, P., and Liao, J. K. (1995)
J. Biol. Chem., 270, 14214-14219.
36.Raychaudhuri, B., Dweik, R., Connors, M. J.,
Buhrow, L., Malur, A., Drazha, J., Arroligi, A., Erzurum, S. C.,
Kavurum, M. S., and Thomassen, M. J. (1999) Am. J. Respir. Cell Mol.
Biol., 21, 311-316.
37.Thomassen, M. J., Buhrow, L. T., Connors, M. J.,
Kaneko, F. T., Erzurum, S. C., and Kavuru, M. S. (1997) Am. J.
Respir. Cell Mol. Biol., 17, 279-283.
38.Kang, J. L., Park, W., Pack, I. S., Lee, H. S.,
Kim, M. J., Lim, C.-M., and Koh, Y. (2002) J. Appl. Physiol.,
92, 795-801.
39.Kang, J. L., Lee, K., and Castranova, V. (2000)
Mol. Cell. Biochem., 215, 1-9.
40.Blackford, J. A., Jones, W., Dey, R. D., and
Castranova, V. (1997) J. Toxicol. Environ. Health, 51,
203-218.
41.Castranova, V., Huffman, L. J., Judy, D. J.,
Bylander, J. E., Lapp, L. N., Weber, S. L., Blackford, J. A., and Dey,
R. D. (1998) Environ. Health Perspect., 106 (Suppl. 5),
1165-1169.
42.Srivastava, K. D., Rom, W. N., Jagirdar, J., Yie,
T.-A., Gordon, T., and Tchou-Wong, K.-M. (2002) Am. J. Respir. Crit.
Care Med., 165, 527-533.
43.Mikawa, K., Nishina, K., Tamada, M., Takao, Y.,
Maekawa, N., and Obara, H. (1998) Crit. Care Med., 26,
905-911.
44.Kristof, A. S., Goldberg, P., Laubach, V., and
Hussain, S. N. (1998) Am. J. Respir. Crit. Care Med.,
158, 1883-1889.
45.Quinlan, T. R., BeruBe, K. A., Hacker, M. P.,
Taatjes, D. J., Timblin, C. R., Goldberg, J., Kimberly, P.,
O'Shaugnessy, P. O., Hemenway, D., Torino, J., Jiminez, L. A., and
Mossman, B. T. (1998) Free Rad. Biol. Med., 24,
778-788.
46.Dorger, M., Allmeling, A.-M., Kiefmann, R.,
Munzing, S., Messmer, K., and Krombach, F. (2002) Toxicol. Appl.
Pharmacol., 181, 93-105.