
|
Modality of Cell Death Induced by Foscan®-Based Photodynamic Treatment in Human Colon Adenocarcinoma Cell Line HT29S. Marchal, L. Bezdetnaya*, and F. GuilleminUnite de Recherche en Therapie Photodynamique, Centre Alexis Vautrin, 54511 Vandoeuvre-les-Nancy, Cedex, France; fax: (33) 3-83-44-76-35; E-mail: l.bolotine@nancy.fnclcc.fr* To whom correspondence should be addressed.
|
Received April 30, 2003
Apoptosis induced by photodynamic therapy (PDT) is considered to be an important factor defining the treatment outcome. Nevertheless, the relevance of apoptotic events in overall cell death should be established for every given photosensitizer. The present study addresses the contribution of Foscan® (meta-tetra(hydroxyphenyl)chlorine; mTHPC) photosensitized apoptosis in overall cell death in a model of cultured HT29 adenocarcinoma cells. Early events of cell death were assessed by the evaluation of mitochondrial response to mTHPC-mediated PDT, cytochrome c release and membrane depolarization. Apoptosis was measured through the activity of caspase-3 and the binding of the fluorescent conjugate Ca2+-dependent protein Annexin-V on membrane externalized phosphatidylserine at 2, 4, and 24 h post-PDT. Immediately after mTHPC-PDT, from 28 to 57% cells exhibited cytochrome c release concomitantly with mitochondrial membrane depolarization for light doses inducing more than 90% overall cell death. The maximum of caspase-3 activation (12-fold more than control) was reached 24 h after irradiation at fluence inducing 90% cell death (LD90). The corresponding measurement of apoptotic cells (12% of Annexin-V bound cells) confirmed the mild and delayed apoptotic response of HT29 cells to mTHPC-PDT.
KEY WORDS: photodynamic therapy, mTHPC, necrosis, apoptosis, cell death
Photodynamic therapy (PDT) is a promising therapeutic strategy for the treatment of superficial, in situ and micro-invasive tumors. PDT is based on the administration of tumor-localizing chemicals (photosensitizers) with successive non-ionizing illumination of the tumor bed. PDT induces localized tumor destruction via the photochemical generation of cytotoxic singlet oxygen (1O2). Two principal mechanisms should be considered in PDT-mediated tumor eradication: direct tumor cell damage via necrotic or apoptotic pathway and microvascular injury.
The ability of PDT to generate apoptotic cells is acknowledged to be an important factor in the PDT treatment efficacy. The PDT-induced apoptotic response is characterized by a rapid cytochrome c release from the mitochondria leading to the inhibition of respiration [1], the activation of caspase-3, which is a key player in the execution phase of apoptosis [2], and chromatin condensation as early 1-2 h after irradiation [3-7]. Apoptosis has been also detected in tumor bearing mice after PDT [8].
Foscan® (meta-tetra(hydroxyphenyl)chlorine; mTHPC) is a second generation photosensitizer with favorable photochemical and immunological properties [9-11]. Presently, mTHPC is applied for the head and neck cancers [12] and has been studied for basal cell carcinoma [13] and locoregional breast cancer recurrences [14].
Despite that mTHPC has been considered as one of the most active photosensitizers studied to date [15], there are only a few studies on mTHPC-photosensitized modalities of cell death. Two recent studies of Chen et al. [3, 16] reported the rapid release of cytochrome c, chromatin condensation, and DNA cleavage shortly after mTHPC photosensitization of two leukemic cell lines, thus assuming an extensive apoptosis.
The present article focuses on the mitochondrial events after HT29 cells photosensitization with mTHPC. Early events of apoptosis were assessed by a light dose response analysis of cytochrome c release from mitochondria, depolarization of the mitochondrial membrane, activity of caspase-3, and the binding of the fluorescent conjugate Ca2+-dependent protein Annexin-V (Ann-V) on membrane externalized phosphatidylserine.
MATERIALS AND METHODS
Chemicals. mTHPC was kindly supplied by Biolitec Pharma Ltd. (UK). mTHPC stock solution was in methanol. Further dilution was performed in phenol red free RPMI 1640 medium supplemented with 2% fetal calf serum to reach a final mTHPC concentration of 1.5 µM. Phycoerythrin-conjugated Annexin-V (PE-Ann-V), caspase-3 fluorogenic substrate DEVD-7-amino-4-trifluoromethylcoumarin (AFC), and caspase-3 inhibitor DEVD-CHO were obtained from BD Pharmingen (USA). SYTOX-->Green was provided by Molecular Probes (The Netherlands). RPMI 1640 medium, glutamine/penicillin/streptomycin, trypsin-EDTA, and phosphate-buffered saline solutions (PBS) were purchased from Life Technologies (USA). Fetal calf serum (FCS) was supplied from Costar France (France). All other chemicals were obtained from Sigma Chemical Co. (USA).
Cell culture. HT29 human adenocarcinoma cells were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated FCS, 1% penicillin (10,000 IU), streptomycin (10,000 µg/ml), and 1% of 200 mM glutamine. Cells were kept at 37°C in a 5% CO2 humidified atmosphere, trypsinized, and reseeded into fresh medium every 7 days. Four days before treatment, 3*104 cells/ml were seeded in a 25-cm2 flask.
Photodynamic treatment. Logarithmically growing HT29 cells were incubated with 1 µg/ml mTHPC solution in RPMI supplemented with 2% FCS for 3 h. After two consecutive washings, cells were irradiated (650 nm) at fluences ranging from 0.06 to 1.92 J/cm2.
Clonogenic assay. Cells were collected from the 25-cm2 flask immediately after PDT and seeded in triplicate into 6-well plates according to a technique previously described [17]. Briefly, a layer consisting of 1 ml of 0.5% molten agar (Bacto agar, Difco, USA) in culture medium was poured in each well. Over this bottom layer, 103 cells were plated in 1 ml culture medium containing 0.3% agar. Cultures were incubated at 37°C with 5% CO2 in air for 14 days. Colonies composed of more than 50 cells were counted with an automatic image analysis program AnalySiS 3.1 (AnalSys, France).
Measurements of the membrane potential DeltaPsim. The green fluorescent probe 5,5´,6,6´-tetrachloro-1,1´,3,3´-tetraethylbenzimidazolylcarbocyanine iodide, JC-1 (Molecular Probes), exists as a monomer at low membrane potential and, at higher potentials, JC-1 forms red fluorescent aggregates. The use of this probe in the measurement of DeltaPsim after PDT has been described previously [18]. Trypsinized cells (5*105 cells) were centrifuged at 400g, the cell pellet was resuspended in 1 ml medium containing 1 µl of JC-1 (final concentration 5 µg/ml), and the resulting suspension was measured by flow cytometry (FACSCalibur) after a 15-min incubation at 37°C. Aggregate fluorescence (lambdaex 488 nm, lambdaem 590 nm), detected in fluorescence channel FL2 with a 585 ± 42 nm band pass, can easily be separated from monomer fluorescence (lambdaex 488 nm, lambdaem 527 nm) detected in fluorescence channel FL1 with a 530 ± 30 nm band pass.
Measurements of cytochrome c release. Cytochrome c release was estimated by labeling digitonin-permeabilized cells with APO2.7 phycoerythrin (PE)-conjugated monoclonal antibody (Beckman Coulter, France) [19]. Trypsinized ((0.5-1)*106) cells were permeabilized with 0.1 µg/ml digitonin for 20 min at 4°C, washed, and labeled with PE-APO2.7 (dilution 1 : 5) for 15 min at room temperature. After washing, cells were measured by flow cytometry using excitation at 488 nm and fluorescence emission at 585 ± 42 nm (FL2).
Quantification of caspase-3 activity. For each experimental point, cells obtained from three flasks or spheroids obtained from three Petri dishes were collected by scraping and added to the supernatant. After centrifugation, the pellet was washed twice in PBS and stored at -20°C until use. The dry pellet was resuspended in 1 ml lysis buffer composed with 10% sucrose, 20 mM Hepes, 0.1% Chaps, 2 mM dithiothreitol, 1 mM EDTA, 1 µg/ml pepstatin, 1 µg/ml leupeptin, and 100 µg/ml phenylmethylsulfonyl fluoride (PMSF), pH 7.4. The cell lysate was incubated on ice for 30 min, sonicated twice for 10 sec, and centrifuged (10 min, 10,000g). The supernatant (800 µl) was incubated with 200 µl (6 µM) of highly specific caspase-3 substrate--DEVD-AFC (Asp-Glu-Val-Asp (DEVD) conjugated with 7-amino-4-trifluoromethylcoumarin (AFC))--in lysis buffer at 37°C for 1 h. The specificity of caspase-3 activity was verified in the reaction of enzymatic activity inhibition by adding 0.05 µM DEVD-CHO for 2 h. The released fluorescent product was measured spectrofluorimetrically (lambdaex/lambdaem 400/450-550 nm; Perkin-Elmer L225 9051, USA). The obtained data were normalized per 1 mg protein. Protein concentrations were estimated in cell lysates by using the Bio-Rad assay. The final results were expressed as the ratio between the experimental and the control (drug, no light) normalized values.
Apoptotic cell fractions assessed by flow cytometry. The use of fluorescein isothiocyanate-labeled Annexin-V (FITC-Ann-V) and propidium iodide (PI) to evaluate apoptotic and necrotic cells, respectively, in suspension of growing cells is now a common practice [20]. A modification of this method was employed in the present study. Namely, an overlap in fluorescence bands between PI (lambdaem 620 nm) and mTHPC (lambdaem 650 nm) was avoided by staining of necrotic cells by SYTOX-->Green probe (lambdaem 523 nm). High-affinity nucleic acid probe SYTOX-->Green easily penetrates cells with compromised plasma membranes and does not cross the membranes of living cells. Apoptotic cells were measured by labeling of membrane externalized phosphatidylserine with PE-Ann-V (lambdaem 575 nm). Culture medium containing floating cells and trypsinized cells were centrifuged and counted. SYTOX-->Green (40 nM) in 100 µl RPMI 1640 was added to 105 cells for 15 min at 4°C. Cells were then washed with RPMI and resuspended in 100 µl binding buffer (10 mM Hepes, 140 mM NaCl, 2.5 mM CaCl2, pH 7.4), and 5 µl of PE-Ann-V was added for 15 min at 4°C. Before flow cytometry analysis, the cell suspension was diluted with 400 µl binding buffer. Fluorescence emission of PE-Ann-V and SYTOX-->Green after 488 nm laser excitation was detected in channel FL2 (585/42 nm bandpass) and channel FL1 (530/30 nm bandpass), respectively, on a FACSCalibur flow cytometer using a compensation setting (FL2-%FL1, 45%).
RESULTS
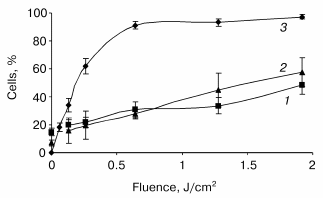
Photoinduced cytochrome c release and mitochondrial membrane depolarization. Figure 1 displays the fluence dependent cytochrome c (cyt c) release and alterations of DeltaPsim immediately after irradiation. These data are plotted with the photocytotoxicity curve. At fluences above 0.64 J/cm2, that induced more than 90% cell death, we observed a progressive increase in the number of cells exhibiting both the leakage of cyt c and loss of DeltaPsim. At the highest fluence applied, cyt c escaped from mitochondria of about 50% of cells with a concomitant dissipation of the mitochondrial membrane potential.
Assessment of PDT induced apoptotic features in monolayer HT29 cell line. Caspase-3 activation is considered as a key element in the apoptosis execution program. The post-PDT kinetics of caspase-3 mediated proteolytic cleavage of DEVD motif were assessed at fluences corresponding to 60% (LD60), 90% (LD90), and 97% (LD97) cell photokilling as measured by clonogenic assay. An increase in enzymatic activity of caspase-3 was observed during the whole 24 h post-PDT incubation period with the better caspase-3 activation (ten- and twelve-fold above the level of untreated cells) for the lower fluences of 0.24 and 0.64 J/cm2 (Fig. 2). For these fluences, a caspase-3 activity reached a maximum 24 h after PDT. The kinetic profiles were however different. A sharp increase in caspase activity within the first 4 h followed by its slight variations was registered for the fluences corresponding to LD90 and LD97, whereas the lowest fluence demonstrated a progressive increase in enzymatic activity. The addition of 0.05 µM DEVD-CHO for 2 h completely inhibited the enzymatic reaction (data not shown).Fig. 1. Post-irradiation effect on mitochondrial membrane depolarization (1), cytochrome c release (2), and loss of clonogenicity (3) in mTHPC photosensitized HT29 cells. Data were obtained immediately after irradiation. The percentages of cells exhibiting the loss of clonogenicity were counted 15 days after PDT. Results are the mean ± SEM of at least three independent experiments.
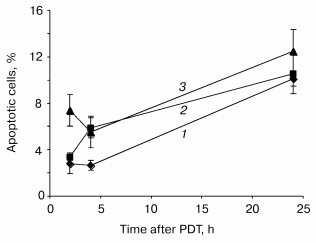
Disruption of the membrane phospholipid asymmetry with subsequent externalization of phosphatidylserine has been identified as one of the early and prominent features of apoptosis. Quantification of cells with membrane phosphatidylserine externalization was assessed by flow cytometry techniques. About 3% of control cells (drug, no light) were labeled with PE-Ann-V. Following treatment, a maximum of apoptotic cells was registered 24 h post-PDT whatever fluence used (Fig. 3). Similar to caspase-3 activation, we did not observe a relationship between number of apoptotic cells and light fluences.Fig. 2. Relative expression of protease activity in mTHPC photosensitized HT29 cells. Cells were exposed to 1.5 µM mTHPC for 3 h and irradiated at 0.24 (LD60, 1), 0.64 (LD90, 2), or 1.92 J/cm2 (LD97, 3) of red light (lambda = 652 nm). At the indicated times after PDT, cells were collected and lysed and 800 µl of protein extract were incubated with DEVD-AFC as described in “Materials and Methods”. Data are shown as the mean ± SEM from at least three independent experiments.
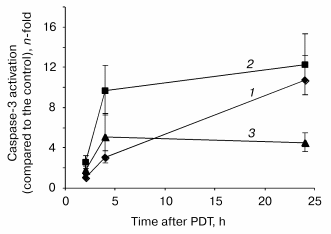
Fig. 3. Time course of apoptosis in response to PDT in HT29 cells. Cells were exposed to 1.5 µM mTHPC for 3 h and irradiated at 0.24 (LD60, 1), 0.64 (LD90, 2), or 1.92 J/cm2 (LD97, 3) of red light (lambda = 652 nm). At the indicated times after PDT, the cells were collected, stained with PE-Ann-V, and analyzed by flow cytometry. Data are shown as the mean ± SEM from at least three independent experiments.
DISCUSSION
Neoplastic tissues respond universally to PDT under appropriate conditions (high sensitizer concentration and light dose). However, the modalities of cell death provided by photodynamic treatment are still under investigation. Both apoptotic and necrotic cell death have been implicated in examples of photocytotoxicity with different sensitizers and cell lines.
Mitochondria play a key role in the pathways to cell death either by mitochondria proteins involved in the apoptotic process or by the loss of functionality resulting in ATP depletion [21]. Thus, mitochondrial damage is suspected to be the major cause of photocytotoxicity [22]. The earliest events that occur within seconds after PDT are the release of cyt c from the mitochondrial intermembrane space to the cytosol simultaneously [7, 23, 24], as observed in our study, or independently [18, 19] from the dissipation of the DeltaPsim.
Compared with the nearly complete release of cyt c about 15 min post phthalocyanine 4 (PC 4) photosensitization [4], mTHPC-PDT did not induce a massive release of cyt c immediately after irradiation (Fig. 1). This result suggests that mitochondria are probably not the primary lethal target of mTHPC-PDT. It is assumed that the initial cell photodamage sites are very close to those of singlet oxygen formation and strictly related to the distribution of the photosensitizer in the cell. Our previous studies performed in adenocarcinoma cells indicated a diffuse intracellular distribution of mTHPC fluorescence outside the nucleus with a strong preference for endoplasmic reticulum and Golgi apparatus [25, 26].
Apoptosis was evaluated by measuring the caspase-3 activity and by the binding of Ann-V on externalized phosphatidylserine after mTHPC photosensitization. Several characteristics are noticeable in the post-PDT activation of caspase-3 (Fig. 2). First, the level of caspase-3 activation (about 12-fold above the control level) is consistent with values reported in a response to PDT in adherent cell lines such as HCT-116, HeLa, MCF-7c3 [5, 27, 28], and is considerably lower compared to PDT-treated lymphocytic cells (more than 100-fold above the control level) [4, 6, 29]. This difference could arise from highly expressed constitutive caspase-3 in lymphocytic cells [2] and could partially explain an extreme sensitivity of leukemic cells to apoptotic cell death opposite to human tumor adherent cell line. Further, the lowest level of DVDase activation at the highest fluence applied could be indicative of the inhibiting of apoptotic process in favor of necrosis [6, 30]. The rapid shift from apoptotic to necrotic death may arise from an excess of oxidative damage [15, 31, 32], and/or the complete abrogation of cellular energy metabolism [33]. Another particularity observed in our study is the delayed activation of caspase-3 (4-24 h) that is not consistent with typical for PDT rapid apoptotic program execution. The moderate caspase activation and delayed apoptosis following PDT was demonstrated in Bcl-2 or Bcl-xL transfected HeLa and HL-60 cells photosensitized with verteporfin (BPD-MA) [34]. Since HT29 cells were reported to contain high levels of Bcl-xL protein [35], the latter might exert a repressive role on the apoptotic process induced by mTHPC photosensitization in HT29 cells.
The quantification of apoptotic cells by Ann-V binding on membrane externalized phosphatidylserine reveals a maximum of 12% of apoptotic cells 24 h after mTHPC photosensitization (Fig. 3). These results are consistent with the delayed and low activation of the caspase-3 mentioned above.
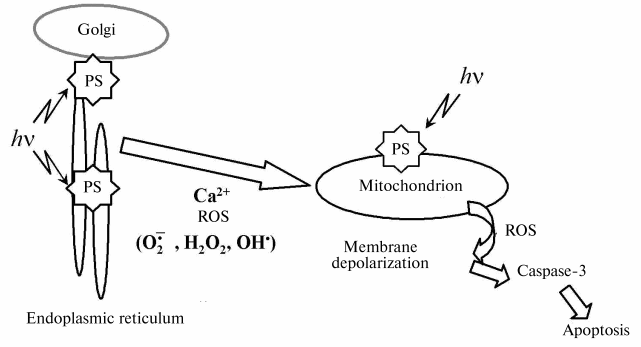
In conclusion, we have demonstrated that mitochondrial membrane damage is strongly implicated in an apoptotic cell death mediated by mTHPC-PDT. Figure 4 outlines the central role played by mitochondria in photodynamic-induced apoptotic pathway.
This work was supported by Alexis Vautrin Cancer Center Research Funds, French Ligue Nationale Contre le Cancer. We gratefully acknowledge Biolitec Pharma Ltd. for providing the mTHPC.Fig. 4. Mitochondrial events following photosensitization (ROS, reactive oxygen species; PS, photosensitizer).
REFERENCES
1.Varnes, M. E., Chiu, S. M., Xue, L. Y., and
Oleinick, N. L. (1999) Biochem. Biophys. Res. Commun.,
255, 673-679.
2.Cohen, G. M. (1997) Biochem. J., 326,
1-16.
3.Chen, J. Y., Mak, N. K., Yow, C. M., Fung, M. C.,
Chiu, L. C., Leung, W. N., and Cheung, N. H. (2000) Photochem.
Photobiol., 72, 541-547.
4.Chiu, S., Evans, H. H., Lam, M., Nieminen, A., and
Oleinick, N. L. (2001) Cancer Lett., 165, 51-58.
5.Granville, D. J., Carthy, C. M., Jiang, H., Shore,
G. C., McManus, B. M., and Hunt, D. W. (1998) FEBS Lett.,
437, 5-10.
6.He, J., Whitacre, C. M., Xue, L. Y., Berger, N. A.,
and Oleinick, N. L. (1998) Cancer Res., 58, 940-946.
7.Kessel, D., and Luo, Y. (1999) Cell Death
Differ., 6, 28-35.
8.Colussi, V. D., Feyes, D. K., Mulvihill, J. W., Li,
Y. S., Kenney, M. E., Elmets, C. A., Oleinick, N. L., and Mukhtar, H.
(1999) Photochem. Photobiol., 69, 236-241.
9.Melnikova, V. O., Bezdetnaya, L. N., Potapenko, A.
Y., and Guillemin, F. (1999) Radiat. Res., 152,
428-435.
10.Coutier, S., Bezdetnaya, L., Marchal, S.,
Melnikova, V., Belitchenko, I., Merlin, J. L., and Guillemin, F. (1999)
Br. J. Cancer, 81, 37-42.
11.Bonnett, R., White, R. D., Winfield, U.-J., and
Berenbaum, M. (1989) Biochem. J., 261, 277-280.
12.Biel, M., D'Cruz, A., and McCaffrey, T. (2002)
Proc. Asco., 38th Ann. Meet., Orlando, 379a.
13.Baas, P., Saarnak, A. E., Oppelaar, H., Neering,
H., and Stewart, F. A. (2001) Br. J. Dermatol., 145,
75-78.
14.Wyss, P., Schwarz, V., Dobler-Girdziunaite, D.,
Hornung, R., Walt, H., Degen, A., and Fehr, M. (2001) Int. J.
Cancer, 93, 720-724.
15.Dougherty, T. J., Gomer, C. J., Henderson, B. W.,
Jori, G., Kessel, D., Korbelik, M., Moan, J., and Peng, Q. (1998) J.
Natl. Cancer Inst., 90, 889-905.
16.Chen, J. Y., Mak, N. K., Wen, J. M., Leung, W.
N., Chen, S. C., Fung, M. C., and Cheung, N. H. (1998) Photochem.
Photobiol., 68, 545-554.
17.Merlin, J. L., Azzi, S., Lignon, D., Ramacci, C.,
Zeghari, N., and Guillemin, F. (1992) Eur. J. Cancer,
28A, 1452-1458.
18.Chiu, S. M., and Oleinick, N. L. (2001) Br. J.
Cancer, 84, 1099-1106.
19.Carthy, C. M., Granville, D. J., Jiang, H., Levy,
J. G., Rudin, C. M., Thompson, C. B., McManus, B. M., and Hunt, D. W.
(1999) Lab. Invest., 79, 953-965.
20.Van Engeland, M., Nieland, L. J. W., Ramaekers,
F. C. S., Schutte, B., and Reutelingsperger, C. P. M. (1998)
Cytometry, 31, 1-9.
21.Bernardi, P., Scorrano, L., Colonna, R.,
Petronilli, V., and Di Lisa, F. (1999) Eur. J. Biochem.,
264, 687-701.
22.Morgan, J., and Oseroff, A. R. (2001) Adv.
Drug Deliver. Rev., 49, 71-86.
23.Vantieghem, A., Xu, Y., Declercq, W.,
Vandenabeele, P., Denecker, G., Vandenheede, J. R., Merlevede, W., de
Witte, P. A., and Agostinis, P. (2001) Photochem. Photobiol.,
74, 133-142.
24.Lam, M., Oleinick, N. L., and Nieminen, A. L.
(2001) J. Biol. Chem., 276, 47379-47386.
25.Teiten, M. H., Bezdetnaya, L., Morliere, P.,
Santus, R., and Giuillemin, F. (2003) Br. J. Cancer, 88,
146-152.
26.Melnikova, V. O., Bezdetnaya, L. N., Bour, C.,
Festor, E., Gramain, M. P., Merlin, J. L., Potapenko, A., and
Guillemin, F. (1999) J. Photochem. Photobiol. B, 49,
96-103.
27.Xue, L. Y., Chiu, S. M., and Oleinick, N. L.
(2001) Exp. Cell Res., 263, 145-155.
28.Matroule, J. Y., Carthy, C. M., Granville, D. J.,
Jolois, O., Hunt, D. W. C., and Piette, J. (2001) Oncogene,
20, 4070-4084.
29.Kessel, D., Luo, Y., Mathieur, P., and Reiners,
J. J., Jr. (2000) Photochem. Photobiol., 71, 196-200.
30.Luo, Y., and Kessel, D. (1997) Photochem.
Photobiol., 66, 479-483.
31.Vantieghem, A., Assefa, Z., Vandenabeele, P.,
Declercq, W., Courtois, S., Vandenheede, J. R., Merlevede, W., de
Witte, P., and Agostinis, P. (1998) FEBS Lett., 440,
19-24.
32.Dellinger, M. (1996) Photochem.
Photobiol., 64, 182-187.
33.Noodt, B. B., Rodal, G. H., Wainwright, M., Peng,
Q., Horobin, R., Nesland, J. M., and Berg, K. (1998) Int. J.
Cancer, 75, 941-948.
34.Granville, D. J., Jiang, H., An, M. T., Levy, J.
G., McManus, B. C., and Hunt, D. W. C. (1998) FEBS Lett.,
422, 151-154.
35.Merchant, A. K., Loney, T. L., and Maybaum, J.
(1996) Oncogene, 13, 2631-2637.