
|
REVIEW: Exploiting Oxidative Stress and Signaling in Chemotherapy of Resistant NeoplasmsN. F. Schor1,2,3,5*, V. E. Kagan3,4, Ye Liang1,5, Ch. Yan5, Yu. Tyurina4, V. Tyurin4, and K. D. Nylander5Departments of Pediatrics1, Neurology2, Pharmacology3, and Environmental and Occupational Toxicology4, University of Pittsburgh, Pittsburgh, USA5Pediatric Center for Neuroscience, Children's Hospital of Pittsburgh, 3705 Fifth Avenue, Pittsburgh, PA 15213, USA; fax: 412-692-6165; E-mail: nfschor@pitt.edu * To whom correspondence should be addressed.
|
Received April 30, 2003
Neural crest tumors of childhood are particularly resistant to apoptosis induction by chemotherapeutic agents. Mechanisms of resistance include altered glutathione handling that accompanies up-regulation of Bcl-2 and its relatives. We have designed and tested in preclinical model systems approaches to this problem. These approaches include adjunctive use of oxygen radical-generating neurotransmitter analogs taken up by these neural crest tumor cells with scavenging (i.e., “rescue”) agents that are selective for normal neural crest and the use of reduction-dependent prodrugs of apoptosis-inducing agents. Promising prototypes for these conceptual approaches include, respectively, adjunctive use of the oxygen radical generator, 6-hydroxydopamine, with the normal cell-selective antioxidant, Tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl), and use of the reduction-dependent chemotherapeutic prodrug neocarzinostatin.
KEY WORDS: oxidative stress, chemotherapeutic agents, apoptosis, neural crest tumor cells
Neuroblastoma is the most common extra-cranial tumor of childhood. Unfortunately, 65% of children with neuroblastoma present clinically with disease that has already metastasized to distant tissues. For these children, the 5-year survival rate is somewhere between 5 and 20% after maximal surgery, chemotherapy, and radiation therapy. For patients with neuroblastoma and other difficult to treat malignant neoplasms, the challenge is to develop therapies that target the tumor without harming normal tissues [1]. We have exploited the expression of dopamine receptors and the overexpression of Bcl-2 by neuroblastoma cells in an effort to design tumor-selective, free radical-generating chemotherapeutic strategies for metastatic neuroblastoma.
CATECHOLAMINE UPTAKE SYSTEM: A GATEWAY FOR SELECTIVE
CHEMOTHERAPY
Neuroblastomas are derived from neurons of the sympathetic nervous system. As such, they express a catecholamine uptake system, and can take up dopamine against a concentration gradient. Many previous reports have demonstrated the exquisite sensitivity of neuroblastoma cells to 6-hydroxydopamine (6-OHDA), an oxygen radical-generating analog of dopamine that is taken up through the catecholamine uptake system [2-4]. Although the use of 6-OHDA as a chemotherapeutic agent for neuroblastoma has been suggested [2, 4], 6-OHDA effects chemical sympathectomy by killing normal sympathetic neurons [2, 5] and thereby causing intolerable toxicity. We have studied the adjunctive use of 6-OHDA with the normal cell-selective free radical scavenger and superoxide dismutase mimic, Tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl) [6].
Tempol has been proposed to function as a superoxide dismutase mimic, carbon- and peroxy-based radical scavenger, and inhibitor of the generation of hydroxyl radical [7-9]. By mechanisms that may involve differential oxidation states of the environment, Tempol is selectively protective against the toxicity of free radicals in normal tissues as opposed to tumors [6, 10].
Protective effects of Tempol against 6-OHDA toxicity. We tested the efficacy and examined the mechanism of action of Tempol in mice treated with 6-OHDA administered by intraperitoneal injection. Pretreatment of A/J mice with Tempol (250 mg/kg) prevented the mortality seen with 6-OHDA alone (350 or 400 mg/kg). Our in vitro mechanistic studies make it likely that processes other than dismutation of superoxide play an important role in the detoxification of 6-OHDA by Tempol. The decay of the Tempol ESR signal during incubation with 6-OHDA is the same under aerobic and anaerobic conditions, and is not influenced by co-incubation with superoxide dismutase. This implies that direct reaction of Tempol with 6-OHDA accounts for the observed decay in the Tempol ESR signal. The effects of catalase and peroxidase on this system are, however, dependent on the degree of oxygenation, indicating that direct reaction of Tempol with 6-OHDA is not the only operative detoxification mechanism [6].
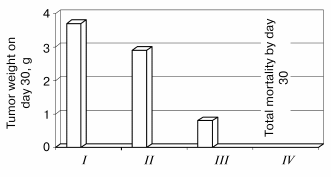
Antitumor effectiveness of adjunctive treatment with 6-OHDA and Tempol. As is shown in Fig. 1, 6-OHDA remains an effective anti-neuroblastoma agent in the presence of Tempol. While 6-OHDA alone is prohibitively toxic, it is rendered non-toxic by adjunctive administration of Tempol. The adjunctive regimen reduces both tumor incidence and growth rate in mice injected subcutaneously with Neuro-2a murine neuroblastoma cells one day prior to initiation of the adjunctive treatment regimen [6].
Fig. 1. Effects of Tempol (250 mg/kg) on the anti-neuroblastoma effects of 6-OHDA (400 mg/kg). Neuro-2a murine neuroblastoma cells were xenografted subcutaneously into A/J mice on day 0. Vehicle (I), Tempol (II), Tempol + 6-OHDA (III), or 6-OHDA (IV) were injected intraperitoneally 10 min apart in a single adjunctive treatment on day 1. Tumor weight was determined on day 30.
Bcl-2: A GENERAL MECHANISM OF RESISTANCE TO CHEMOTHERAPY
Recent studies make it clear that altered levels of Bcl-2 and the related protein Bcl-XL contribute to the resistance of neuroblastomas to chemotherapy [11, 12]. The discovery and development of chemotherapeutic strategies that overcome drug resistance would contribute greatly to the conquest of this common and deadly disease.
Studies in our laboratories and those of others have suggested that Bcl-2 exerts its effects at least in part by a mechanism that includes a shift in the cellular capacity for glutathione (GSH) turnover [13-16]. A GSH-related mechanism for Bcl-2 activity is particularly plausible in light of other studies that link both the initiating and subsequent steps involved in the apoptotic process to exposure of the cell to reactive oxygen species [17-19] and studies implicating release of cytochrome c from the mitochondrion with a resultant breakdown in electron transport and increased formation of reactive oxygen species [20, 21].
We have pursued a strategy for overcoming Bcl-2-mediated chemoresistance that takes advantage of the increase in cellular free sulfhydryl content by using chemotherapeutic agents that require reduction by GSH for their activity. Our initial studies focused on the efficacy of one such group of agents, the enediynes, in tumor cells that were genetically engineered to overexpress bcl-2 [22].
Neocarzinostatin (NCS) is an enediyne DNA-cleaving natural product that induces apoptosis in neural crest tumor cells in culture [23, 24]. Like many other naturally-occurring enediynes, NCS is actually a prodrug that requires sulfhydryl activation for efficacy. As such, the cytotoxicity of NCS has been demonstrated, within the range of physiological manipulations performed, to vary directly with the GSH content of the cell [25-27]. This information led to our prediction that, contrary to the case for other chemotherapeutic agents studied, overexpression of bcl-2 and the resulting shift in GSH handling in the cell would potentiate the induction of apoptosis by NCS.
We subsequently showed that, in PC12 rat pheochromocytoma cells that have been bcl-2- or mock-transfected, bcl-2 overexpression does indeed potentiate the apoptosis-inducing activity of NCS [22, 28]. In this same system, bcl-2 overexpression decreases apoptosis induction by the non-enediyne, cisplatin, and synthetic and natural enediynes that do not require reductive activation by GSH, indicating that GSH is important in the potentiation of NCS-induced apoptosis by Bcl-2. In addition, inhibition of GSH synthesis completely abolishes the difference in NCS sensitivity between cells that do and do not overexpress bcl-2. The GSH-dependent enediyne prodrugs are therefore the only class of drug that has been demonstrated to work best in those tumor cells that have become resistant to other known chemotherapeutic agents.
Our more recent studies suggest that potentiation of NCS-induced apoptosis by Bcl-2 overproduction is dependent, not only on altered metabolism of GSH, but also on caspase 3-mediated cleavage of Bcl-2 [29]. The resulting cleavage product of this anti-apoptotic protein is, in fact, pro-apoptotic. The relationship between Bcl-2 cleavage and altered metabolic handling of GSH is not known, but it is intriguing that caspase 3 activation, like NCS activation, has been demonstrated to be sulfhydryl reduction-dependent. It is not clear how Bcl-2, a mitochondrial protein that should prevent the release of cytochrome c from the mitochondrion and thereby block activation of cytosolic caspase 3, should be cleaved in potentiated fashion by caspase 3 in Bcl-2-transfected PC12 cells, unless intramitochondrial, activated NCS facilitates cytochrome c release despite the presence of Bcl-2.
NCS and related enediynes have previously been proposed for clinical use in a variety of human cancers. Initial clinical trials of NCS were hampered by anaphylactic responses to its non-covalently bound protein component, ironically a portion of the NCS structure with no relationship to its efficacy. Since that time, NCS protein has been rendered immunologically inert by coupling to a maleic acid-based polymer (SMANCS), and is in clinical use in Japan and China primarily for hepatocellular carcinoma. The related enediyne, calecheamicin, has been coupled to antibodies to tumor-specific antigens and has been proposed for use as targeted therapy for breast cancer. No prior studies have taken specific advantage of the need for sulfhydryl activation of these compounds or have proposed identification of particular subgroups of patients and/or tumors for which these compounds might present an improved therapeutic index.
It is critically important that we understand the mechanism by which bcl-2 overexpression leads to altered GSH handling, increased cleavage of Bcl-2 after NCS treatment, and, ultimately, increased susceptibility to NCS-induced apoptosis. These determinations will facilitate the development of more globally efficacious pharmacological agents for bcl-2-overexpressing, chemoresistant cancer and will allow physicians to predict which tumors will or will not respond to treatment with NCS and related, sulfhydryl activation-dependent drugs.
Mechanistic studies of the relationship between bcl-2 expression and the increase in the cellular concentration of GSH. The induction and enactment of apoptosis have been associated with the generation of reactive oxygen species. In keeping with this association, overproduction of the anti-apoptosis protein, Bcl-2, is associated in some systems with increased tolerance to oxidant stress [13-15], an increase in the cellular content of reducing species [13], and prevention of the release of cytochrome c from the mitochondrion [20, 21]. However, in contradistinction to these studies, others have reported a pro-oxidant activity of Bcl-2 [30].
GSH is one of several antioxidant species the metabolism of which is thought to be altered by Bcl-2 overproduction. We have previously discussed the implications of this alteration for apoptosis induction during cancer chemotherapy with GSH-dependent agents [22] and in the course of neurodegenerative disease [28]. In the studies described below, we examined the metabolism of GSH in mock- and bcl-2-transfected cell lines. We compared and contrasted results of these studies performed in PC12 rat pheochromocytoma and MCF-7 human breast cancer cell lines.
Differential GSH accumulation after N-acetylcysteine treatment. Incubation of bcl-2-transfected PC12 cells with the GSH precursor N-acetylcysteine (NAcCys, 10 mM) for up to 8 h results in a GSH level 5-times baseline (p < 0.002) by 4 h of treatment. In contrast, similar incubation of mock-transfected PC12 cells results in only a 1.6-fold increase in intracellular GSH (p < 0.002) that is apparent by 1 h and plateaus at that level for 4 h. Without additional NAcCys supplementation, the GSH content of bcl-2-transfected cells declines and the difference in GSH content between the two transfectants loses statistical significance (p > 0.05) by 12 h after addition of NAcCys to the medium.
Unlike the case for PC12 cells, bcl-2 transfection does not alter the response of MCF-7 cells to NAcCys. Analogous treatment of both mock- and bcl-2-transfected MCF-7 cells with NAcCys results in GSH content kinetics that resemble those seen with mock-transfected PC12 cells, and there was no significant difference between the MCF-7 bcl-2- and mock-transfectants in this regard.
GSH accumulation in bcl-2-overexpressing PC12 cells results from enhancement of the capacity for GSH turnover. The NAcCys-induced GSH accumulation seen in bcl-2-transfected PC12 cells could result from augmented GSH turnover. As such, we examined the effects of bcl-2 transfection on the ability of PC12 cells to turnover GSH (so-called “functional thiol reserves” [31]) in response to treatment with NCS, a known “consumer” of GSH. We have done this by measuring the generation of glutathionyl radical [32] and the decline in GSH content [33] resulting from irreversible trapping of the glutathionyl radical after 1-h incubation with NCS. We measured glutathionyl radical formation by trapping it with the spin trapping agent, 5,5-dimethylpyrroline-1-oxyl (DMPO), and measuring the trapped GS-DMPO adduct and the consequent decrease in GSH. In this assay, the larger the rate of GS-DMPO formation and GSH depletion, the higher the capacity for GSH turnover in the system being examined [32].
Treatment of bcl-2-transfected PC12 cells with NCS (0.4 nmol/106 cells) results in the trapping of approximately 4 nmol/mg protein (p < 0.005) of glutathionyl radical and a decline in GSH content of 1.4 nmol/mg protein (p < 0.0005) over 1 h. In sharp contrast, treatment of mock-transfected PC12 cells with NCS results in no significant change in the cumulative generation of glutathionyl radical or the cellular content of GSH. Parallel studies of the effect of NCS treatment on cellular protein thiol content revealed no significant change in either mock- or bcl-2-transfected PC12 cells.
Further studies demonstrated that GSH accumulation in bcl-2-transfected PC12 cells is not the result of increased activity of either glutathione reductase or gamma-glutamylcysteine synthetase or decreased GSH efflux.
ROLES OF Bcl-2 AND GSH IN THE MECHANISM BY WHICH INCREASED bcl-2
EXPRESSION LEADS TO AN INCREASE IN CELLULAR SENSITIVITY TO THE
ENEDIYNES
Correlation of altered GSH handling with potentiation of apoptosis. Our previous studies demonstrated the paradoxical increase in sensitivity of PC12 cells to NCS afforded by bcl-2 transfection ([22]; concentration inducing apoptosis in 50% of cells (IC50) decreases from >0.06 to 0.01 µM). This potentiation of NCS toxicity was abrogated by prior incubation of PC12 cells with buthionine sulfoximine, an inhibitor of GSH synthesis. The difference in effect of bcl-2 transfection on GSH handling in PC12 and MCF-7 cells led us to predict that bcl-2 transfection would not potentiate and might even prevent apoptosis in MCF-7 cells treated with NCS.
As predicted, bcl-2 overexpression in MCF-7 cells did not potentiate death induced by NCS (IC50 increases from 0.0025 ± 0.0005 to 0.0045 ± 0.0009 µM). This result was borne out and the cell death induced by NCS in MCF-7 transfectants determined to be apoptotic in studies done by manual counting of adherent versus floating cells, propidium iodide staining with fluorescence microscopic analysis, and flow cytometry after 7-AAD staining [29, 34]. The small protective effect of bcl-2 overexpression in this system did not quite reach statistical significance (p between 0.05 and 0.1). However, at every concentration tested, the survival of the MCF-7 bcl-2 transfectants exceeded that of the mock transfectants.
In light of the accumulation of GSH seen only in bcl-2-transfected PC12 cells after incubation with NAcCys, and the concentration-dependent role of GSH in activation of NCS [25-27, 35], we hypothesized that pretreatment of bcl-2-transfected PC12 cells with NAcCys would result in further potentiation of NCS-induced apoptosis, while the response of mock-transfected PC12 cells and mock- and bcl-2-transfected MCF-7 cells to NCS would remain unaltered by NAcCys [16]. NAcCys treatment (4 h, 10 mM) of bcl-2-transfected PC12 cells prior to treatment with NCS results in a twofold shift in the concentration-response curve of such cells in the direction of increased potency of NCS (p < 0.002). In contrast, the concentration-response curve of bcl-2-transfected MCF-7 cells to NCS is unaffected by NAcCys treatment (IC50 of 0.0017 ± 0.0002 µM without NAcCys and 0.0018 ± 0.0002 µM with NAcCys; p > 0.05). The sensitivity to NCS of mock-transfected cells of both types is unaffected by NAcCys exposure.
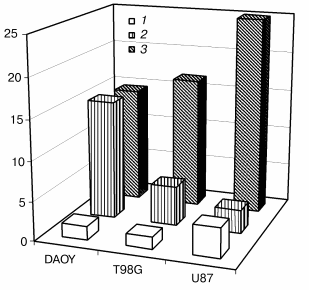
Furthermore, in an effort to verify this relationship in cell lines derived from other tumor types and in non-transfected cells with intrinsically different Bcl-2 contents, we have performed a single experiment to determine the native contents of GSH and Bcl-2 and the IC50 for apoptosis induction by NCS in three different brain tumor cell lines. Figure 2 demonstrates that among DAOY medulloblastoma cells and T98G and U87 glioblastoma cells, high Bcl-2 content correlates with high GSH content and low IC50 (i.e., high sensitivity) for apoptosis induction by NCS. Although this experiment involves cell lines that undoubtedly differ from one another in ways other than their content of Bcl-2, it underscores the likelihood that our findings in the “cleaner” PC12/MCF-7 studies can be generalized to native tumor cells derived from different tissues.
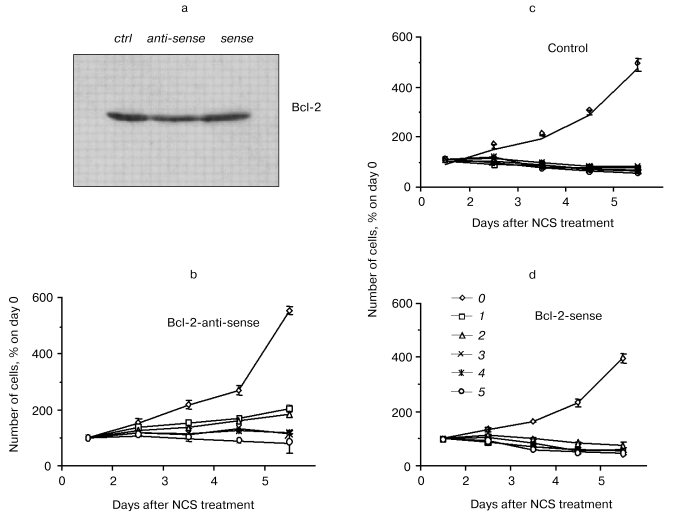
Bcl-2 as mediator of the potentiation of NCS-induced apoptosis. The role of Bcl-2 in the potentiation of NCS-induced apoptosis was directly tested in studies involving treatment of bcl-2-transfected PC12 cells with antisense oligonucleotide to bcl-2 (from 24 h prior to NCS treatment through the conclusion of the study; Liang, Srinivas, and Schor, preliminary results). Control treatments for these studies included a sense oligonucleotide and vehicle without oligonucleotide. Figure 3 shows the decrease in Bcl-2 level after treatment with antisense oligonucleotide, but not with vehicle or sense oligonucleotide. Subcellular fractionation studies indicated that all of the small decrease in Bcl-2 is accounted for in the mitochondria. Figure 3 also demonstrates that antisense treatment and consequent decreased mitochondrial Bcl-2 abrogate the potentiation of NCS-induced apoptosis in cells that overexpress bcl-2. The concentration of NCS required to completely suppress the increase in culture growth (shown in our previous publications to correlate with apoptosis induction [22, 29]) increases from 100 ng/ml under control and sense treatment conditions to 500 ng/ml under the antisense treatment condition. Coupled with our GSH studies, these results support the notion that potentiation of NCS-induced apoptosis results from a Bcl-2-induced shift in the GSH turnover capacity of the cell with increased functional thiol reserves and consequent potentiation of activation of this GSH-dependent chemotherapeutic prodrug. They further suggest that mitochondrial Bcl-2 constitutes the relevant pool of this protein for this phenomenon.Fig. 2. Correlation of GSH content (10-15 mol/cell (1)), relative Bcl-2 content (3), and IC50 (nM) for NCS (2) in three neural tumor lines in culture.
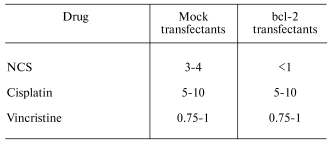
Exploiting these mechanisms in the therapy of tumors established from bcl-2-overexpressing, chemotherapy-resistant cells. Nude mice (n = 4 for each transfectant at each NCS dose) were administered subcutaneous injections of mock- or bcl-2-transfected PC12 cells. Subcutaneous implants of bcl-2-transfected PC12 cells more readily generate gross tumors than their mock-transfected counterparts. That is, the tumors, that form from them, are palpable earlier and ultimately larger than those formed from implanted mock-transfected PC12 cells. Nonetheless, as is the case for in vitro treatment of bcl-2- and mock-transfected PC12 cells, implants from bcl-2-transfected cells are more susceptible to NCS (0-5 mg/kg)-induced inhibition of tumor growth rate than are implants from mock-transfected cells (table).Fig. 3. Effects of an antisense construct to bcl-2 on Bcl-2 content (Western blot (a)) and sensitivity to NCS in bcl-2-transfected PC12 cells in culture (b-d). Bcl-2 content decreases after antisense treatment. a) Antisense treatment also results in decreased sensitivity to NCS treatment (b) as compared to vehicle (c) or bcl-2 sense construct (d) treatment. NCS concentration (ng/ml): 0) 0; 1) 100; 2) 200; 3) 300; 4) 400; 5) 500.
IC50 (mg/kg body mass) for day 30 tumor size reduction
Note: Tumor growth was assessed in 5-7-week-old male NIH athymic mice
injected subcutaneously with 106 mock- or bcl-2-transfected
PC12 cells into the left flank on day 0 of each study. Either NCS
(0-5 mg/kg), or cisplatin (0-5 mg/kg), or vincristine
(0-1.25 mg/kg) was administered intraperitoneally on day 1
(n = 4 for each dose). Mice were examined daily for grossly
visible tumor, and, once tumors appeared, they were assessed by
measurement of the largest and smallest diameter of each tumor at
multiple time points during the month following implantation. Tumor
volume was calculated as the product of the largest diameter and the
square of the smallest diameter. Mean tumor volume for the four mice in
each group was calculated for each day of measurement assuming a tumor
volume of 0 for those mice in which a tumor was not palpable. For each
treatment group, the IC50 for day 30 tumor size reduction
was defined as the drug dose at which the day 30 mean tumor volume was
half that of the simultaneously saline-injected group. In most cases,
the IC50 falls between two actually administered doses; it
is therefore expressed in the table as the dose range between which it
falls. Saline-injected mice were used for control [29].
An analogous study performed with intraperitoneal administration of cisplatin (0-25 mg/kg) demonstrated comparable depression of tumor growth rate in murine xenografts of mock- and bcl-2-transfected PC12 cells (table). At doses of cisplatin below 10 mg/kg, no depression of tumor growth rate was seen in either xenografted transfectant. In no case was Bcl-2-mediated potentiation of the anti-tumor effect seen. At doses of cisplatin above 10 mg/kg, lethal toxicity of the drug, evident before tumors were grossly detectable in control animals, precluded evaluation of the effect of higher-dose cisplatin on tumor growth.
While NCS is a cell cycle phase-specific, proliferation-dependent drug, cisplatin is cell cycle-non-specific and proliferation-independent. To exclude the possibility that the difference in in vivo sensitivity to NCS between mock- and bcl-2-transfected PC12 cells was merely the result of the faster growth rate of tumors in the latter transfectants, we also examined tumor growth in xenograft-bearing nude mice treated with vincristine (0-1.25 mg/kg), another cell cycle-specific, proliferation-dependent chemotherapeutic agent. As was the case for cisplatin, and in contrast to the case for NCS, mock- and bcl-2-transfected PC12 cell xenografts exhibited comparable sensitivity to vincristine (table). This makes it unlikely that differential sensitivity to NCS is merely the result of the more rapid cycling of bcl-2-transfected cells.
Neuroblastoma is the single most common solid tumor of childhood. It arises from those cells of the neural crest that become the sympathetic component of the peripheral nervous system. With currently available combinations of surgery, radiation, and chemotherapy, children with metastatic neuroblastoma have only a 20% probability of long-term survival [36]. Recent attempts at less conventional therapies involve the use of autologous bone marrow transplantation with or without prior marrow “purging” to remove residual tumor cells. Currently available data suggest that it prolongs survival somewhat, but does not alter ultimate survival figures [37]. The dismal prognosis for many children with neuroblastoma and the relatively common nature of this disease make imperative the development of new therapeutic approaches to this tumor.
Resistance to chemotherapy and prohibitive toxicity to normal tissues of otherwise effective chemotherapy constitute the two most difficult problems in treating cancer in general and neuroblastoma in particular. Targeted strategies that exploit the particular characteristics of the tumor cell and cell type hold the promise of overcoming both of these issues. In the case of patients with neuroblastoma, taking aim at the neurotransmitter receptors on the surface of these cells, and overcoming Bcl-2-mediated chemotherapeutic resistance may change the poor prognosis for the majority of these patients.
The studies described in this review were funded by the National Cancer Institute (CA74289), the National Institute of Neurological Disease and Stroke (NS38569 and NS41297), and the Carol Ann Craumer Endowment of Children's Hospital of Pittsburgh.
REFERENCES
1.Schor, N. F. (1999) J. Neuro-Oncol.,
41, 159-166.
2.Chelmicka-Szorc, E., and Arnason, B. G. W. (1976)
Cancer Res., 36, 2382-2384.
3.Reynolds, C. P., and Perez-Polo, J. R. (1981)
Neurosci. Lett., 26, 151-155.
4.Schor, N. F. T. (1987) Cancer Res.,
47, 5411-5414.
5.Thoenen, H., and Tranzer, J. P. (1968)
Naunyn-Schmiedebergs Arch. Exp. Pathol. Pharmacol., 261,
271-288.
6.Purpura, P., Westman, L., Will, P., Eidelman, A.,
Kagan, V. E., Osipov, A. N., and Schor, N. F. (1996) Cancer
Res., 56, 2336-2342.
7.Chateauneuf, J., Lusztyk, J., and Ingold, K. U.
(1988) J. Org. Chem., 53, 1629-1632.
8.Samuni, A., Mitchell, J. B., Krishna, C. M.,
Samuni, U., and Russo, A. (1991) Free Rad. Res. Commun.,
12/13, 197-194.
9.Samuni, A., Winkelsberg, D., Pinson, A., Hahn, S.
M., Mitchell, J. B., and Russo, A. (1991) J. Clin. Invest.,
87, 1526-1530.
10.Hahn, S. M., Tochner, Z., Murali Krishna, C.,
Glass, J., Wilson, L., Samuni, A., Sprague, M., Venzon, D., Glatstein,
E., Mitchell, J. B., and Russo, A. (1992) Cancer Res.,
52, 1750-1753.
11.Dole, M., Nunez, G., Merchant, A. K., Maybaum,
J., Rode, C. K., Bloch, C. A., and Castle, V. P. (1994) Cancer
Res., 54, 3253-3259.
12.Dole, M., Jasty, R., Cooper, M. J., Thompson, C.
B., and Nunez, G. (1995) Cancer Res., 55, 2576-2582.
13.Kane, D. J., Sarafian, T. A., Anton, R., Hahn,
H., Gralla, E. B., Valentine, J. S., Ord, T., and Bredesen, D. E.
(1993) Science, 262, 1274-1277.
14.Albrecht, H., Tschopp, J., and Jongeneel, C. V.
(1994) FEBS Lett., 351, 45-48.
15.Tyurina, Y. Y., Tyurin, V. A., Carta, G., Quinn,
P. H., Schor, N. F., and Kagan, V. E. (1997) Arch. Biochem.
Biophys., 344, 412-413.
16.Schor, N. F., Rudin, C. M., Hartmann, A.-R.,
Thompson, C. B., Tyurina, Y. Y., and Kagan, V. E. (2000)
Oncogene, 19, 472-476.
17.Ratan, R. R., Murphy, T. H., and Baraban, J. M.
(1994) J. Neurosci., 14, 4385-4392.
18.Sandstrom, P. A., Mannie, M. D., and Buttke, T.
M. (1994) J. Leukoc. Biol., 55, 221-226.
19.Forrest, V. J., Kang, Y. H., McClain, D. E.,
Robinson, D. H., and Ramakrishnan, N. (1994) Free Rad. Biol.
Med., 16, 675-684.
20.Yang, J., Liu, X., Bhalla, K., Kim, C. N.,
Ibrado, A. M., Cai, J., Peng, T.-I., Jones, D. P., and Wang, X. (1997)
Science, 275, 1129-1132.
21.Kluck, R. M., Bossy-Wetzel, E., Green, D. R., and
Newmeyer, D. D. (1997) Science, 275, 1132-1136.
22.Cortazzo, M. H., and Schor, N. F. (1996)
Cancer Res., 56, 1199-1203.
23.Hartsell, T. L., Yalowich, J. C., Ritke, M.,
Martinez, A. J., and Schor, N. F. (1995) J. Pharm. Exp. Ther.,
275, 479-485.
24.Hartsell, T. L., Hinman, L. M., Hamann, P. R.,
and Schor, N. F. (1996) J. Pharm. Exp. Ther., 277,
1158-1166.
25.Beerman, T. A., Poon, R., and Goldberg, I. H.
(1977) Biochim. Biophys. Acta, 475, 294-306.
26.DeGraff, W. G., and Mitchell, J. B. (1985)
Cancer Res., 45, 4760-4762.
27.Schor, N. F. (1992) J. Clin. Invest.,
89, 774-781.
28.Schor, N. F., Tyurina, Y. Y., Fabisiak, J. P.,
Tyurin, V. A., Lazo, J. S., and Kagan, V. E. (1999) Brain Res.,
831, 125-130.
29.Liang, Y., Nylander, K. D., and Schor, N. F.
(2002) Mol. Pharmacol., 61, 142-149.
30.Steinman, H. M. (1995) J. Biol. Chem.,
270, 3487-3490.
31.Hubel, C. A., Kagan, V. E., Kisin, E. R.,
McLaughlin, M. K., and Roberts, J. M. (1997) Free Rad. Biol.
Med., 23, 596-609.
32.Stoyanovsky, D. A., Goldman, R., Jonnalagadda, S.
S., Day, B. W., Claycamp, H. G., and Kagan, V. E. (1996) Arch.
Biochem. Biophys., 330, 3-11.
33.Langmuir, M. E., Yang, J.-R., LeCompte, K. A.,
and Durand, R. E. (1996) Fluorescence Microscopy and Fluorescent
Probes, Plenum Press, N. Y., pp. 229-234.
34.Liang, Y., Yan, C., and Schor, N. F. (2001)
Oncogene, 20, 6570-6578.
35.DeGraff, W. G., Russo, A., and Mitchell, J. B.
(1985) J. Biol. Chem., 260, 8312-8315.
36.Bonilla, M. A., and Cheung, N.-K. V. (1994)
Cancer Invest., 12, 644-653.
37.Johnson, F. L., and Goldman, S. (1993)
Hematol. Oncol. Clin. N. Amer., 7, 647-662.