
|
REVIEW: Enhancement of Nitric Oxide and Superoxide Generations by alpha-Tocopheryl Succinate and Its Apoptotic and Anticancer EffectsK. Fukuzawa*, K. Kogure, M. Morita, S. Hama, S. Manabe, and A. TokumuraFaculty of Pharmaceutical Sciences, Tokushima University, Tokushima 770-8505, Japan; fax: +81-88-633-9572; E-mail: fukuzawa@ph.tokushima-u.ac.jp* To whom correspondence should be addressed.
|
Received April 30, 2003
Tocopheryl succinate (TS), a succinyl ester of alpha-tocopherol (alpha-T), has been reported to have various biological activities. In this communication, we review the current findings about TS including our recent studies of its effects on nitric oxide (NO) and superoxide (O2-) generations implicated in cancer and atherosclerosis. First, we investigated the effect of TS on NO production in vascular smooth muscle cells (VSMC) under atherosclerosis-like conditions using lipopolysaccharide (LPS) and interferon-alpha (IFN). TS enhanced LPS/IFN-dependent NO production, but alpha-T itself did not. The enhancement by TS of NO production was inhibited by alpha-T but not by antioxidants such as ascorbic acid and 2[3]-t-butyl-4-hydroxyanisole (BHA). TS enhanced the amount of protein kinase Calpha (PKCalpha) in VSMC, and PKC inhibitors inhibited TS-enhanced NO production, suggesting that the enhancing effect of TS on NO production is caused by up-regulation of PKC. Second, we found that TS induced apoptosis in VSMC associated with increase in O2- generation via NADPH-dependent oxidase. We further observed that a mouse breast cancer cell line C127I was more susceptible for TS-induced apoptosis than a mouse breast normal cell line NmuMG, and that superoxide dismutase, alpha-T, and BHA inhibited TS-caused morphological cell damage in C127I. From these results, O2- itself and/or other reactive oxygen species are assumed to associate with TS-induced cell toxicity, and antioxidative defense systems are supposed to be lowered in cancer cells. Finally, we found that intravenous injection of TS vesicles completely inhibited the growth of melanoma cells B16-F1 inoculated on the back of hairless mice and enhanced their survival time.
KEY WORDS: alpha-tocopheryl succinate, vitamin E, nitric oxide, superoxide, apoptosis, atherosclerosis, cancer cells, anticancer drug
Vitamin E (alpha-tocopherol) is a well-known lipid-soluble antioxidant. Recently, unlike the redox-active alpha-tocopherol (alpha-T), the redox-inactive alpha-tocopheryl succinate (TS), an amphiphilic succinyl ester of alpha-T (see Fig. 1) has attracted much attention [1]. TS is a naturally occurring compound first isolated from a green barley extract that stimulates release of prolactin and growth hormone from pituitary cells in vitro [2]. Later, various biological activities of TS such as inactivation of transcriptional factor nuclear factor kappaB (NF-kappaB) [3-6], suppression of the cell growth [7-13], induction of apoptosis [14-30], promotion of differentiation [31-33], and cell cycle arrest [10, 13] were reported. In this communication, we review our current understanding of the molecular basis for the effects of TS implicated in our recent findings about it [34-37]: 1) TS enhanced NO production by increasing inducible NO synthase (iNOS) in vascular smooth muscle cells (VSMC) under atherosclerosis-like conditions [34]; 2) TS induced apoptosis, which is associated with increase in O2- generation by NADPH-oxidase [35]; 3) cancer cells are more susceptible than normal cells to TS-induced apoptosis, possibly due to lowered antioxidative defense systems [36]; 4) TS inhibited the growth of melanoma in hairless mice in vivo and prolonged their survival time [37].
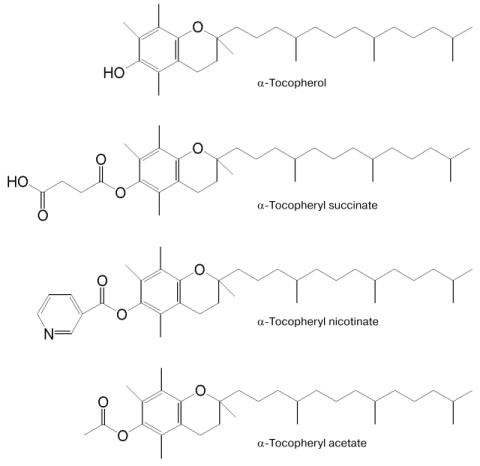
Fig. 1. Structures of alpha-tocopherol (alpha-T) and its derivatives--alpha-tocopheryl succinate (TS), alpha-tocopheryl acetate (TA), and alpha-tocopheryl nicotinate (TN).
ENHANCEMENT BY alpha-TOCOPHERYL SUCCINATE OF NITRIC OXIDE PRODUCTION INDUCED BY LIPOPOLYSACCHARIDE AND INTERFERON-gamma IN RAT VASCULAR SMOOTH MUSCLE CELLS
In VSMC, NO is known to play a critical role in vasodilatory function and atherosclerosis processes [38], and inducible NO production in the VSMC system has been well studied [39]. In atherosclerotic plaque, cytokines such as tumor necrosis factor-alpha (TNF-alpha) and interleukin-1 (IL-1) secreted from macrophages and foam cells have been implicated in the pathogenetic events [40]. On the other hand, since these cytokines are responsible for iNOS expression through NF-kappaB activation, they are supposed to elicit the NO-dependent vasodilation to improve the decreased blood flow in the vascular lesions. As TS was recently reported to induce NO production and iNOS expression in U937 human monoblasts through activation of NF-kappaB [41], we investigated in the VSMC system in detail for the effect of TS on NO production to obtain useful information about its therapeutic possibilities on vascular diseases like atherosclerosis. To investigate the enhancing effect of TS on NO production under atherosclerosis-like conditions, we used the system containing lipopolysaccharide (LPS) known as a stimulant of iNOS expression through similar signaling cascade to these cytokines [40, 42]. Interferon-gamma(IFN), which was also reported to be secreted from T-cells in the lesions of atherosclerosis, was further used together with LPS to strengthen LPS function.
Transfer of TS from Culture Medium to VSMC
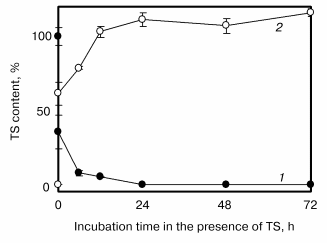
Prior to the main experiments, we investigated the transfer of TS (50 µM) from the culture medium to the cells using HPLC [35]. As shown in Fig. 2, TS contents in the cells increased biphasically with increase in the incubation period. The cellular contents of TS increased at first immediately up to about 60% just after addition of TS to culture medium and then gradually. Most of the TS treated was observed in the cell fraction 12 h after the addition, but its hydrolysis product, alpha-T, was not observed both in cultured medium and cells, indicating that TS was not hydrolyzed under our experimental conditions.
TS Enhances NO Production Induced by LPS and IFN in VSMCFig. 2. Time-dependent alpha-tocopheryl succinate (TS) transfer from culture medium to vascular smooth muscle cells [35]. TS contents in the medium (1) and cells (2) were quantified using HPLC. The concentration of TS in the culture medium at 0 time was 50 µM. Values are means ± SD (n = 3).
A typical condition of NO production in our experiments was as follows [34]. The VSMC (1*106 cells) isolated from rat were cultured for 24 h in medium with 10% fetal calf serum and further cultured for 24 h in medium containing TS without serum. Then LPS and IFN were added at final concentrations of 10 µg/ml and 100 U/ml, respectively, and after incubation for 48 h, cells were subjected to various assays. In the absence of LPS/IFN, 10 µM TS alone did not induce NO production, although Kim et al. [41] reported that TS itself induced NO production. In the presence of LPS/IFN, however, TS enhanced NO production about 2-fold that induced by LPS/IFN alone. Furthermore, TS enhanced the expression of LPS/IFN-induced iNOS protein measured by western blotting, as well as NO production. Accordingly, we concluded that the enhancing effect of TS on LPS/IFN-induced NO production is caused by enhancement of iNOS protein induction.
LPS and IFN have been reported to stimulate NO production through independent signal pathways. The former stimulates cells by activating NF-kappaB and the latter by the interferon regulatory factor-1 (IRF-1) [42-45]. Next, we examined which signal pathway of LPS or IFN was stimulated with TS. The enhancing effect of TS was not observed in the presence of IFN alone even at a high concentration. However, the enhancing effect increased with increase in the concentration of LPS alone but was relatively low compare with that in the presence of both LPS and IFN. These results suggest that TS participates in the pathway stimulated with LPS, and IFN is necessary for expansion of the TS effect.
alpha-T and its derivatives alpha-tocopheryl acetate (TA) and alpha-tocopheryl nicotinate (TN) (see Fig. 1), even at 50 µM, did not enhance the LPS/IFN-induced NO production in VSMC in contrast to TS. However, they lowered the TS-enhanced NO generation and iNOS expression induced by LPS/IFN. Antioxidants such as butylhydroxyanisole (BHA) and ascorbic acid (AsA) did not affect the TS-enhanced NO production, indicating that active oxygens and/or free radicals do not participate in the TS effect, and that the lowering effect of alpha-T is not mediated by its antioxidative reaction.
Implication of Protein Kinase for TS-Enhanced LPS/IFN-Induced NO Production in VSMC
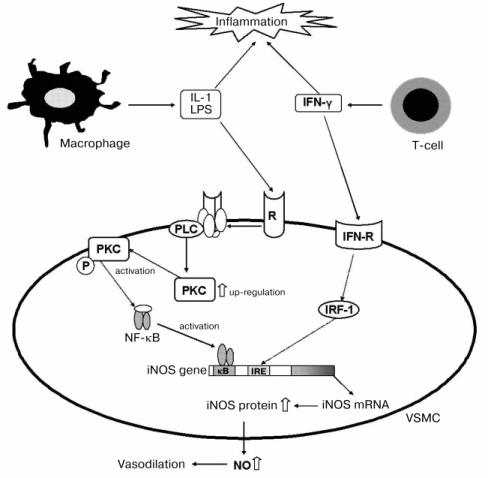
Since TS was suggested to affect mainly the signal pathway stimulated with LPS, we examined the effects of inhibitors of PKA and PKC on TS-enhanced NO production [34]. The PKA inhibitors KT-5720 and H8 had no effect, but the PKC inhibitors Ro31-2880 and GF109203X significantly inhibited NO production in the combination system of 10 µM TS with LPS/IFN, although they did not affect apparently LPS/IFN-induced NO production in the system without TS. Addition of 10 µM TS to the LPS/IFN-system enhanced the amount of PKCalpha, but no change of PKCalpha involved with and without TS was observed in the LPS/IFN treated cells. The amounts of other proteins related with the LPS-stimulated signal pathway, such as TRAF6 and MyD88, were not affected by the addition of TS. These results suggested that TS induced up-regulation of PKCalpha. This suggestion was supported by the findings that enhancements by TS of NO production and iNOS expression were inhibited by alpha-T, which was reported to decrease PKCalpha activity through activation of protein phosphatase-2A (PP2A) in smooth muscle cells [46-48]. From these results, we proposed the molecular mechanism in Fig. 3 of TS-enhanced NO generation by iNOS, expression of which was increased via increase in PKC in VSMC under the atherosclerotic inflammatory condition.
It is interesting that alpha-T and TS, having very similar structure, showed opposite effects on PKC in our study, although Neuzil et al. [1, 23, 24] reported inhibition by both TS and alpha-T of PKC activity through PP2A activation. From the recent reports that TS induced apoptosis by causing lysosomal membrane instability [49], and that alpha-T prevented the injurious effects and changes of signaling molecule functions through membrane alterations induced by bioactive phospholipids [50, 51], it was supposed that an amphiphilic TS with negatively charged terminal carboxyl moiety alters the membrane structure and the consequent up-regulation of PKC, and alpha-T stabilizes the TS-mediated membrane disruption.Fig. 3. Molecular mechanism of TS-enhanced NO production under atherosclerosis-like conditions using LPS and IFN [34]. White arrows indicate quantitative increase by TS. VSMC, vascular smooth muscle cells; PLC, phospholipase C; PKC, protein kinase C; PKC-P, activated protein kinase C; IFN, interferon-gamma; R, receptor; IFN-R, interferon receptor; IRF-1, interferon regulating factor-1.
SUPEROXIDE IS RESPONSIBLE FOR APOPTOSIS INDUCED BY
alpha-TOCOPHERYL SUCCINATE
It has been found that TS induces apoptosis in various cell lines such as human breast cancer cells, neuroblastoma cells, and lymphoblastoid cells [12, 14-27]. Multiple signaling pathways have been suggested to play a role in the TS-induced apoptosis. These may involve transforming growth factor-beta (TGF-beta), PKC, Fas, and the mitogen-activated protein kinase (MAPK) signalings [12, 14-19, 21, 26-30]. Furthermore, it was also reported that the dysfunction of mitochondria, lysosomal instability, and activation of caspase cascades were caused in the process of TS-induced apoptosis [19, 22, 23, 27, 49]. However, the trigger event of TS-induced apoptosis is still obscure. In this study, we examined the effect of TS on rat VSMC to obtain further information about the cell toxicity of TS.
Cytotoxic and Apoptotic Effects of TS on Cultured VSMC
The VSMC (1*106 cells) isolated from rat were cultured for 24 h in medium with 10% fetal calf serum, and further cultured in medium containing TS without serum for 48-96 h. Lactate dehydrogenase (LDH) release from VSMC, which is a typical parameter of cell toxicity, was slowly induced by TS in a concentration-dependent manner; on 96 h treatment, 50 µM TS caused about 60% release of total LDH [35]. TS also concentration- and time-dependently caused DNA fragmentation, which is known as a typical apoptotic event. The TS-induced LDH release and DNA fragmentation of VSMC were inhibited by exogenous addition of superoxide dismutase (SOD), but not by catalase, indicating that O2-, but not H2O2, would play an important role in the TS toxicity. alpha-T also inhibited the cytotoxic effect of TS but AsA did not.
Increased O2- Generation by NADPH-oxidase Is Responsible for TS-Induced Apoptosis
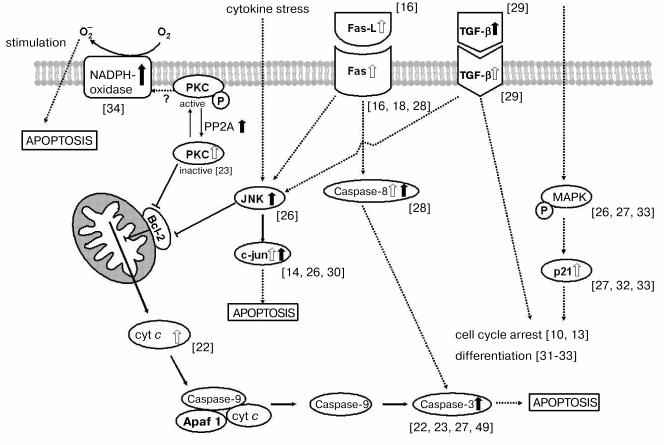
NADPH oxidase is well known to be an O2- generating system in various cells [52]. And O2- generated by NADPH oxidase is reported to contribute to apoptosis in various cells such as sympathetic neurons and human leukemia cells [53, 54]. Exogenous addition of NADPH in TS-treated VSMC caused significant increase in O2- generation measured by the cytochrome c reduction method. The reduction of cytochrome c was inhibited by SOD. Addition of diphenyleneiodonium (DPI) or 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF), which are specific inhibitors of flavin-containing oxidases such as NADPH oxidase, also inhibited the reduction of cytochrome c, although the inhibitions were not complete [35]. The coexistence of AEBSF also inhibited the LDH release and DNA fragmentation induced with TS. These results suggest that the exogenous O2- generation via NADPH-dependent flavin-containing oxidase system is responsible for the TS-induced cell toxicity. Figure 4 shows the multiple signaling pathways that play a role in the TS-induced apoptosis.
Fig. 4. Molecular mechanism of TS-triggered apoptosis via the TGF-beta, Fas, and JNK/MAPK signaling pathways and the mitochondrial membrane permeability transition pathway. White and black arrows indicate TS-induced increases in amount and activity, respectively. TGF-beta, transforming growth factor beta; PKC, protein kinase C; PP2A, protein phosphatase 2A; cyt c, cytochrome c; Fas-L, Fas-ligand; JNK, c-Jun-end kinase; c-Jun, transcription factor; MAPK, mitogen-activated protein kinase; Apaf, apoptotic proteinase-activating factor.
APOPTOTIC AND ANTICANCER EFFECTS OF alpha-TOCOPHERYL SUCCINATE
Cancer Cells Are More Susceptible to TS-Induced Cytotoxicity Than Normal Cells
Neuzil et al. reported that TS showed higher cell toxicity toward various cancer cells than normal cells [23, 24, 55]. However, the molecular mechanism of high susceptibility of cancer cells to TS-induced toxicity is unclear. Since TS induced apoptosis in VSMC is associated with increase in O2- generation via NADPH-oxidase as above mentioned, we investigated whether activation of O2- generation is also a trigger feature of TS-induced apoptosis in other cell systems, especially cancer cells, and why the toxicity of TS is high in cancer cells [36]. We used two cell lines derived from the same species of animal and tissue, a mouse breast cell lines NmuMG and a mouse breast cancer cell line C127I, and compared their susceptibility to TS. In cancer cells, the concentration of TS required for apoptosis was 50 µM, but in normal cells, a higher concentration (over 100 µM) was required. Exogenous addition of SOD (2000 units/ml) inhibited TS-induced apoptosis and increased survival of C127I cells, but catalase (2000 units/ml) did not. Microscopic images indicated that 50 µM TS-caused morphological C127I cell damage was prevented by addition of 50 µM alpha-T, 50 µM butylhydroxyanisole (BHA), and SOD. These results suggest that O2- itself and reactive oxygen species derived from O2- and/or free radicals are associated with TS toxicity, and the high susceptibility of cancer cells to TS toxicity is due to failure of their antioxidative defense systems.
Anticancer Effect of TS
We and others have shown that TS is a potent inducer of apoptosis in a multitude of malignant cells while it appears largely nontoxic towards normal cells in vitro [7, 12, 19, 23, 24, 36, 55]. We proposed that one possible cause was due to failure of antioxidative defense systems of cancer cells [36] as described above. Neuzil [1] postulated that because apoptosis induced by TS, an agent with low pK value, is more efficient at low pH [49], TS preferentially kills tumor cells, as tumore interstitium is acidic but that of normal tissues features neutral pH [53], and that protonated (uncharged) TS can freely diffuse into cancer cells, while deprotonated (charged) TS would not be readily taken up by cells of normal tissues characterized by neutral pH. Recently, it was reported that intraperitoneal administration of TS inhibited the growth of human colon cancer cells HCT116, human breast cancer cells MDA-MB-231, and mouse melanoma cells B16-F10 inoculated into nude mice in vivo [11, 55, 57]. The proportion of apoptotic cells in tumors of mice treated with TS is significantly higher than that of mice treated without TS in vivo [55, 57], suggesting that the inhibitory effect of TS reported on the growth of cancer cells in vivo is due to the induction of apoptosis in tumors.
In this study, because the reported intraperitoneal (i.p.) administration of TS dissolved in an organic solvent is not a general therapeutic procedure for humans, we attempted intravenous (i.v.) administration of TS vesicles, which could be useful for efficient delivery to tumor tissue with vigorous angiogenesis like liposomes [58], and investigate the preventing effect of TS on growth of mouse melanoma cells B16-F1 inoculated on the back of hairless mice [37].
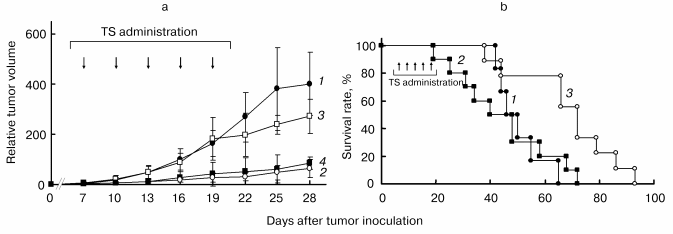
Figure 5a shows the preventive effect of i.v. administration of TS (TS-vesicle i.v.) and i.p. administration of TS dissolved in dimethyl sulfoxide (TS i.p.) on growth of mouse melanoma cells B16-F1 inoculated on the back of hairless mice. TS-vesicle i.v. almost completely prevented tumor growth in all mice even at day 28, although the administration was stopped on day 19. The percentage inhibition of tumor growth in the TS-vesicle i.v. group was about 85%. On the other hand, the inhibiting effect in the TS i.p. group was not complete, i.e., it significantly prevented tumor growth in half of the mice, but did not have a preventive effect in the other half. It is unclear why TS i.p. did not affect all the mice in the same way. Possibly, this was due to a difference in delivery efficiency of TS to the tumor on each administration.
Figure 5b shows the percentage survival of tumor-inoculated mice treated with and without TS. The percentage of TS-untreated control mice decreased from day 42, and on day 65, all mice in the control group were dead. The decrease of the survival in the TS i.p. group started a little earlier than that in the control group, and the mean survival time in the TS i.p. group was 44.5 days, which was also shorter than that (50.3 days) in the control group, although tumor growth of half the mice in the TS i.p. group was prevented almost completely. However, the survival time of mice in the TS-vesicle i.v. group was elongated about 1.4-fold (68.4 days) from the control group. Accordingly, administration of TS-vesicle i.v. not only prevented growth of the tumor, but also prolonged the life of tumor-inoculated mice.Fig. 5. Preventive effect of TS on the growth of mouse melanoma B16-F1 inoculated on the back of hairless mice in vivo (a), and increasing effect of TS on their survival percentage (b) [37]. a: 1) Control group (n = 6); 2) TS-vesicle i.v. group (n = 9); 3, 4) TS i.p. group (n = 10). TS i.p. group was divided into two classes: 3) no effect class (TS i.p., n = 5); 4) tumor growth-prevented class (TS i.p., n = 5). Values are means ± S. D. Statistical significances among the groups except for that between the TS i.p. group (4) and TS-vesicle i.v. group (2) were recognized. b: 1) Control group (n = 6); 2) TS i.p. group (n = 10); 3) TS-vesicle i.v. group (n = 9). Administration points of TS were indicated by arrows. The dosage of TS at one administration was 10 µmol/mouse.
We suppose that the prevention of tumor growth is caused by apoptosis induced by TS, but the life-prolonging effect of TS-vesicle i.v. is due to another mechanism. In vivo experiments reported previously [11, 55, 57] were performed using nude mice which lacked a thymus, but in this study we used hairless mice with a thymus. Therefore, the difference in the life prolonging effect of TS might be caused by the difference in the animals examined. TS was reported to activate CD4+ T-helper cells [59], which are known to play an important role in the immune response against tumors [60]. As TS was observed in thymus of mice treated with TS-vesicle i.v. in this study, it is supposed that TS in thymus is responsible for the life-prolonging effect due to activation of the T cell immune system in the thymus. Consequently, it is suggested that TS-vesicles are a good new type of anticancer agent, and our findings are useful for therapeutic studies of TS.
REFERENCES
1.Neuzil, J. (2002) Biochem. Biophys. Res.
Commun., 293, 1309-1313.
2.Badamchian, M., Spangelo, B. L., Bao, Y., Hagiwara,
Y., Hagiwara, H., Ueyama, H., and Goldstein, A. L. (1994) J. Nutr.
Biochem., 5, 145-150.
3.Suzuki, Y. J., and Packer, L. (1993) Biochem.
Biophys. Res. Commun., 193, 277-283.
4.Suzuki, Y. J., and Packer, L. (1993) Biochem.
Mol. Biol. Int., 31, 693-700.
5.Erl, W., Weber, C., Wardemann, C., and Weber, P. C.
(1997) Am. J. Physiol., 273, H634-H640.
6.Nakamura, T., Goto, M., Matsumoto, A., and Tanaka,
I. (1998) BioFact., 7, 21-30.
7.Fariss, M. W., Fortuna, M. B., Everett, C. K.,
Smith, J. D., Trent, D. F., and Djuric, Z. (1994) Cancer Res.,
54, 3346-3351.
8.Simmons-Menchaca, M., Qian, M., Yu, W., Sanders, B.
G., and Kline, K. (1995) Nutr. Cancer, 24, 171-185.
9.Djuric, Z., Heilbrun, L. K., Lababidi, S.,
Everett-Bauer, C. K., and Fariss, M. W. (1997) Cancer Lett.,
111, 133-139.
10.Turley, J. M., Ruscetti, F. W., Kim, S. J., Fu,
T., Gou, F. V., and Birchenall-Roberts, M. C. (1997) Cancer
Res., 57, 2668-2675.
11.Malafa, M. P., and Neitzel, L. T. (2000) J.
Surg. Res., 93, 163-170.
12.Turley, J. M., Funakoshi, S., Ruscetti, F. W.,
Kasper, J., Murphy, W. J., Longo, D. L., and Birchenall-Roberts, M. C.
(1995) Cell Growth Differ., 6, 655-663.
13.Ni, J., Chen, M., Zhang, Y., Huang, J., and Yeh,
S. (2003) Biochem. Biophys. Res. Commun., 300,
357-363.
14.Qian, M., Kralova, J., Yu, W., Bose, H. R., Jr.,
Dvorak, M., Sanders, B. G., and Kline, K. (1997) Oncogene,
15, 223-230.
15.Yu, W., Heim, K., Qian, M., Simmons-Menchaca, M.,
Sanders, B. G., and Kline, K. (1997) Nutr. Cancer, 27,
267-278.
16.Turley, J. M., Fu, T., Ruscetti, F. W., Mikovits,
J. A., Bertolette, D. C., III, and Birchenall-Roberts, M. C. (1997)
Cancer Res., 57, 881-890.
17.Yu, W., Sanders, B. G., and Kline, K. (1997)
Nutr. Cancer, 27, 92-101.
18.Yu, W., Israel, K., Liao, Q. Y., Aldaz, C. M.,
Sanders, B. G., and Kline, K. (1999) Cancer Res., 59,
953-961.
19.Neuzil, J., Svensson, I., Weber, T., Weber, C.,
and Brunk, U. T. (1999) FEBS Lett., 445, 295-300.
20.Pussinen, P. J., Lindner, H., Glatter, O.,
Reicher, H., Kostner, G. M., Wintersperger, A., Malle, E., and Sattler,
W. (2000) Biochim. Biophys. Acta, 1485, 129-144.
21.Israel, K., Yu, W., Sanders, B. G., and Kline, K.
(2000) Nutr. Cancer, 36, 90-100.
22.Yamamoto, S., Tamai, H., Ishisaka, R., Kanno, T.,
Arita, K., Kobuchi, H., and Utsumi, K. (2000) Free Rad. Res.,
33, 407-418.
23.Neuzil, J., Weber, T., Schroder, A., Lu, M.,
Ostermann, G., Gellert, N., Mayne, G. C., Olejnicka, B.,
Negre-Salvayre, A., Sticha, M., Coffey, R. J., and Weber, C. (2001)
FASEB J., 15, 403-415.
24.Neuzil, J., Weber, T., Terman, A., Weber, C., and
Brunk, U. T. (2001) Redox Rep., 6, 143-151.
25.Neuzil, J., Weber, T., Gellert, N., and Weber, C.
(2001) Br. J. Cancer, 84, 87-89.
26.Yu, W., Liao, Q. Y., Hantash, F. M., Sanders, B.
G., and Kline, K. (2001) Cancer Res., 61, 6569-6576.
27.Bang, O. S., Park, J. H., and Kang, S. S. (2001)
Biochem. Biophys. Res. Commun., 288, 789-797.
28.Wu, K., Li, Y., Zhao, Y., Shan, Y. J., Xia, W.,
Yu, W. P., and Zhao, L. (2002) World J. Gastroenterol.,
8, 982-986.
29.Charpentier, A., Simmons-Menchaca, M., Yu, W.,
Zhao, B., Qian, M., Heim, K., Sanders, B. G., and Kline, K. (1996)
Nutr. Cancer, 26, 237-250.
30.Zhao, Y., Wu, K., Xia, W., Shan, Y. J., Wu, L.
J., and Yu, W. P. (2002) World J. Gastroenterol., 8,
782-786.
31.You, H., Yu, W., Sanders, B. G., and Kline, K.
(2001) Cell Growth Differ., 12, 471-480.
32.You, H., Yu, W., Munoz-Medellin, D., Brown, P.
H., Sanders, B. G., and Kline, K. (2002) Mol. Carcinog.,
33, 228-236.
33.Lee, J. K., Jung, J. C., Chun, J. S., Kang, S.
S., and Bang, O. S. (2002) Mol. Cells, 13, 125-129.
34.Kogure, K., Morita, M., Hama, S., Nakashima, S.,
Tokumura, A., and Fukuzawa, K. (2002) Eur. J. Biochem.,
269, 2367-2372.
35.Kogure, K., Morita, M., Nakashima, S., Hama, S.,
Tokumura, A., and Fukuzawa, K. (2001) Biochim. Biophys. Acta,
1528, 25-30.
36.Kogure, K., Hama, S., Manabe, S., Tokumura, A.,
and Fukuzawa, K. (2002) Cancer Lett., 186, 151-156.
37.Kogure, K., Manabe, S., Hama, S., Tokumura, A.,
and Fukuzawa, K. (2003) Cancer Lett., 192, 19-24.
38.Jeremy, J. Y., Rowe, D., Emsley, A. M., and
Newby, A. C. (1999) Cardiovasc. Res., 43, 580-594.
39.Hecker, M., Cattaruzza, M., and Wagner, A. H.
(1999) Gen. Pharmacol., 32, 9-16.
40.Ross, R. (1999) New Engl. J. Med.,
340, 115-126.
41.Kim, S.-J., Bang, O.-S., Lee, Y.-S., and Kang,
S.-S. (1998) J. Cell Sci., 111, 435-441.
42.Zhang, F. X., Kirschning, C. J., Mancinelli, R.,
Xu, X. P., Jin, Y., Faure, E., Mantovani, A., Rothe, M., Muzio, M., and
Arditi, M. (1999) J. Biol. Chem., 274, 7611-7614.
43.Saura, M., Zaragoza, C., Bao, C., McMillan, A.,
and Lowenstein, C. J. (1999) J. Mol. Biol., 289,
459-471.
44.Chen, C. C., Wang, J. K., and Lin, S. B. (1998)
J. Immunol., 161, 6206-6214.
45.Chen, C. C., Chiu, K. T., Sun, Y.-T., and Chen,
W. C. (1999) J. Biol. Chem., 274, 31559-31564.
46.Ozer, N. K., and Azzi, A. (2000)
Toxicology, 148, 179-185.
47.Azzi, A., and Stocker, A. (2000) Progr. Lipid
Res., 39, 231-255.
48.Azzi, A., Breyer, I., Feher, M., Ricciarelli, R.,
Stocker, A., Zimmer, S., and Zingg, J. M. (2001) J. Nutr.,
131, 378S-381S.
49.Neuzil, J., Zhao, M., Ostermann, G., Sticha, M.,
Gellert, N., Weber, C., Eaton, J. W., and Brunk, U. T. (2002)
Biochem. J., 362, 709-715.
50.Ghosh, P. K., Vasanji, A., Murugesan, G., Eppell,
S. J., Graham, L. M., and Fox, P. L. (2002) Nature Cell Biol.,
4, 894-900.
51.Kogure, K., Nakashima, S., Tsuchie, A., Tokumura,
A., and Fukuzawa, K. (2003) Chem. Phys. Lipids,
126, 29-38.
52.Babior, B. M. (1999) Blood, 93,
1464-1476.
53.Hiraoka, W., Vazquez, N., Nieves-Neira, W.,
Chanock, S. J., and Pommier, Y. J. (1998) Clin. Invest.,
102, 1961-1968.
54.Tammariello, S. P., Quinn, M. T., and Estsu, S.
(2000) J. Neurosci., 20, RC53.
55.Weber, T., Lu, M., Andera, L., Lahm, H., Gellert,
N., Fariss, M. W., Korinek, V., Sattler, W., Ucker, D. S., Terman, A.,
Schroder, A., Erl, W., Brunk, U. T., Coffey, R. J., Weber, C., and
Neuzil, J. (2002) Clin. Cancer Res., 8, 863-869.
56.Kozin, S. V., Shkarin, P., and Gerweck, L. E.
(2001) Cancer Res., 61, 4740-4743.
57.Malafa, M. P., Fokum, F. D., Mowlavi, A.,
Abusief, M., and King, M. (2002) Surgery, 131, 85-91.
58.Allen, T. M. (1997) Drugs, 54,
8-14.
59.Kidao, S., Sanders, B. G., and Kline, K. (1993)
Biotechnol. Ther., 4, 117-132.
60.Ossendorp, F., Toes, R. E. M., Offringa, R., van
der Burg, S. H., and Melief, C. J. M. (2000) Immunol. Lett.,
74, 75-79.