REVIEW: Molecular Architecture of Bacteriophage T4
V. V. Mesyanzhinov1*, P. G. Leiman2, V. A. Kostyuchenko1, L. P. Kurochkina1, K. A. Miroshnikov1, N. N. Sykilinda1, and M. M. Shneider1
1Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, Moscow 117997, Russia; fax: (7-095) 336-6022; E-mail: vvm@ibch.ru2Department of Biological Sciences, Purdue University, West Lafayette, Indiana 47907-1392, USA; fax: +1 (765) 496-1189; E-mail: leiman@purdue.edu
* To whom correspondence should be addressed.
Received July 9, 2004
In studying bacteriophage T4--one of the basic models of molecular biology for several decades--there has come a Renaissance, and this virus is now actively used as object of structural biology. The structures of six proteins of the phage particle have recently been determined at atomic resolution by X-ray crystallography. Three-dimensional reconstruction of the infection device--one of the most complex multiprotein components--has been developed on the basis of cryo-electron microscopy images. The further study of bacteriophage T4 structure will allow a better understanding of the regulation of protein folding, assembly of biological structures, and also mechanisms of functioning of the complex biological molecular machines.
KEY WORDS: bacteriophage T4, baseplate, tail sheath, infection mechanisms, virus structure, protein structure, cryo-electron microscopy, X-ray analysis, 3D reconstruction
Bacteriophage T4 Escherichia coli is one of the primary tools of molecular biology; it is widely used for studying the mechanisms controlling assembly and morphogenesis of complex biological structures [1, 2]. The principles involved in T4 assembly can be encountered in a broader range of cell biology processes including cytoskeleton biology, signaling pathways, transcription, protein trafficking, and in assembly of other viruses.
The virion of bacteriophage T4 has extreme structural complexity and consists of a 1195 Å long, 860 Å wide capsid and a 1000 Å long, 210 Å diameter contractile tail. The baseplate with attached long and short fibers is located in the distal part of tail (Fig. 1, see color insert). The T4 genome (168,903 base pairs) has 289 open reading frames encoded proteins, eight tRNA genes, and at least two other genes that encode small, stable RNAs of unknown function [3, 4]. More than 150 T4 genes have been characterized, but only 62 genes are “essential”. The T4 virion is constructed of more than 2000 protein subunits that represent about 50 different gene products. Of these, 24 gene products are involved in the head assembly, 22 in the tail assembly, and six in formation of the tail fibers. More than 10 other genes code for scaffolding, catalytic, and chaperone-like proteins that assist polypeptide chain folding and protein assembly, but which are not present in the assembled virion particle. T4 head, tail, long tail fibers, and whiskers are assembled via four independent pathways and joined together to form a mature infectious virion [5].
Most T4 structural proteins are synthesized simultaneously during the latter half of the infectious cycle, and the order of their assembly is controlled at the level of protein-protein interactions [6]. However, T4 genetic system appears to be tuned to produce components in fixed relative amounts with tails and tail fibers in excess over capsids [7].Fig. 1. Morphogenesis of the bacteriophage T4 virion [2]. The overall assembly pathway can be divided into three independent stages: head, tail, and long tail fiber assembly. The chaperons and other catalytic proteins are indicated in brackets near the protein, or assembly step, that requires the chaperon. Known protein stoichiometries are given as subscripts. Crystal structures of structural proteins are shown as “ribbon” drawings.
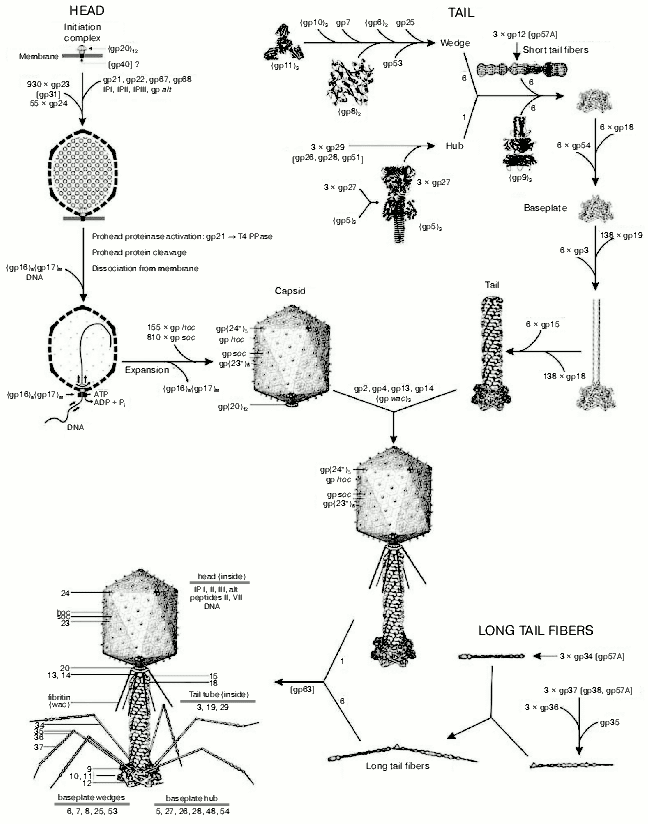
Currently X-ray crystallography, cryo-electron microscopy (cryo-EM) and three-dimensional (3D) image reconstruction are widely used for investigation of virus structure [8-10]. To study phage T4 in collaboration with M. G. Rossmann (Purdue University, USA), we have used the methods of structural biology [11-14]. This review summarizes the current knowledge of the T4 structure and points out the most significant recent results in this area. We also present data on the individual T4 proteins whose spatial structures are known at atomic resolution, and discuss how the virus infects cells.
Revealing of the quasi-atomic structure of the T4 baseplate--a multiprotein nanomachine that controls host cell recognition and attachment, tail sheath contraction, and viral DNA ejection into a cell--is very important. The T4 baseplate employs a novel structural element, a rigid molecular cell-puncturing device, containing a lysozyme domain [11, 12]. Its function is to penetrate the outer cell membrane and to disrupt the peptidoglycan layer for subsequent injection of viral DNA into the host cell.
HEAD STRUCTURE AND ASSEMBLY
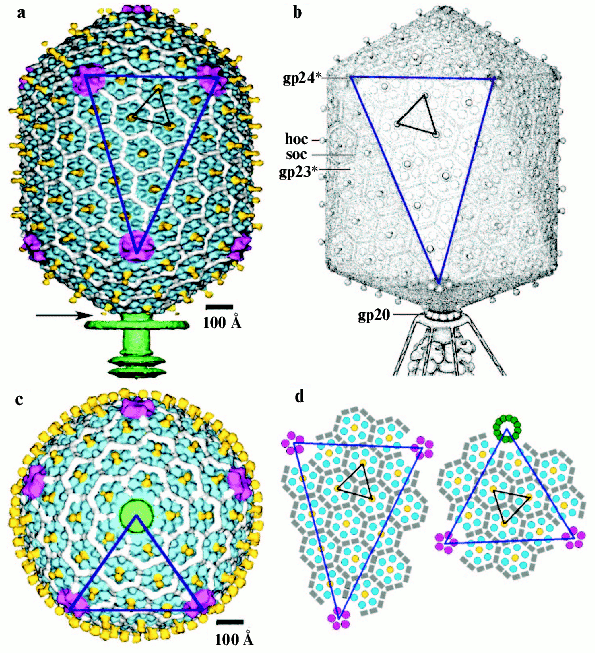
The head of phage T4, or capsid, is a prolate icosahedron elongated along a fivefold axis and is composed of more than 1500 protein subunits encoded by at least twelve genes (Table 1). This wild-type capsid is structurally characterized by triangulation number T = 13 [15]. The T value represents the number of subunits in the asymmetric cell of the icosahedron, where each facet contains 3T subunits [16]. Discrete increments of axial elongation can be characterized geometrically by a Q number. The number of capsomers in that case is 5(T + Q) + 2. The Q number of the wild-type T4 capsid is 20 [13] (Fig. 2, see color insert). In total, the mature T4 capsid contains 930 subunits of gp23* (* indicates a protein proteolytically processed during capsid maturation) and 55 subunits of gp24*. Pentamers of gp24* occupy eleven vertices of the icosahedron [13, 17], and gp20 forms a unique portal vertex required for DNA packaging and subsequent attachment of the tail. The T4 capsid shell is decorated on the outside with gphoc (highly antigenic outer capsid protein) and gpsoc (small outer capsid protein) [18]. The latter two proteins enhance head stability, but they are non-essential for head morphogenesis and they both are absent in the closely related phage T2.
Table 1. Protein compositions of the
bacteriophage T4 prohead and mature head*
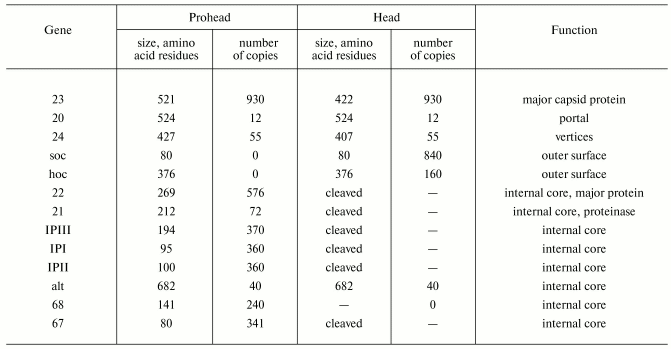
*Modified from [13, 122].
The T4 head assembly occurs via a number of discrete intermediate stages (Fig. 1). First, a DNA-free precursor, or prohead is assembled; then the prohead is processed proteolytically with phage protease (gp21). Next, the genomic DNA is packaged in a process that requires energy of ATP. Finally, head maturation is completed by adding of a few proteins. Studies of T4 head morphogenesis have resulted in the discovery of novel assembly mechanisms: 1) capsid assembles on the basis of a transition internal structure--the prohead scaffolding core--that is eliminated from the assembled prohead; 2) the assembly pathway is regulated by limited proteolysis; 3) packaging DNA into a preformed prohead occurs using energy of ATP hydrolysis; 4) large-scale conformational changes of the assembled capsid proteins increase the stability and internal volume of the capsid. These assembly mechanisms are characteristic for many viruses, including eukaryotic viruses.Fig. 2. Structure of bacteriophage T4 head [13]. The model with parameters Tend = 13 laevo, Tmid = 20, h1 = 3, k1 = 1, h2 = 4, and k2 = 2 is compared with the previously proposed model (Tend = 13 laevo, Tmid = 21, h1 = 3, k1 = 1, h2 = 3, and k2 = 3) [4, 9]. The facet triangles are shown in blue and the basic triangles are shown in black. a) Shaded surface representation of the cryo-electron microscopy reconstruction viewed perpendicular to the fivefold axis. Gp23* is shown in blue, gp24* is in purple, soc is in white, hoc is in yellow, and the tail is in green. b) Model of the previously proposed T4 head structure (adapted from [4]). c) View of the reconstruction along the fivefold axis with the portal vertex; the tail has been cut away at the level shown by the black arrow in (a). Proteins are colored as described for (a). d, left) Schematic representation of the distribution of proteins in the elongated midsection facet. d, right) Schematic representation of an end-cap facet. Proteins are colored as described for (a) except the soc molecules are shown as gray rectangles. Panels (a) and (c) were prepared using the programs DINO (www.dino3d.org) and POVRAY (www.povray.org).
Folding of major capsid protein gp23 is controlled by the host GroEL chaperonin in cooperation with T4 gp31, which completely substitutes E. coli GroES [19, 20]. In spite of low sequence identity (below 15%), gp31 of T4 and GroES of E. coli both have similar folds, assembled to heptamers. Presumably, the volume of the GroEL/GroES chamber is insufficient to accommodate a gp23 monomer and hence gp23 requires gp31, which might provide a folding chamber of larger volume [21].
An assembly of the T4 prohead starts with the formation of the gp20-gp40 membrane-spanning initiation complex at the inner side of the cytoplasmic membrane [22]; subsequently, other proteins attach to the gp20-gp40 complex and form a prohead, consisting of the major core protein (gp22) and minor head proteins, gpalt, gp21, gp67, gp68, and internal proteins IPI, IPII, and IPIII. Gp40 is not present in the assembled heads, and 12 subunits of gp20 form a portal for DNA packaging and tail attachment [23] (Table 1). Gp23 is assembled around core surface and forms a procapsid [24, 25].
Molecular mechanisms determining the shape and size of the T4 prohead are not clearly understood. Mechanisms, based on geometric constraints of interaction of internal core and shell, have been discussed [26, 27]. In contrast to these models, the “kinetic” model proposed that length regulation is based on the relative kinetics of assembly of the core and the prohead shell [28].
The major capsid protein gp23 has morphogenetic properties. Several studies have reported two large mutation classes in gene 23: pt mutations (petit) lead to the production of isometric heads and ptg mutations (petit-giant) produce both petite and giant capsids [27]. The ptg mutations were mapped at 10 different loci arranged in three rather narrow clusters of gene 23: cluster 1 (residues 66 to 97), cluster 2 (residues 268 to 287), and cluster 3 (residues 457 to 461) [29]. These three regions of gp23 have been shown to have homology with gp24. A class of point mutations in gene 23, called 24-bypass mutations, compensate for deficiency in gp24 by substituting it with gp23 [30]. Isometric and intermediate-length heads are associated only with mutations in genes 22, 67, and 68 coding core proteins [31-34]. All intermediate and isometric capsids have the same width as normal heads [27]. The following model was proposed for the normal prolate T4 core: the length of the core is determined by the way in which six filamentous strands of gp22 molecules form a prolate ellipsoid [35]. The polypeptide chain of gp22 probably forms the coiled coil strands [36].
A catalytic core component, gp21, reveals a homology to the active site of serine proteases [37]. Upon completion of the assembly of the T4 prohead, inactive gp21 zymogen is converted to the active T4 PPase, which, in turn, degrades the scaffold proteins into small peptides and then carries out the limited proteolysis of gp23 and gp24. Gp24 might be the proteinase activator, acting when it occupies the vertices of the assembling prohead [24, 30]. The T4 gene lip (late inhibitor of proteinase) that is located between the head gene 24 and the baseplate gene 25 might also participate in regulation of T4 PPase activity [38].
The T4 PPase specifically cleaves the 65-residue-long amino-terminal “Delta-piece” of the gp23 molecule [39]. A small piece of the gp24 sequence (2.2 kD) is also cleaved, but the portal protein gp20 is not cleaved. The T4 PPase catalyzes all cleavages at a consensus Leu(Ile)-XXX-Glu recognition site [27]. Most small peptides produced by proteolysis are expelled from the prohead. After cleavage of the gp23 Delta-piece, the near-hexagonal capsid lattice expands from 112 to 130 Å, thus increasing the capsid volume by ~50% [15]. Concomitant with this expansion of the gp23* lattice, which involves rotation of the gp23* protomers, their profound rearrangements and translocation of at least two distinct epitopes occur from one capsid surface to the other and thereby exposing the hoc and soc binding sites [40].
DNA PACKAGING
The proteolytically processed prohead released from the cytoplasmic membrane is capable of the packaging of replicated concatameric DNA. This process requires the terminase (packaging-linkage) proteins with ATPase activity that link DNA molecule into a mature prohead at the portal vertex (gp20), act enzymatically during translocation of DNA, and cut DNA following packaging. Two partially overlapping T4 late genes, 16 and 17, code the terminase proteins; the phage T4 terminase holoenzyme subunit apparently consists of multiple copies of the large subunit, gp17, and a small subunit, gp16 [41]. The large subunit possesses the prohead-binding and putative DNA-translocation ATPase activities, while the small subunit regulates the large gp17 subunit activities and conveys specific DNA recognition [42-45]. Gp17 interacts with multiple proteins, including gp16, gp20, gp32, and gp55 [46, 47] and presumably DNA; it binds and hydrolyses ATP. The interaction with gp16 is most important for conversion of gp17 to a catalytic form.
The length of the T4 packaged DNA is proportional to the volume of the head. Mutants with altered lengths of the capsid are capable of DNA packaging [26]. Isometric heads contain only about 70% of the genome, whereas the giants can contain more than a dozen of the genomic sequences repeated along a single dsDNA molecule. The termination of packaging is probably regulated by a mechanical stress applied to the portal complex. It has been proposed that the DNA packaging apparatus represents a rotary motor powered by ATP hydrolysis [48, 49]. The DNA is a movable central spindle of the motor, surrounded by a dodecameric portal protein to which the ATPase terminase complex is attached.
Finally, one molecule of hoc and six molecules of soc per gp23* capsomer are bound to T4 capsid [41]. The head maturation is completed by gp13, gp14, gp2, and gp4; these proteins presumably bind to the portal vertex formed by gp20 dodecamer, and they are required for subsequent attachment of the tail. The tail does not attach to the empty head, and probably a DNA-mediated interaction is involved in the head-tail joining process.
THREE-DIMENSIONAL (3D) T4 HEAD RECONSTRUCTION
Cryo-electron microscopy and three-dimensional image reconstruction methods have been applied recently to study the isometric T4 capsids. 3D maps of the empty isometric T4 capsids with and without gpsoc [50] and DNA-filled capsids [51] have been determined at 27 and at 15 Å resolution, respectively. The capsid diameter varies from ~970 Å along the fivefold axes to ~880 Å along the threefold and twofold axes. T4 isometric capsid is composed of 155 gp23* hexamers and 11 gp24* pentamers. Gphoc is a balloon-shaped molecule extended to ~50 Å away from the shell surface; its protruding part is composed of two domains: a rounded base (~19 Å high) and a globular head (~20 Å wide, 24 Å high). Every gpsoc molecule binds between two gp23* subunits but does not bind around the gp24* pentamers. The failure of gp24* to bind gpsoc provides a possible explanation for the property of osmotic shock resistance [52]. It was speculated that gp24* mutant resistant to osmotic shock might have acquired the ability to bind gpsoc, thus stabilizing the capsid.
3D-Reconstruction has been determined at 22 Å resolution recently for the wild phage T4 capsid forming a prolate icosahedron [13]. The T4 capsid surface has a hexagonal lattice conforming to the triangulation number T 13 and Q 20. The 3D map of capsid surface is covered with hexamers of gp23* and pentamers of gp24* as well as gpsoc and gphoc (Fig. 2). The shape and dimensions of gp23* hexamers probably are similar to those of other phages, for example HK97 [53]. The major capsid protein gp23* forms a hexagonal lattice with a separation of ~140 Å between hexamer centers. The protrusions corresponding to the gp24* in the 11 pentagonal vertices along fivefold axes are larger than gp23* hexamers. A dodecamer gp20 of the portal vertex has mushroom shape and its density is analogous to that of the bacteriophages SPP1 [54] and phi29 [49] connector assemblies.
Although the structure of the end caps is closely similar to the structure of the T4 isometric capsid, there is an additional layer of gp23* capsomers in midsection. The total number of gp23* monomers in T4 capsid is 930 for Q 20. One gphoc molecule is attached to the center of each gp23* hexamer, hence the total number of gphoc monomers in the T4 capsid is 155. Gphoc forms a continuous mesh that encircles the gp23* hexamers. One gpsoc molecule binds between two gp23* subunits but not between gp23* and gp20 and, therefore, gpsoc does not bind around the gp24* pentamers; therefore, the total number of gpsoc copies is 810.
TAIL STRUCTURE
Products of at least 22 genes are involved in assembly of the T4 phage tail (Table 2) that uses the energy of the sheath contraction for DNA ejection into the host cell. The assembly pathway of the tail is based on strictly ordered sequential interactions of proteins [55-61].
Table 2. Proteins of the bacteriophage T4
tail*
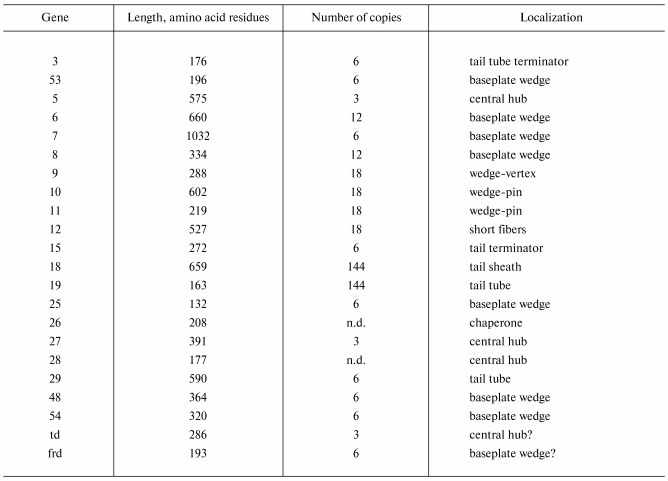
Note: n.d., not determined.
*Modified from [62].
The baseplate is a remarkably complex multiprotein structure of the tail that serves as a control unit of virus infection. The baseplate is composed of ~150 subunits of at least 16 different gene products, many of which are oligomeric, and assembled from six identical wedges that surround a central hub [62]. The T4 gp11 (the short tail fiber connecting protein), gp10, gp7, gp8, gp6, gp53, and gp25 combine sequentially to built up a wedge. The central hub is formed by gp5, gp27, and gp29 and probably gp26 and gp28.
Assembly of the baseplate is completed by attaching gp9 and gp12 forming the short tail fibers, and also gp48 and gp54 that are required to initiate polymerization of the tail tube, a channel for DNA ejection that is constructed of 138 copies of gp19 [14]. The length of the tail tube is probably determined by the “ruler protein” or template, gp29 [63]. The tail tube serves as a template for assembly of 138 copies of gp18 that form the contractile tail sheath. In the absence of the tail tube, gp18 assembles into long polysheaths with a structure similar in several aspects to the contracted state [64, 65]. Both the tail tube and the tail sheath have helical symmetry with a pitch of 40.6 Å and successive subunits repeated every 17.7° [66, 67]. Assembled tail sheath represents a metastable supramolecular structure, and sheath contraction is an irreversible process. During contraction the length of tail sheath decreases from 980 to 360 Å and its outer diameter increases from 210 to 270 Å [62].
The assembly of the tail is completed by a gp15 hexamer that binds to the last gp18 ring of the tail sheath [68]. The assembled tail associates with the head after DNA packaging. Then six gpwac (fibritin) molecules attach to the neck of the virion forming a ring embracing it (“collar”) and thin filaments protruding from the collar (“whiskers”) that help with attachment of the phage particle to other fibrous proteins, the long tail fibers.
THE STRUCTURES OF PHAGE FIBERS
Certain viruses, like adeno- and reoviruses, as well as many bacteriophages use fibrous proteins to recognize their host receptors. T4 has three types of fibrous proteins: the long tail fibers, the short tail fibers, and whiskers. The long tail fibers, which are ~1450 Å long and only ~40 Å in diameter, are primary reversible adsorption devices [69, 70]. Each fiber consists of the rigid proximal halves, encoded by gene 34, and the distal ones, encoded by genes 36 and 37. These halves are connected by gp35 that forms a hinge region and interacts with gp34 and gp36 [71, 72]. The proteins that form the long tail fibers are homotrimers, except for gp35 that assembles as a monomer [72]. The N-terminus of gp34 forms the baseplate-binding bulge, and the C-terminus of gp37 binds to a cell lipopolysaccharide (LPS) receptor.
Two phage-encoded chaperones, gp57A and gp38, are required for assembly of both long tail fiber proximal and distal parts. Gp38 is a structural component for closely related T2 phage distal part of long tail fiber; it binds to the tip of gp37 and is responsible for receptor recognition [73]. Gp57A is also required for assembly of short tail fibers. Another two assembly-assisted proteins, gp63 and gpwac, participate in the attachment of the long tail fibers to the baseplate.
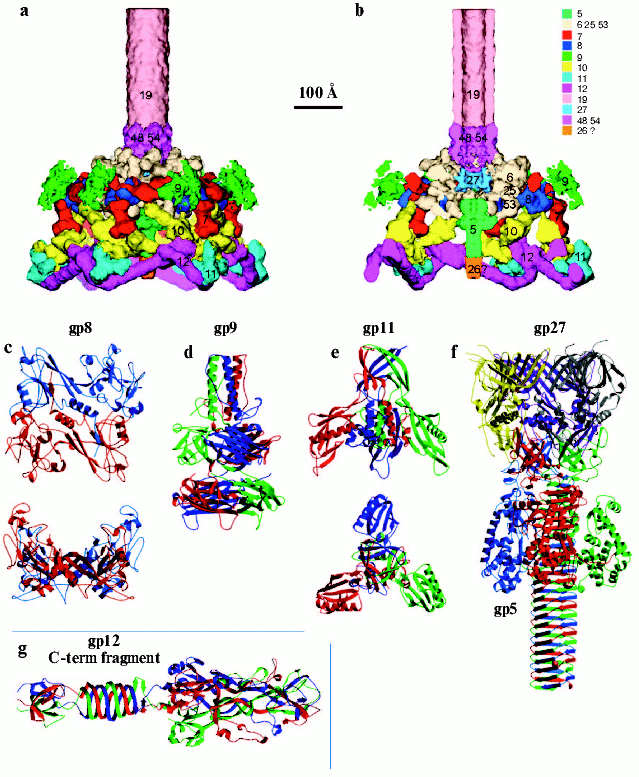
Structure of short tail fibers. The short tail fiber is a club-shaped molecule ~340 Å long consisting of a parallel, in-register assembled trimer of gp12 of 527 residues per polypeptide chain [74, 75]. Short tail fibers are attached to baseplate by the N-terminal thin part, while the globular C-terminus binds to the host cell LPS receptors [75]. The structure of this domain of the short tail fibers was recently determined by X-ray crystallography [76, 77] (Fig. 3g, see color insert). Initially, a 33 kD proteolytic fragment consisting of residues 85-396 and 518-527 was crystallized, but residues 85-245 were invisible in the crystal structure [76]. The ordered residues, 246-289, revealed a new folding motif, which is composed of intertwined strands. Residues 290-327 form a central right-handed triple beta-helix, a structure reminiscent of, but different from the adenovirus triple beta-spiral [78].
The treatment of short tail fibers by trypsin in the presence of zinc ions resulted in a 45 kD fragment [77]. X-Ray crystallography of this fragment at 1.5 Å resolution reveals the structure of the C-terminal part of the molecule that has a novel “knitted” fold, consisting of three extensively intertwined gp12 monomers, and interacts with LPS. The intertwining of the receptor binding domain represents a case of a 3D “domain swapping” phenomena found in several proteins [79, 80]. Residues 399-472, containing beta-strands F-M and alpha-helix III have been “swapped” to the neighboring monomer, leading to cyclic trimerization. The C-terminal domain has a metal-binding site that contains a Zn ion coordinated by six His residues in a stable octahedral conformation. Since the short tail fibers have no enzymatic activity, unlike the trimeric P22 phage tailspike [81], zinc is probably necessary for proper assembly and stability of the gp12 trimer.Fig. 3. Structure of the baseplate according to cryo-electron microscopy reconstruction at 12 Å resolution [12] and crystal structures of its six proteins [11, 76, 113, 118, 122]. a) Side view of the complex of baseplate and tail tube (about one third of the tube is shown). Proteins of the complex are shown in different colors and labeled with their respective gene numbers. b) Cross-section of the interior structure of dome, including the central complex, is shown. c) Structure of gp8 dimer in two perpendicular projections [122]. d) Structure of gp9 trimer [113]. e) Structure of gp11 trimer in two perpendicular projections [118]. f) Structure of gp5-gp27 trimer complex [11]. g) Structure of trimer of gp12 C-terminal fragment (residues 250-527) [76]. Each monomer in the protein structure is shown in a different color (c-g).
Fibritin structure. T4 fibrous protein, gpwac (whisker antigen control) or fibritin, is attached to the neck formed by gp13 and gp14, the collar and whiskers. Fibritin is a homotrimer assembled in parallel and attached to the T4 neck by the N-terminal domain. The C-terminal domain is a “foldon”, a unit required for fibritin folding and initiation of protein assembly. It was shown that the foldon provides correct alignment of three polypeptide chains [82, 83]. The foldon is a protein unit, which forms on the initial steps of folding [84] and often remains intact after it is transferred into other proteins [85-88].
Fibritin belongs to a specific class of accessory proteins acting in the phage assembly as a bi-complementary template accelerating the connection of the distal parts of long tail fibers to their proximal parts [89]. Being a structural component of the mature phage particle [90], fibritin also works as a primitive molecular sensor [91]. Under conditions unfavorable for phage growth (low temperature), fibritin holds the long fibers in a fixed position, raised to the tail and capsid, keeping virus particles noninfectious.
The sequence of the gpwac polypeptide chain of 487 residues is composed of heptad repeats (a-b-c-d-e-f-g)n, with the a and d positions preferentially occupied by apolar residues and the e and g positions often occupied by charged residues [92]. Such a motif, a characteristic feature of alpha-helical coiled coils proteins, is widely distributed [93]. A major part of the gpwac sequence (residues 50-460) divides into 13 continuous coiled-coil regions that are linked by the short segments without heptad repeats [82].
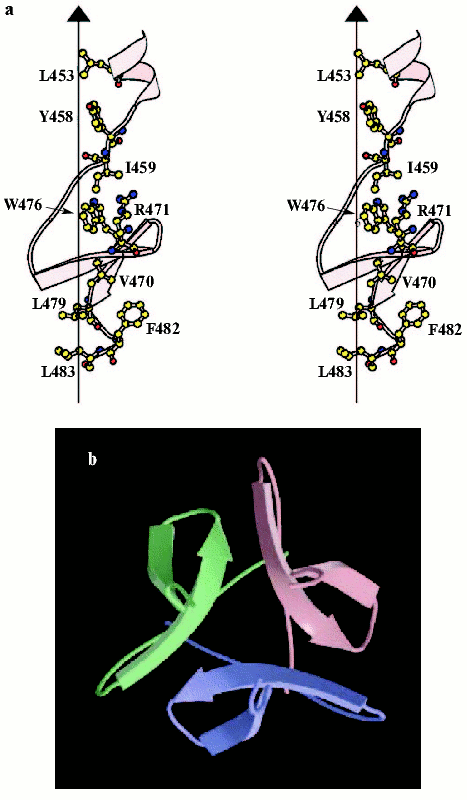
It is difficult to crystallize the entire-length fibritin, but several N-terminally truncated mutants have been successfully crystallized, and structures of three of them, fibritin E (Fig. 4, see color insert), M, and NCCP have been solved at atomic resolution [94-97]. Fibritin E is a truncated molecule comprising the last 119 residues, and fibritin M of the last 74 residues per monomer. The structures confirmed successfully early predictions for fibritin as a parallel trimeric coiled-coil with a small beta-structural domain at the C-terminus. Three coiled-coil segments of fibritin E are separated by loops: residues Gly386-Gly391 form the first loop (L10) and the second loop (L11) contains the residues Asn404-Gly417 [95]. Loops in fibritin E do not interrupt coiled-coil continuation--the helical segments started at position d and ended at position a of a heptad repeat. The C-terminal foldon is composed of the last 30 residues that form two short beta-strands in each subunit stabilized in the trimer by extensive hydrogen bonds and hydrophobic and some polar interactions within and between subunits. The fibritin foldon is absolutely necessary for proper protein folding in vivo and in vitro [95, 98, 99]. The foldon has been used recently to design fibritin NCCP variant containing N-terminal region, first coiled-coil fragment, and intact foldon [97].
The fibritin foldon has been successfully used for engineering of stable chimera proteins including the T4 phage fibers [100, 101], collagen fragments [102, 103], human adenovirus type 5 fiber protein [104, 105], and a stable trimeric form of a 140 kD protein of HIV-1 [106]. Engineering of such de novo proteins can be used for a variety of applications, including the study of receptor-ligand recognition.Fig. 4. C-Terminal domain (foldon) of fibritin E. a) Stereo diagram of the C-terminal domain. The side chains shown are those located in the hydrophobic interior formed at the interface between three symmetry-related subunits. The vertical line shows the trimer axis; atoms are shown in standard colors. b) Ribbon diagram of the C-terminal domain looking along the trimer axis, each subunit being shown in a different color (adapted from [95]).
Baseplate structure. Practically every assembled T4 particle is able to infect an E. coli host cell. The baseplate is the control center of the viral infectivity, and understanding of the baseplate structure, a multiprotein machine, is a challenging problem. Earlier the approximate positions of gp9, gp11, and short tail fibers were established by analysis of negative-stained EM images of baseplate [107]. Using chemical cross-links and gold-labeled antibodies, the contacts between neighboring baseplate proteins were studied [108, 109]. However, the details of baseplate structure remained unclear. Below we represent the data about baseplate proteins with known atomic structure.
Gp9 is a structural protein, which connects the long tail fibers [110]. The association of the proximal part of the long tail fiber with gp9 is helped by the chaperone-like protein, gp63 [111]. After attachment of the long tail fibers to the LPS receptors, gp9 initiates a baseplate structural transition to a six-pointed star with subsequent tail sheath contraction and injection of the T4 DNA into the cell. Gp9 also has a stabilizing effect on the baseplate, preventing its abortive triggering [110]. Another of the gp9 functions is to allow for the “up” and “down” movements of the long tail fibers [69]. In the “down” position, the long tail fibers can oscillate freely and are able to interact with their cell surface receptors. Gp9 probably determines the collective behavior of the long tail fibers by fixing the hinge angle around which the fibers can oscillate. At acidic pH or low temperature the long tail fibers are retracted into the “up” configuration and are close to the tail sheath of the virus particle; they are not able to interact with cell receptors.
Recombinant gp9 is functionally active and is able to incorporate into gp9- defective particles converting them in vitro into infectious phage [112]. The structure of gp9 has been solved to 2.3 Å resolution by X-ray crystallography [113]. The functionally active protein, like phage P22 tail-spike protein [114], is an SDS-resistant trimer, assembled in parallel, with overall dimensions of 60 × 60 × 130 Å (Fig. 3d). The gp9 polypeptide chain of 288 amino acid residues forms a trimer with three discrete domains: N-terminal coiled-coil (residues Met1-Gln59), middle (Ile60-Pro164), and C-terminal (Ala175-Gln288). The N-terminal sequence (Met1-Thr20) has an irregular secondary structure, which folds down antiparallel in the direction of middle domain. The sequence Thr20-Asn40 forms a parallel coiled-coil structure. The middle domain is a seven-stranded beta-sandwich and represents a unique protein fold. The C-terminal domain is an eight-stranded, antiparallel beta-barrel with a slight resemblance to the jelly-roll virus capsid structure found in numerous plant and animal viruses [115, 116]. Residues Met167-Ser173 connect the middle domain with the C-terminal domain by an extended chain that may provide mobility of the domains relative to each other. The charge distribution on the C-terminal surface shows three symmetry related clusters of high negative charge arising from the sequence Glu243-Thr-Glu-Glu-Asp-Glu that might be required to bind the long tail fiber to the gp9.
The N-terminal and middle domains are on the same side of the trimer threefold axis, while the C-terminal domain has been rotated near Pro174 by ~120° around the triad axis relative to the N-terminal and middle domains. The change in position of the C-terminal domain gives rise to a swapping domain topology of the three adjacent monomers in the gp9 trimeric protein [79].
Gp11 protein is required for attachment of the short tail fibers to the baseplate and is localized at the vertices of the hexagonal rim of the baseplate dome [107]. Gp11 forms an equimolar complex with gp10, (gp10)3-(gp11)3 [68]. The crystal structure of recombinant gp11 has been determined to 2.0 Å resolution [117] (Fig. 3e). The polypeptide chain of gp11 has three domains: the first is the N-terminal alpha-helical domain (residues 12-64), the second is the middle or distal domain--“finger” (residues 80-188), and the third is formed by residues 65-79 and 189-219. Gp11 is a homotrimer with approximate dimensions 78 × 78 × 72 Å. A search for folds homologous to gp11 using the program DALI [118] showed no significant similarity to any other structure in the Protein Data Bank (PDB). The N-terminal of gp11 forms in the trimer a parallel coiled coil that is located at the center of the molecule and is surrounded by the three finger domains and the three C-terminal domains. The finger domain is a seven-stranded, antiparallel, skewed beta-roll containing one long alpha-helix. The C-terminal domains of gp11 monomers generate a structure whose topology is similar to the fibritin foldon [95].
Gp8 is also a structural protein of the baseplate wedge [55]. The gp8 monomer has 334 residues [119], and gp8 is a dimer in solution [108, 120]. At the stage of wedge assembly, gp8 binds to the (gp11)3-(gp10)3-gp7 complex creating a binding site for gp6 [58]. Mutations in several baseplate proteins, including gp8 but excluding the long and short tail fibers, can influence the host preferences of T4 phage for different E. coli strains [121]. A 2.0 Å resolution X-ray structure of gp8 shows that the two-domain protein forms a dimer in which each monomer consists of a three-layered beta-sandwich with two loops, each containing an alpha-helix at the opposite sides of the sandwich [122] (Fig. 3c). The monomer structure can be divided into two domains: residues 1-87 and 246-334 forming domain I and residues 88-245 forming domain II. A search for folds homologous to gp8 showed no significant similarity to any other structure in the PDB.
Gp5 and gp27 have been identified as the structural proteins of the baseplate central hub forming a stable complex [57]. Gp5 contains the lysozyme domain that is required for digestion of the cellular peptidoglycan layer during the initial stages of infection [123]. Gp5 has 40% sequence identity with the cytoplasmic soluble T4 lysozyme coded by gene e [124]. After incorporation into the baseplate, gp5 undergoes a self-cleavage between Ser351 and Ala352. Both resulting parts, the N-terminal (gp5*) and the C-terminal (gp5C), remain in the phage particle [125]. Thus, gp5-gp27 complex consists of nine polypeptide chains--(gp27-gp5*-gp5C)3. The gp5 C-terminal domain, gp5C, is an SDS-resistant trimer containing 11 ValXGlyXXXXX octapeptide repeats.
The structure of the gp5-gp27 complex has been determined to 2.9 Å resolution [11] (Fig. 3f); it resembles a 190 Å long torch, the gp27 trimer forming the torch's cylindrical “head”. Three gp27 monomers are assembled into a hollow cylinder, with internal and external diameters of about 30 and 80 Å, respectively, that encompass the three N-terminal domains of gp5* to which the trimeric gp5C is attached. The C-terminal parts of the three gp5C chains intertwine and fold into a rigid beta-helical prism, and the middle lysozyme domains surrounded the amino end of the prism.
The gp27 monomer of 319 residues is folded into four domains. Two domains (residues 2-111, 207-239, and 307-368) create a toroidal structure with a pseudo-sixfold-symmetry at the top of the gp5-gp27 complex torch. Although there is no significant sequence similarity between these domains (4% sequence identity only), they have the same fold and their external surfaces are mostly hydrophobic and the charge distributions are similar. Thus, the gp27 trimer can form an interface between the hexameric baseplate and trimeric gp5.
The internal and external surfaces of the gp27 cylinder match the dimensions of the tail tube, suggesting that the gp27 trimer may serve as an extension to the tail tube and forms an channel for DNA ejection into the cell. The other two gp27 domains, formed by residues 112-206, 240-306, and 359-376, bind to the two adjacent N-terminal domains of gp5*.
The gp5* monomer has two domains--N-terminal and lysozyme. The N-terminal beta-structural domain (residues 1-129) has an oligonucleotide/oligosaccharide-binding (OB) site [126] that might facilitate binding of gp5* to oligosaccharides of the periplasmic peptidoglycan layer for its digestion by the lysozyme domain. The gp5 lysozyme domain is located on the periphery of the central beta-helix. The structures of the gp5 lysozyme domain and lysozyme e domain [127] are similar, and both enzymes have identical residues in their active sites; this suggests that the enzymatic mechanism is the same and probably both molecules have a common evolutionary origin. Activation of lysozyme domains in (gp5*-gp5C)3 structure occurs after its dissociation from the complex.
The most remarkable part of gp5 is the triple-stranded beta-helix. Three chains of gp5C wind around a central crystallographic threefold axis and create an equilateral triangular prism 110 Å long and 28 Å in diameter. The beta-strands run roughly orthogonal to the prism axis and each face of the prism has a slight left-handed twist (-3° per beta-strand). The width of the prism face narrows gradually from 33 Å at the amino end to 25 Å at the C-terminal end that creates a pointed needle-like structure. This narrowing is caused by a decrease in length of the beta-strands and by the Met554 and Met557 residues, which break the octapeptide repeat near the tip of the helix.
The first five beta-strands of gp5C (residues 389-435) form an antiparallel beta-sheet, creating one of the three faces of the prism. The succeeding 18 gp5C beta-strands form a three-start beta-helix, and each chain makes six complete turns along the helix length. These intertwined strands (residues 436-575) generate a remarkably smooth continuation of the non-intertwined N-terminal beta-sheet prism. A similar beta-sheet prism domain has been observed in the central portion of the phage P22 tailspike protein [81, 128]. However, unlike the hydrophobic interior of the P22 tailspike, the interior of gp5 beta-sheet prism is mostly polar.
The octapeptide sequence of the intertwined part of the beta-helix (residues a through h) has Gly at position a, Asn or Asp at position b, Val at position g, and polar or charged residues at position h. Residues b through g form the extended beta-strands that run at an angle of 75° with respect to the helix axis, whereas the Gly residues at position a and residues at position h kink the polypeptide chain by ~130° clockwise. The inside of the beta-helix is progressively more hydrophobic toward its C-terminal tip. The middle part of the helix has at least 42 water molecules. The helix is stabilized by two ions situated on the threefold symmetry axis. Three Lys454 residues coordinate an anion, which is probably a phosphate (HPO42-). The other ion is a cation with a van der Waals radius larger than 2 Å, coordinated by three Glu552 residues. Potential candidates are Na, Mg, K, and Ca. This ion is buried in the hydrophobic environment where it neutralizes the negative charge of Glu552 residues, and it is presumably required for proper folding of the protein.
The triple-stranded beta-helix fold has also been observed in the structure of the bacteriophage T4 short tail fibers [76]. Although the ribbon diagrams of the gp5C and gp12 beta-helices look very similar, their structures have different properties. The gp5C beta-helix has a well-defined, octapeptide repeat with a common motif, whereas the gp12 helix lacks a repeating sequence motif. The inside of the gp12 helix is mainly hydrophobic, whereas the inside of the gp5C helix presents hydrophobic, polar, and charged patches. The gp5C helix is designed to be a mechanically more rigid structure than the gp12 helix.
The structure of the gp5-gp27 complex suggests its function. The trimeric gp5-gp27 complex forms the central hub of the hexagonal baseplate. The gp27 cylinder is an extension of the tail tube that is continued by the three N-terminal domains of gp5 and terminated with a sharp tip formed by the beta-helix. The three lysozyme domains surround the beta-helix and are inactive until the infection starts. The 30 Å diameter pore formed by the gp27 cylinder is large enough to accommodate a dsDNA molecule passing through it.
3D-Reconstruction of tail tube-baseplate complex. A 3D structure based on a cryo-EM reconstruction of the baseplate has been determined to 12 Å resolution [12]. Based on the 3D reconstruction data, it is possible to identify precisely the location of six oligomeric proteins whose atomic structures are known--gp9, gp8, gp11, gp12, gp5, and gp27. In addition, using previous genetic, biochemical, and structural data another four structural proteins (gp7, gp10, gp48 or gp54, and gp19) were identified and their overall shapes determined.
Contrary to the earlier planar models of the baseplate as a hexagonal structure [62, 107], the cryo-EM reconstruction shows that the baseplate is dome-shaped; it is about 270 Å long and 520 Å at its widest diameter around the dome's base (Fig. 3, a and b). The helical tail tube extends 940 Å from the top of the baseplate and it has an outer diameter of 96 Å and an inner diameter of 43 Å. The rim of the dome is formed by six symmetry-related, arrow-like fibrous proteins ~340 Å long and ~40 Å thick. At the center of the dome, along the sixfold axis, is a needle-like structure ~105 Å long and 38 Å wide. It was previously identified as belonging to the gp5-gp27 complex, a cell-puncturing device [11].
The crystal structures of the individual oligomeric proteins fit well into the cryo-EM structures of the native baseplate [12]. Thus, the regulation of the baseplate assembly is apparently not based mainly on conformational changes of assembled proteins but is most probably the result of generation of new sites of interaction of the proteins. Identification and positioning of the proteins with known X-ray structures (gp8, gp9, gp11, gp12, and gp5-gp27) in the cryo-EM density by visual inspection was relatively easy. However, fitting of the gp5-gp27 complex into the cryo-EM density of the baseplate showed additional density at the tip of the gp5 beta-structure, suggesting the presence of another protein with an approximate molecular mass of 23 kD. This density may correspond to the hub protein gp26 of 23.4 kD, whose presence in the baseplate remains uncertain.
The atomic structure of the C-terminal fragment (residues 246-527) of the short tail fiber is strongly fitting in arrow-like filaments around the rim of the dome (Fig. 5, see color insert). The shaft of the “arrow” is kinked in the middle of the filament, changing its direction by ~90°, bending around gp11, and its N-terminus is attached to the next counterclockwise-associated arrowhead. Thus, the short tail fibers package under the baseplate, forming a garland, and their total length is ~340 Å including an 80 Å long “arrowhead” domain. The stability of the hexagonal baseplate is maintained by the interactions of the short tail fibers with each other and with gp11. In the absence of the short tail fibers, the baseplate could easily switch to a star-shaped conformation [107]. On initiation of infection, the dissociation of the garland of short tail fibers should trigger the structural reorganization of the baseplate.
The structure of the three-fingered gp11 molecule [117] was recognized as being located at the vertices of the hexagonal rim of the baseplate dome. The gp11 trimer associates with the short tail fibers and a domain of another protein that was identified as gp10, which is clamped between the three fingers of gp11. The kink of each fiber is attached to the space between the central head domain and one of the fingers of the gp11 trimer. This arrangement is consistent with the baseplate assembly pathway in which the binding of the gp11 trimer is a prerequisite for the attachment of the short tail fibers to the baseplate [57]. Secondary structure of the central part of the gp12 C-terminal domain has topological similarity to the C-terminal domain of gp11 [76]. Therefore, gp11 may also perform the function of gp12--binding of the cellular LPS.Fig. 5. Stabilization of native conformation of baseplate by short tail fibers and gp9, the long tail fibers attachment protein [12]. a) Gp9 is fitted into a region the shape of which shows the range of possible protein orientations. Midpoint (green line) and two extreme (red and blue lines) projected positions are shown. b) Gp9 structure as C-alpha-backbone in midpoint position. The lines indicate the movements of the long tail fibers. c) Six short tail fibers form the garland, and gp11 is shown as C-alpha-backbone.
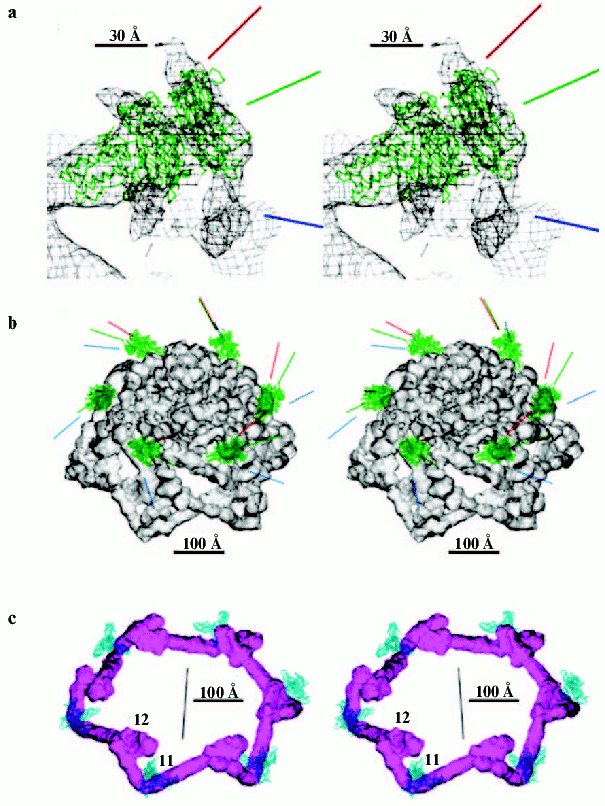
The structure of the gp8 dimer [122] located on the inside of the upper part of the baseplate dome was clearly recognizable in the baseplate 3D reconstruction.
The gp9 trimer is required for attachment of the long tail fibers to the baseplate [107]. In Fig. 5 there is visible a region of fragmented density on the upper outer edge of the baseplate, ~180 Å from the baseplate axis, corresponding to gp9 trimer [113]. The overall shape of the gp9 density suggests that the protein can pivot by up to ~55° about a radial axis of the baseplate, perpendicular to the three-fold axis of gp9. The N-terminal coiled-coil domain of gp9 is associated with identified gp7 in the upper rim of the baseplate. The C-terminal domain of gp9 forms the collinear long tail fiber attachment site.
Negatively stained EM micrographs showed that the baseplate had six pins at the vertices of the plate, running roughly parallel to the long axis of the tail [107]. These pins were associated with gp7, gp8, gp10, and gp11 [59]. The 3D map has a density at each vertex that could be ascribed as the pins, running from the top of the dome near gp9, connecting with gp8, and then to gp11. Parts of this density that have a triangular cross-section were assigned to be the gp10 trimer, which interacts with the three gp11 fingers, the N-terminal end of one short tail fiber molecule, and the C-terminal part of another short tail fiber molecule. The volume of the uninterpreted density in each pin corresponds to ~280 kD, consistent with three gp10 subunits and one copy of gp7. The component of the pin density associated with gp8 on the exterior of the baseplate [59] was assigned to gp7 [12].
Gp54 of 35 kD and gp48 of 40 kD are necessary for initiation of tail tube and tail sheath assembly [57, 129]. Attachment of gp48 precedes attachment of gp54 to the baseplate [130]; moreover, gp48 forms a 100 Å diameter platform on top of the baseplate, whereas attachment of gp54 to the baseplate does not change its morphology. Hence, density at the top and outside of the dome, where the tail tube and baseplate connect, is likely to be the site of gp54 and gp48. The volume of this density suggests that the associated monomer might have a molecular mass of ~33 kD, which would be consistent with the presence of at least gp48 or gp54. This protein connects the end of the tail tube with the top of the gp27 cylinder, a part of the cell-puncturing device. Furthermore, this protein creates a cap to the gp27 cylinder, and thus also could act as a stopper for DNA at the end of the tail tube.
The remaining unassigned proteins in the baseplate are gp6 (molecular mass 74 kD, with six or twelve copies per baseplate), gp25 (molecular mass 5.1 kD, six copies per baseplate), and gp53 (molecular mass 23 kD, six copies per baseplate). Removal of all the assigned protein density from the baseplate-tail tube reconstruction gives a total volume per wedge in the center of the baseplate corresponding to a molecular mass of 166 kD. They form a platform on top of the baseplate, which resembles a layer of the tail sheath subunits, gp18 [130]. The proteins in the uninterpreted density presumably provide the nucleation site for the tail sheath polymerization.
The tail tube is a rather featureless cylindrical structure composed of six-fold symmetric, 40.6 Å thick rings. In contrast to the results obtained by Duda [131], the tail tube in 3D reconstructions has no density in its internal channel [12]. This channel density could correspond to gp29, the ruler protein [63], which might have been lost during purification of baseplate-tail tube complexes.
BACTERIOPHAGE T4 AS INFECTION DEVICE
Short treatment of bacteriophage T4 with 3 M urea resulted in the transformation of the baseplate to a star-shape and subsequent tail sheath contraction. These structural rearrangements are analogous to phage transformation during infection of the cell [132]. Cryo-EM micrographs of 2580 phage particles with contracted tails treated a short time with 3 M urea were taken and digitized [133]. The star-shaped baseplate is significantly flatter and wider than that in the dome-shaped conformation; upon completion of the switch, the baseplate diameter increases from 520 to 610 Å and the height decreases from 270 to 120 Å [133]; the two structures consist of the same proteins (Fig. 6, see color insert). Although the overall conformational transition of the baseplate is quite large, the shapes of the proteins and their domains have changed little in the two states and appeared to move as rigid bodies. In the dome-shaped structure, the short tail fibers are associated collinearly with a domain of gp10 [12]. In the star-shaped baseplate this domain (~80 Å) points down towards the host cell surface. The structure of gp7 in the star-shaped baseplate resembles that in the dome-shaped and is composed of three large domains; the overall shapes of these domains are almost unchanged in the two conformations, but their orientations are somewhat different. The largest, dumbbell-looking domain, connecting gp10 to gp8, swivels from an almost vertical to a nearly horizontal orientation. As a result, the major arrow-like domain of gp10 changes its orientation from being at ~45° with respect to the baseplate sixfold axis to nearly orthogonal to it. Compared to the native conformation, these rearrangements lead to flattening and widening of the star-shaped baseplate [14]. The proximal part of the long tail fibers emanates from the star-shaped baseplate; long tail fibers are attached collinearly with gp9 and have also a lateral interaction with gp11, removing it from short tail fibers. The orientation of the gp11 trimer is also changed. In the native baseplate, the gp11 trimer axis forms ~144° angle with respect to the baseplate sixfold axis; in the star-shaped structure, the gp11 axis has ~48° angle.
The 3D structures of the T4 baseplate and its proteins [11, 12, 133] lead to a predicted mechanism of infection that might relate to the infection processes in many Myo- and Siphoviridae bacteriophages. The T4 phage initiates infection of E. coli by recognizing the LPS cell surface receptors with the distal ends of its long tail fibers. The recognition signal is then transmitted to the baseplate attachment protein, gp9, and then to the baseplate itself. Depending on the environmental conditions, the long tail fibers can be extended or retracted and aligned along the tail sheath [69, 133]. Such changes are consistent with the observed variable orientation of gp9, where the trimeric long tail fibers attach to gp9 collinearly with the gp9 threefold axis. The recognition of the LPS receptor by the tips of at least three long tail fibers can be transmitted to the baseplate as mechanical tension via primary changes in the gp9 conformation. It is known that the structure of gp9 can be affected by its environment. For example, the crystal structure of gp9 at pH 7.5 [113] when compared to the crystal structure at pH 4.0 (S. V. Strelkov, V. V. Mesyanzhinov, unpublished) shows a rotation by ~20° and shift by ~3 Å of the C-terminal domain relative to the middle domain.Fig. 6. Structure of the contracted T4 tail [14] (side view (a), with an inclination (b), cross-section (c), from the bottom (d)). Each protein or complex is labeled with their respective gene number and indicated by color: spring green, gp5; red, putative gp7; dark blue, gp8; green, gp9; yellow, putative gp10; cyan, gp11; magenta, gp12; salmon, gp19; sky blue, gp27; pink, putative gp48 or gp54; beige, gp6 + gp25 + gp53; orange, putative gp26.
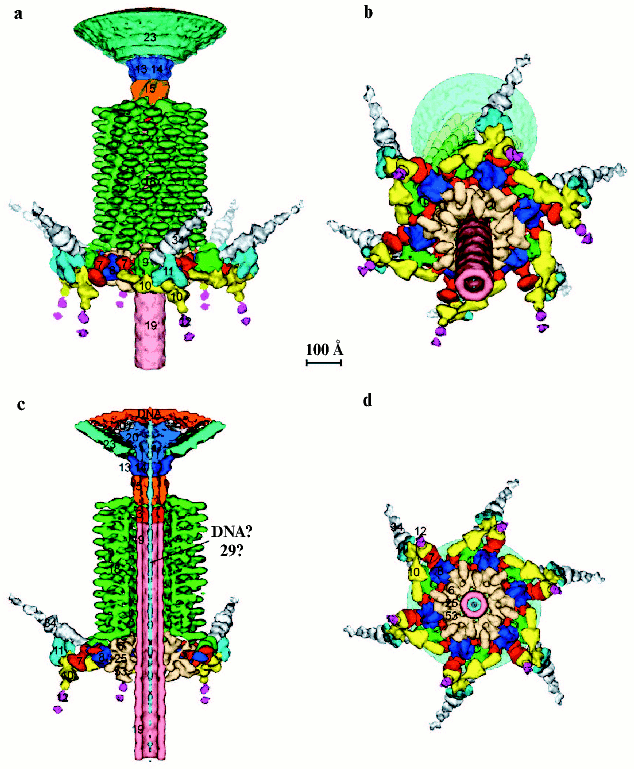
The mechanical forces to the gp9 might have a substantial impact, transmitting the signal first to gp7 and gp8, then to gp10, and eventually to gp11; a rotation of gp11 around its three-fold axis would direct the short tail fibers arrowheads toward the host cell surface that allow them to bind the LPS receptors. The long tail fibers should bind laterally to the gp11 trimers, grab them, and pull the pins outwards, which should lead to dissociation of the short tail fibers from gp11 and subsequently should break the garland arrangement. The dissociation of the short tail fibers garland allows the baseplate “pins” to move outward, expanding it from a ~520 to a ~740 Å diameter star-shaped structure. The impact of the gp9 conformational changes on the surrounding gp7 and gp8 molecules should also affect the central platform on the top of the dome and associated tail sheath, causing tail sheath contraction and conformational changes of gp18, which proceeds as a domino effect [65].
This mechanical force is transmitted through the gp27-gp5 N-terminal domain complex to the beta-helix needle, causing the latter to puncture the outer membrane. As the tail sheath contraction progresses, the beta-helix needle spans the entire ~40 Å width of the outer membrane, thereby enlarging the pore in the membrane. Subsequently, when the beta-helix rigid needle comes into contact with the periplasmic peptidoglycan layer, it should dissociate from the tip of the tail tube, thus activating the gp5 lysozyme domain that dissociates from the gp27 cylinder and locally digests the cell wall, permitting the tail tube to contact the cytoplasmic membrane. The gp27 trimer, sitting on the tip of the tail tube, probably interacts with a specific receptor on the membrane, and this event initiates release of DNA from the phage head through the tail tube into the host cell. The involvement of the receptor in transfer of the T4 DNA into the infected cell is supported by the rate of ~4000 bp/sec [134], which is much higher than the rate for passive transfer.
Early works of S. Brenner [135], A. Klug [136, 137], and also B. F. Poglazov and his collaborators [138, 139] were basic ones for structural investigations of bacteriophage T4. Application of modern high resolution methods of structural biology has made it possible to extend knowledge of phage T4 assembly regulation, its architecture, and virus infectivity mechanism. The 3D structure of individual T4 proteins, whose spatial structures have been solved recently by X-ray crystallography at atomic resolution and represented new folds, will be useful (for example, fibritin) in designing of new proteins and their complexes for bio- and nanotechnology. Future crystallographic studies of capsid and core proteins should help us to understand principles of head morphogenesis, mechanisms of assembly pathway regulation, and the DNA packaging process.
The T4 bacteriophage particle and its components are attractive objects for development of electron microscopy methods and image reconstruction. T4 tail sheath in extended form was the first biological structure whose 3D reconstruction was solved using negatively stained images [136]. The 3D structures at high resolution of cryo-electron microscopy images of such multi-protein complexes (~8.8 MD) like the T4 baseplate, and localization of individual proteins in it, lead us to establish in detail how virus infects the cell [14, 140]. Mechanistic prediction of the infection process and structural results obtained allow the use of mutagenesis of individual genes to confirm principles and details of the T4 infection machine, and also to understand the mechanism of tail sheath contraction. Studying of gp18 mutants unable to form polysheaths should probably allow us to solve the atomic structure of the protein [141].
Also, the T4 particle is an elegant system for development of correct methods for fitting of X-ray crystallographic data at atomic resolution into 3D reconstructed images obtained by cryo-electron microscopy. This is now especially important for interpretation of 3D reconstructed maps of many biological structures.
The authors are grateful to professor M. G. Rossmann for his interest in the structure of bacteriophage T4 and cooperation over a long period of time. We also thank V. D. Knorre for help in preparation of the manuscript. Researches of the laboratory are supported by grants of the Russian Foundation for Basic Research (Nos. 03-04-48652 and 02-04-50012), Howard Hughes Medical Institute (55000324), and Human Frontiers Science Program (RGP-28/2003) (in co-cooperation with M. G. Rossman and F. Arisaka).
REFERENCES
1.Kellenberger, E. (1990) Eur. J. Biochem.,
190, 233-248.
2.Leiman, P. G., Kanamaru, S., Mesyanzhinov, V. V.,
Arisaka, F., and Rossmann, M. G. (2003) Cell Mol Life Sci.,
60, 2356-2370.
3.Kutter, E., Guttman, B., Batts, D., Peterson, S.,
Djavakhishvili, T., Stidham, T., Arisaka, F., Mesyanzhinov, V., Ruger,
W., and Mosig, G. (1993) in Genomic Maps (O'Brien, S. J., ed.)
Cold Spring Harbor Laboratory Press, N. Y., pp. 1-27.
4.Miller, E. S., Kutter, E., Mosig, G., Arisaka, F.,
Kunisawa, T., and Ruger, W. (2003) Microbiol. Mol. Biol. Rev.,
67, 86-156.
5.Wood, W. B., Edgar, R. S., King, J., Lielausis, I.,
and Henninger, M. (1968) Fed. Proc., 27, 1160-1166.
6.Kellenberger, E. (1976) Phylos. Trans. R. Soc.
Lond. B. Biol. Sci., 276, 3-13.
7.Wood, W. B. (1980) Q. Rev. Biol., 55,
353-367.
8.Baker, T. S., Olson, N. H., and Fuller, S. D.
(1999) Microbiol. Mol. Biol. Rev., 63, 862-922.
9.Fuller, S. D., Berriman, J. A., Butcher, S. J., and
Gowen, B. E. (1995) Cell, 81, 715-725.
10.Heymann, J. B., Cheng, N., Newcomb, W. W., Trus,
B. L., Brown, J. C., and Steven, A. C. (2003) Nat. Struct.
Biol., 10, 334-341.
11.Kanamaru, S., Leiman, P. G., Kostyuchenko, V. A.,
Chipman, P. R., Mesyanzhinov, V. V., Arisaka, F., and Rossmann, M. G.
(2002) Nature, 415, 553-557.
12.Kostyuchenko, V. A., Leiman, P. G., Chipman, P.
R., Kanamaru, S., van Raaij, M. J., Arisaka, F., Mesyanzhinov, V. V.,
and Rossmann, M. G. (2003) Nat. Struct. Biol., 10,
688-693.
13.Fokine, A., Chipman, P. R., Leiman, P. G.,
Mesyanzhinov, V. V., Rao, V. B., and Rossmann, M. G. (2004) Proc.
Natl. Acad. Sci. USA, 101, 6003-6008.
14.Leiman, P. G., Chipman, P. R., Kostyuchenko, V.
A., Mesyanzhinov, V. V., and Rossmann, M. G. (2004) Cell, in
press.
15.Aebi, U., Bijlenga, R. K. L., van den Broek, J.,
van den Broek, R., Eiserling, F., Kellenberger, C., Kellenberger, E.,
Mesyanzhinov, V., Muller, L., Showe, M., Smith, R., and Steven, A.
(1974) J. Supramol. Struct., 2, 253-275.
16.Caspar, D. L. D., and Klug, A. (1962) Cold
Spring Harbor Symp. Quant. Biol., 27, 1-24.
17.Olson, N. H., Gingery, M., Eiserling, F. A., and
Baker, T. S. (2001) Virology, 279, 385-391.
18.Ishii, T., Yamaguchi, Y., and Yanagida, M. (1978)
J. Mol. Biol., 120, 533-544.
19.Richardson, A., Landry, S. J., and Georgopoulos,
C. (1998) Trends Biochem. Sci., 23, 138-143.
20.Ang, D., Keppel, F., Klein, G., Richardson, A.,
and Georgopoulos, C. (2000) Ann. Rev. Gen., 34,
439-456.
21.Hunt, J. F., van der Vies, S. M., Henry, L., and
Deisenhofer, J. (1997) Cell, 90, 361-371.
22.Laemmli, U. K., Molbert, E., Showe, M., and
Kellenberger, E. (1970) J. Mol. Biol., 49, 99-113.
23.Hsiao, C. L., and Black, L. W. (1978)
Virology, 91, 15-25.
24.Onorato, L., Stirmer, B., and Showe, M. K. (1978)
J. Virol., 27, 409-426.
25.Showe, M. K., and Black, L. W. (1973) J. Mol.
Biol., 107, 35-54.
26.Lane, T., and Eiserling, F. (1990) J. Struct.
Biol., 104, 9-23.
27.Black, L. W., Showe, M. K., and Steven, A. C.
(1994) in Molecular Biology of Bacteriophage T4 (Karam, J. D.,
ed.) American Society for Microbiology, Washington, D. C., pp.
218-258.
28.Onorato, L., Stirmer, B., and Showe, M. K.
(1978) J. Virol., 27, 409-426.
29.Doermann, A. H., Eiserling, F. A., and Boeher, L.
(1973) J. Virol., 12, 374-385.
30.Donate, L. E., Murialdo, H., and Carrascosa, J.
L. (1990) Virology, 179, 936-940.
31.McNicol, A. A., Simon, L. D., and Black, L. W.
(1997) J. Mol. Biol., 116, 261-283.
32.Paulson, J. R., Lazaroff, S., and Laemmli, U. K.
(1976) J. Mol. Biol., 103, 99-109.
33.Volker, T. A., Gafner, J., Bickle, T. A., and
Showe, M. K. (1982) J. Mol. Biol., 161, 479-489.
34.Volker, T. A., Kuhn, A., Showe, M. K., and
Bickle, T. A. (1982) J. Mol. Biol., 161, 491-504.
35.Keller, B., Dubochet, J., Andrian, M., Maeder,
M., Wurtz, M., and Kellenberger, E. (1988) J. Virol., 62,
2960-2969.
36.Engel, A., van Driel, R., and Driedonks, R.
(1982) J. Ultrastruct. Res., 80, 12-22.
37.Mesyanzhinov, V. V., Sobolev, B. N., Marusich, E.
I., Prilipov, A. G., and Efimov, V. P. (1990) J. Struct. Biol.,
104, 24-31.
38.Keller, B., and Bickle, T. A. (1986) Gene,
49, 245-251.
39.Kaliman, A. V., Khasanova, M. A., Kryukov, V. M.,
Tanyashin, V. I., and Bayev, A. A. (1990) Nucleic Acids Res.,
18, 4277.
40.Parker, M. L., Christensen, A. C., Boosman, A.,
Stockard, J., Young, E. T., and Doermann, A. H. (1984) J. Mol.
Biol., 180, 399-416.
41.Aebi, U., van Driel, R., Bijlenga, R. K. L., Ten
Heggeler, B., van der Broek, R., Steven, A., and Smith, P. R. (1977)
J. Mol. Biol., 110, 687-698.
42.Rao, V. B., and Black, L. W. (1988) J. Mol.
Biol., 200, 475-488.
43.Lin, H., Simon, M. N., and Black, L. W. (1997)
J. Biol. Chem., 272, 3495-3501.
44.Lin, H., and Black, L. W. (1998) Virology,
242, 118-127.
45.Kuebler, D., and Rao, V. B. (1998) J. Mol.
Biol., 281, 803-814.
46.Leffers, G., and Rao, V. B. (2000) J. Biol.
Chem., 275, 37127-37136.
47.Malys, N., Chang, D. Y., Baumann, R. G., Xie, D.,
and Black, L. W. (2002) J. Mol. Biol., 319, 289-304.
48.Hendrix, R. W. (1978) Proc. Natl. Acad.
Sci. USA, 75, 4779-4783.
49.Simpson, A. A., Tao, Y., Leiman, P. G., Badasso,
M. O., He, Y., Jardine, P. J., Olson, N. H., Morais, M. C., Grimes, S.,
Anderson, D. L., Baker, T. S., and Rossmann, M. G. (2000)
Nature, 408, 745-750.
50.Iwasaki, K., Trus, B. L., Wingfield, P. T.,
Cheng, N., Campusano, G., Rao, V. B., and Steven, A. C. (2000)
Virology, 271, 321-333.
51.Olson, N. H., Gingery, M., Eiserling, F. A., and
Baker, T. S. (2001) Virology, 279, 385-391.
52.Leibo, S. P., Kellenberger, E., Kelenberger-van
der Kamp, C., Frey, T. G., and Sternberg, C. M. (1979) J.
Virol., 30, 327-338.
53.Wikoff, W. R., Liljas, L., Duda, R. L., Tsuruta,
H., Hendrix, R. W., and Johnson, J. E. (2000) Science,
289, 2129-2133.
54.Orlova, E. V., Dube, P., Beckmann, E., Zemlin,
F., Lurz, R., Trautner, T. A., Tavares, P., and van Heel, M. (1999)
Nat. Struct. Biol., 6, 842-846.
55.Kikuchi, Y., and King, J. (1975) J. Mol.
Biol., 99, 645-672.
56.Kikuchi, Y., and King, J. (1975) J. Mol.
Biol., 99, 673-694.
57.Kikuchi, Y., and King, J. (1975) J. Mol.
Biol., 99, 695-716.
58.Plishker, M. F., Chidambaram, M., and Berget, P.
B. (1983) J. Mol. Biol., 170, 119-135.
59.Watts, N. R., and Coombs, D. H. (1990) J.
Virol., 64, 143-154.
60.Ferguson, P. L., and Coombs, D. H. (2000) J.
Mol. Biol., 297, 99-117.
61.Zhao, L., Takeda, S., Leiman, P. G., and Arisaka,
F. (2000) Biochim. Biophys. Acta, 1479, 286-292.
62.Coombs, D. H., and Arisaka, F. (1994) in
Molecular Biology of Bacteriophage T4 (Karam, J. D., ed.)
American Society for Microbiology, Washington, D. C., pp. 259-281.
63.Abuladze, N. K., Gingery, M., Tsai, J., and
Eiserling, F. A. (1994) Virology, 199, 301-310.
64.Kellenberger, E., and Boy de la Tour, E. (1964)
J. Ultrastruct. Res., 11, 545-563.
65.Moody, M. F. (1973) J. Mol. Biol.,
80, 613-635.
66.Moody, M. F., and Makowski, L. (1981) J. Mol.
Biol., 150, 217-244.
67.Zhao, L., Kanamaru, S., Chaidirek, C., and
Arisaka, F. (2003) J. Bacteriol., 185, 1693-1700.
68.Kellenberger, E., Bolle, A., Boy de la Tour, E.,
Epstein, R. H., Franklin, N. C., Jerne, N. K., Reale-Scafati, A.,
Sechaud, J., Benet, I., Goldstein, D., and Lauffer, M. A. (1965)
Virology, 26, 419-440.
69.Goldberg, E., Grinius, L., and Letellier, L.
(1994) in Molecular Biology of Bacteriophage T4 (Karam, J. D.,
ed.) American Society for Microbiology, Washington, D. C., pp.
347-356.
70.Wood, W. B., Eiserling, F. A., and Crowther, R.
A. (1994) in Molecular Biology of Bacteriophage T4 (Karam, J.
D., ed.) American Society for Microbiology, Washington, D. C., pp.
282-290.
71.Cerritelli, M. E., Wall, J. S., Simon, M. N.,
Conway, J. F., and Steven, A. C. (1996) J. Mol. Biol.,
260, 767-780.
72.Henning, U., and Hashemolhosseini, S. (1994) in
Molecular Biology of Bacteriophage T4 (Karam, J. D., ed.)
American Society for Microbiology, Washington, D. C., pp. 291-298.
73.Mason, W. S., and Haselkorn, R. (1972) J. Mol.
Biol., 66, 445-469.
74.Makhov, A. M., Trus, B. L., Conway, J. F., Simon,
M. N., Zurabishvili, T. G., Mesyanzhinov, V. V., and Steven, A. C.
(1993) Virology, 194, 117-127.
75.Van Raaij, M. J., Schoehn, G., Burda, M. R., and
Miller, S. (2001) J. Mol. Biol., 314, 1137-1146.
76.Thomassen, E., Gielen, G., Schutz, M., Schoehn,
G., Abrahams, J. P., Miller, S., and van Raaij, M. J. (2003) J. Mol.
Biol., 331, 361-373.
77.Van Raaij, M. J., Mitraki, A., Lavigne, G., and
Cusack, S. (1999) Nature, 401, 935-938.
78.Athappilly, F. K., Murali, R., Rux, J. J., Cai,
Z., and Burnett, R. M. (1994) J. Mol. Biol., 242,
430-455.
79.Liu, Y., and Eisenberg, D. (2002) Prot.
Sci., 11, 1285-1299.
80.Liu, Y., Gotte, G., Libonati, M., and Eisenberg,
D. (2002) Prot. Sci., 11, 371-380.
81.Steinbacher, S., Seckler, R., Miller, S., Steipe,
B., Huber, R., and Reinemer, P. (1994) Science, 265,
383-386.
82.Efimov, V. P., Nepluev, I. V., Sobolev, B. N.,
Zurabishvili, T. G., Schulthess, T., Lustig, A., Engel, J., Haener, M.,
Aebi, U., Venyaminov, S. Yu., Potekhin, S. A., and Mesyanzhinov, V. V.
(1994) J. Mol. Biol., 242, 470-486.
83.Boudko, S. P., Londer, Y. Y., Letarov, A. V.,
Sernova, N. V., Engel, J., and Mesyanzhinov, V. V. (2002) Eur. J.
Biochem., 269, 833-841.
84.Yu, M.-H., and King, J. (1984) Proc. Natl.
Acad. Sci. USA, 81, 6584-6588.
85.Panchenko, A. R., Luthey-Schulten, Z., and
Wolynes, P. G. (1996) Proc. Natl. Acad. Sci. USA, 93,
2008-2013.
86.Inaba, K., Kobayashi, N., and Fersht, A. R.
(2000) J. Mol. Biol., 302, 219-233.
87.Yanagawa, H., Yoshida, K., Torigoe, C., Prak, J.
S., Sato, K., Shirai, T., and Go, M. (1993) J. Biol.
Chem., 268, 5861-5865.
88.Wakasugi, K., Isimori, K., Imai, K., Wada, Y.,
and Morishima, I. (1994) J. Biol. Chem., 269,
18750-18756.
89.Terzaghi, B. E., Terzaghi, E., and Coombs, D.
(1979) J. Mol. Biol., 127, 1-14.
90.Coombs, M. P., and Eiserling, F. A. (1977) J.
Mol. Biol., 116, 375-405.
91.Conley, M. P., and Wood, W. B. (1975) Proc.
Natl. Acad. Sci. USA, 72, 3701-3705.
92.Sobolev, B. N., and Mesyanzhinov, V. V. (1991)
J. Biomol. Struct. Dyn., 8, 953-965.
93.Cohen, C., and Parry, D. A. (1994)
Science, 263, 488-489.
94.Strelkov, S. V., Tao, Y., Rossmann, M. G.,
Kurochkina, L. P., Shneider, M. M., and Mesyanzhinov, V. V. (1997)
Virology, 219, 190-194.
95.Tao, Y., Strelkov, S. V., Mesyanzhinov, V. V.,
and Rossmann, M. G. (1997) Structure, 5, 789-798.
96.Strelkov, S. V., Tao, Y., Shneider, M. M.,
Mesyanzhinov, V. V., and Rossmann, M. G. (1998) Acta
Crystallogr., D54, 805-816.
97.Boudko, S. P., Strelkov, S. V., Engel, J., and
Stetefeld, J. (2004) J. Mol. Biol., 339, 927-935.
98.Letarov, A. V., Londer, Y. Y., Boudko, S. P., and
Mesyanzhinov, V. V. (1999) Biochemistry (Moscow), 64,
817-823.
99.Boudko, S., Frank, S., Kammerer, R. A.,
Stetefeld, J., Schulthess, T., Landwehr, R., Lustig, A., Bachinger, H.
P., and Engel, J. (2002) J. Mol. Biol., 317, 459-470.
100.Miroshnikov, K. A., Marusich, E. I.,
Cerritelli, M. E., Cheng, N., Hyde, C. C., Steven, A. C., and
Mesyanzhinov, V. V. (1998) Prot. Eng., 11, 329-332.
101.Miroshnikov, K. A., Sernova, N. V., Shneider,
M. M., and Mesyanzhinov, V. V. (2000) Biochemistry (Moscow),
65, 1346-1351.
102.Frank, S., Kammerer, R. A., Mechling, D.,
Schulthess, T., Landwehr, R., Bann, J., Guo, Y., Lustig, A., Bachinger,
H. P., and Engel, J. (2001) J. Mol. Biol., 308,
1081-1089.
103.Stetefeld, J., Frank, S., Jenny, M.,
Schulthess, T., Kammerer, R. A., Boudko, S., Landwehr, R., Okuyama, K.,
and Engel, J. (2003) Structure, 11, 339-346.
104.Krasnykh, V., Belousova, N., Korokhov, N.,
Mikheeva, G., and Curiel, D. T. (2001) J. Virol., 75,
4176-4183.
105.Kashentseva, E. A., Seki, T., Curiel, D. T.,
and Dmitriev, I. P. (2002) Cancer Res., 62, 609-616.
106.Yang, X., Lee, J., Mahony, E. M., Kwong, P. D.,
Wyatt, R., and Sodroski, J. (2002) J. Virol., 76,
4634-4642.
107.Crowther, R. A., Lenk, E. V., Kikuchi, Y., and
King, J. (1977) J. Mol. Biol., 116, 489-523.
108.Watts, N. R., and Coombs, D. H. (1990) J.
Virol., 64, 143-154.
109.Watts, N. R., Hainfeld, J., and Coombs, D. H.
(1990) J. Mol. Biol., 216, 315-325.
110.Crowther, R. A. (1980) J. Mol. Biol.,
137, 159-174.
111.Wood, W. B., Conley, M. P., Lyle, H. L., and
Dickson, R. C. (1978) J. Biol. Chem., 253, 2437-2445.
112.Navruzbekov, G. A., Kurochkina, L. P.,
Kostyuchenko, V. A., Zurabishvili, T. G., Venyaminov, S. Y., and
Mesyanzhinov, V. V. (1999) Biochemistry (Moscow), 64,
1266-1272.
113.Kostyuchenko, V. A., Navruzbekov, G. A.,
Kurochkina, L. P., Strelkov, S. V., Mesyanzhinov, V. V., and Rossmann,
M. G. (1999) Struct. Fold. Des., 7, 1213-1222.
114.Betts, S., and King, J. (1999) Struct. Fold.
Des., 7, R131-R139.
115.Rossmann, M. G., and Johnson, J. E. (1989)
Annu Rev. Biochem., 58, 533-573.
116.Nandhagopal, N., Simpson, A. A., Gurnon, J. R.,
Yan, X., Baker, T. S., Graves, M. V., van Etten, J. L., and Rossmann,
M. G. (2002) Proc. Natl. Acad. Sci. USA, 99,
14758-14763.
117.Liljas, L. (1999) Curr. Opin. Struct.
Biol., 9, 129-134.
118.Leiman, P. G., Kostyuchenko, V. A., Shneider,
M. M., Kurochkina, L. P., Mesyanzhinov, V. V., and Rossmann, M. G.
(2000) J. Mol. Biol., 301, 975-985.
119.Holm, L., and Sander, C. (1993) J. Mol.
Biol., 233, 123-138.
120.Shneider, M. M., Boudko, S. P., Lustig, A., and
Mesyanzhinov, V. V. (2001) Biochemistry (Moscow), 66,
693-697.
121.Georgopoulos, C. P., Georgiou, M., Selzer, G.,
and Eisen, H. (1977) Experientia, 33, 1157-1158.
122.Leiman, P. G., Shneider, M. M., Kostyuchenko,
V. A., Chipman, P. R., Mesyanzhinov, V. V., and Rossmann, M. G. (2003)
J. Mol. Biol., 328, 821-833.
123.Nakagawa, H., Arisaka, F., and Ishii, S.
(1985) J. Virol., 54, 460-466.
124.Matthews, B. W., and Remington, S. J. (1974)
Proc. Natl. Acad. Sci. USA, 71, 4178-4182.
125.Kanamaru, S., Gassner, N. C., Ye, N., Takeda,
S., and Arisaka, F. (1999) J. Bacteriol., 181,
2739-2744.
126.Murzin, A. G., and Chothia, C. (1992) Curr.
Opin. Struct. Biol., 2, 895-903.
127.Kuroki, R., Weaver, L. H., and Matthews, B. W.
(1993) Science, 262, 2030-2033.
128.Seckler, R. (1998) J. Struct. Biol.,
122, 216-222.
129.Meezan, E., and Wood, W. B. (1971) J. Mol.
Biol., 58, 685-692.
130.King, J. (1971) J. Mol. Biol.,
58, 693-709.
131.Duda, R. L., Gingery, M., and Eiserling, F. A.
(1986) Virology, 151, 296-314.
132.To, C. M., Kellenberger, E., and Eisenstark, A.
(1969) J. Mol. Biol., 46, 493-511.
133.Kellenberger, E., Stauffer, E., Haner, M.,
Lustig, A., and Karamata, D. (1996) Biophys. Chem., 59,
41-59.
134.Letellier, L., Boulanger, P., de Frutos, M.,
and Jacquot, P. (2003) Res. Microbiol., 154, 283-287.
135.Brenner, S., Streisenger, G., Horne, R. W.,
Champe, S. P., Barnett, L., Benzer, S., and Rees, M. W. (1959) J.
Mol. Biol., 1, 281-292.
136.DeRosier, D., and Klug, A. (1968)
Nature, 217, 130-134.
137.Amos, L., and Klug, A. (1975) J. Mol.
Biol., 99, 51-73.
138.Poglazov, B. F. (1973) Monogr. Dev.
Biol., 7, 1-105.
139.Serysheva, I. I., Tourkin, A. I., Venyaminov,
S. Yu., and Poglazov, B. F. (1984) J. Mol. Biol., 179,
565-569.
140.Rossmann, M. G., Mesyanzhinov, V. V., Arisaka,
F., and Leiman, P. G. (2004) Curr. Opin. Struct. Biol.,
14, 171-180.
141.Poglazov, B. F., Efimov, A. V., Marco, S.,
Carrascosa, J., Kuznetsova, T. A., Aijrich, L. G., Kurochkina, L. P.,
and Mesyanzhinov, V. V. (1999) J. Struct. Biol., 127,
224-230.