REVIEW: Antibodies as Specific Chaperones
D. N. Ermolenko, A. V. Zherdev, and B. B. Dzantiev*
Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, Moscow 119071, Russia; fax: (7-095) 954-2804; E-mail: dzantiev@inbi.ras.ru* To whom correspondence should be addressed.
Received July 6, 2004
Protein folding is often accompanied by formation of non-native conformations leading to protein aggregation. A number of reports indicate that antibodies can facilitate folding and prevent aggregation of protein antigens. The influence of antibodies on folding is strictly antigen specific. Chaperone-like antibody activity may be due to the stabilization of native antigen conformations or folding transition states, or screening of aggregating hydrophobic surfaces. Taking advantage of chaperone-like activity of antibodies for immunotherapy may prove to be a promising approach to the treatment of Alzheimer's and prion-related diseases. Antibody-assisted folding may enhance renaturation of recombinant proteins from inclusion bodies.
KEY WORDS: antibodies, protein aggregation, folding, chaperones, inclusion bodies
Folding of polypeptide chains both in vitro and in vivo does not always proceed in a productive manner and may lead to the formation of misfolded conformations lacking appropriate biological activity. Protein misfolding is often accompanied by aggregation [1, 2]. In recent years it has become clear that quite a number of human diseases, such as Alzheimer's, Huntington's, Parkinson's, and Creutzfeldt-Jakob's diseases are related to misfolding and aggregation of proteins [3] taking place inside or outside cells. Applications of recombinant proteins in biotechnology are often hampered by misfolding of protein expressed in bacterial cells and the formation of the so-called inclusion bodies [1]. Hence, study of causes and mechanisms of protein misfolding and aggregation, as well as the search for techniques favoring productive protein folding in vivo are important tasks. Another task is to find ways to enhance renaturation in vitro of recombinant proteins accumulating during expression in the form of inclusion bodies.
In a living cell specific proteins, called molecular chaperones, interact with polypeptide chains and folding intermediates growing on the ribosome, thus preventing their association and misfolding [4]. Besides chaperones, specific protein macromolecular ligands can also influence the folding of cell proteins. Many proteins in their natural environment are structurally disordered and undergo folding coupled to binding with DNA, RNA, or some protein (E-cattgerin, translation factor eIF4G, inhibitor of cyclin-dependent kinase Cdk2, and many others) [5, 6]. Thus, an unstructured protein can be in an inactive conformation until it meets its specific ligand, and this gives the cell additional possibilities to control protein functions.
Based on these examples of folding related to specific macromolecular ligand binding, one can assume that antibodies can influence the folding of their protein antigens. Antibodies usually possess a high affinity towards antigen molecules, association constants being in the range 10-7-10-10 M, reflecting a significant lowering of the free energy of the system due to binding. This also correlates well with data on changes of hydrogen exchange rate of lysozyme amide groups [7] and those of cytochrome c [8] in complexes with antibodies. A considerable lowering of hydrogen exchange rates was found not only at the epitope location, but also at other regions quite distant from the antibody, pointing to a decrease in dynamic structural fluctuations of protein antigens and stabilization of their native conformations. X-Ray study of several antibody-protein antigen complexes has shown that antibody binding induces conformational changes in labile regions of the antigen molecule [9]. Increased thermal stabilities of specific antibody-antigen protein complexes have been demonstrated for quite a number of enzymes: alpha- and beta-galactosidases [10, 11], catalase [12], hexosaminidases [13], carboxypeptidase [14], and acetylcholinesterase [15].
Many antigenic determinants are formed by amino acid residues located far away from each other in the unfolded protein but brought close upon folding. So-called conformation sensitive or conformation dependent antibodies, recognizing such determinants, have become a widely used tool for studying domain structure, conformation transitions, and folding of proteins [16]. It is known that formation of conformational antigenic determinants may precede the final folding of the native protein. Thus, conformation sensitive monoclonal antibodies (mAb) bind to refolding intermediates of beta2-subunit of bacterial tryptophan synthase from guanidine hydrochloride [17, 18]. Further studies have revealed that this immuno-reactive refolding intermediate possesses characteristics of a “molten globule” in which the secondary structure is fully formed, while the closely packed tertiary structure is absent [19]. The binding of conformation sensitive antibodies to folding intermediates of hen lysozyme [20] and cytochrome c [21, 22] has also been reported. Immuno-reactive folding intermediates, pointing to co-translational character of folding, were discovered in polypeptide chains growing on the ribosome for tryptophan synthase [23], beta-galactosidase [24], hemagglutinin [25], and tailspike protein of phage P22 [26].
EXAMPLES OF CHAPERONE-LIKE ACTIVITY OF ANTIBODIES
Until recently there were no systematic studies on the effect of antibodies on protein folding, publications on the theme being few and far between. Increase in renaturation yield in the presence of specific antibodies was first established for refolding of acetylcholinesterase after its thermal denaturing [27] and for refolding of bovine serum albumin (BSA) from urea [28]. Later antibody induced folding of a subunit fragment of bacterial tryptophan synthase [17], S-protein, fragment of pancreatic ribonuclease A [29], and some other proteins was reported (see table).
Effect of antibodies on enzyme renaturation yield estimated by
restoration of catalytic activity
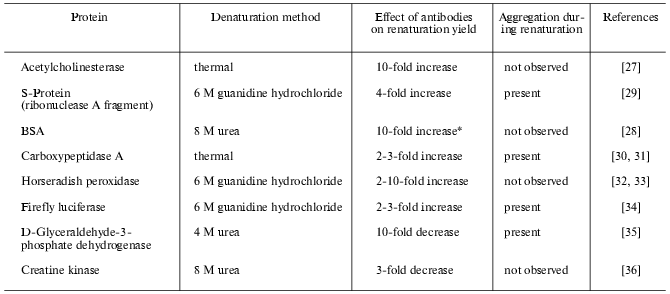
*Immunological activity of the protein was monitored.
The effect of antibodies is strictly specific, nonspecific antibodies being incapable of influencing folding [27, 32]. Some antibodies induce folding while others inhibit it (table). From a panel of several mAb against a single protein antigen, only some could influence folding [29, 32-34]. Moreover, conformation sensitive antibodies against yeast cytochrome c, recognizing the native epitope at early stages of folding, did not change the slow kinetic phase of folding [21], and antibodies recognizing the folding intermediate of hen lysozyme had no effect on restoration of catalytic activity during refolding [20]. Antibodies recognizing a partially folded monomer of phage P22 tailspike protein did not influence the refolding yield of the protein from urea, while antibodies against the native trimer form of phage P22 tailspike protein inhibited trimerization [37]. Evidently, there is no universal mechanism of the effect of antibodies on folding; it is rather dependent on the epitope properties and the folding pathway of each particular protein. It should also be noted that antibodies influenced folding both in cases when there was no aggregation during refolding (BSA [28], horseradish peroxidase [32], creatine kinase [36]), and in cases where aggregation competed with productive folding (S-protein [29], carboxypeptidase [30], firefly luciferase [34], D-glyceraldehyde-3-phosphate dehydrogenase [35]).
It is the ability of antibodies to inhibit protein and peptide aggregation that has recently drawn the attention of researchers to the earlier underestimated chaperone-like activity of antibodies [38-40]. In vitro inhibition of aggregation of beta-amyloid peptide that forms insoluble fibrils and plaques in the brain of Alzheimer patients by mAb against its N-terminal region was first reported in [41]. The same mAb were effective in a partial disaggregation and solubilization of already formed fibrils [42]. Later it was shown that immunization of mice with amyloid peptide results in a drastic reduction of amyloid plaques in Alzheimer disease transgenic model [43, 44]. The immunotherapeutic effect is partially due to Fc-dependent phagocytosis and also to antibody binding proper [45]. It was also demonstrated that mAb against the native form of the cellular form of prion-protein (PrPC) inhibit prion propagation both in vitro [46] and in vivo [47]. The inhibition by antibodies of the formation of a mutant fibril form of human lysozyme observed in patients with systemic amyloidosis was also reported [48].
These data show that antibodies can favor proper folding of the antigen protein and prevent its aggregation. This suggests a chaperone-like activity of antibodies, although in contrast to cellular chaperone proteins the effect of antibodies is strictly specific and involves only the antigen protein. The mechanisms of true chaperones and the effect of antibodies on folding are apparently also different.
Lately the ability of antibodies to influence cis-trans isomerization of proline residues limiting the folding process of many proteins was shown [49]. By immunization with synthetic hapten, mAb capable of catalyzing cis-trans isomerization of proline residues in phage T1 peptides and RNase were obtained. In this case, the antibody-abzyme acts not as a specific chaperone, but as an enzyme also favoring folding.
MECHANISM OF CHAPERONE-LIKE ACTION OF ANTIBODIES
Antibodies can probably influence folding and aggregation of proteins in a number of ways. Antibody may sterically hinder their aggregation upon binding to the antigen molecules. That may be the mechanism of solubilization of beta-amyloid peptide, which is only 42 residues long. Aggregation of beta-amyloid peptide is inhibited both by antibodies raised against the short N-terminal peptide fragment (the first five residues) [50] and those raised against residues 13-28 [50]. Apparently, antibodies against different determinants sterically hinder the conformation prone to aggregation, or screen the surfaces involved in aggregation. A similar mechanism may explain the increase of firefly luciferase refolding yield in the presence of mAb N2E3, which recognizes the determinant accessible for binding only in the denatured state of the antigen [34]. This antibody may bind to a folding intermediate and thus screen hydrophobic regions involved in luciferase aggregation.
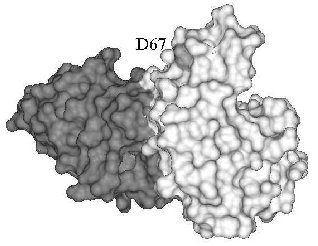
A different mechanism can be invoked for the inhibition of human lysozyme aggregation. Several mutations were found in the gene of the human lysozyme related to systemic amyloidosis, including mutation D67H. The latter markedly reduces lysozyme stability as compared with the wild-type protein, resulting in partial unfolding and aggregation of lysozyme [51, 52]. The mutant lysozyme aggregation was inhibited in the presence of antibody cAb-HuL6 fragment against the native protein [48]. Substituting D67H lowers the thermal stability of lysozyme by 10°C, but in the presence of antibody cAb-HuL6, the melting temperature of the D67H variant is increased by 15°C (5°C higher than for the wild-type lysozyme) [48]. The structure of antibody cAb-HuL6 complexed with native lysozyme was solved by X-ray analysis [48]. It turned out that residue 67, whose mutation leads to aggregation, makes no part of the epitope and is out of contact with the antibody (Fig. 1). The destabilizing effect of D67H substitution is due to disruption of interaction between alpha- and beta-domains of lysozyme [51]. The antibody binds to the C-terminal loop region of beta-domain and C-helix of alpha-domain [48] and in this way compensates for the D67H mutation, stabilizing essential for the native conformation interaction between alpha- and beta-domains of lysozyme.
Antibody induced stabilization of the conformation of a protein may be the cause of not only inhibition of protein aggregation but also of antibody-dependent folding, found for acetylcholinesterase, S-protein, BSA, and horse-radish peroxidase (table). In fact, reactivation of catalase [12] and beta-galactosidase [11, 53, 54], largely inactivated due to destabilizing mutations, was observed in the presence of antibodies against the native enzyme. In both cases, antibodies did not influence the activity of the wild-type protein. The activation during refolding probably results from a shift in equilibrium between unfolded and native conformations of the both enzymes towards folding induced by the antibody. It is worth note that antibodies that reactivated mutant forms of catalase and beta-galactosidase also increased their thermal stability [11, 12, 53].Fig. 1. X-Ray structure of antibody cAb-HuL6 (black) and human wild-type lysozyme (gray) complex [48]. Residue 67 is shown light gray.
Thus, one might expect that antibodies against non-native protein conformations should inhibit folding. Indeed, it was shown that antibodies against apo-form of myoglobin, structurally different from its holo-form, induce heme release from holo-myoglobin [55]. Monoclonal antibodies, binding to denatured creatine kinase [36], and antibodies against dimer intermediate of unfolding of tetrameric D-glyceraldehyde-3-phosphate dehydrogenase [35] inhibited renaturation of these enzymes from urea. As noted above, folding induced by a macromolecular ligand stabilizing the native conformation is not limited to an example of antibody-antigen interaction, but is a general mode of folding and binding coupling common to many proteins.
It cannot be excluded that antibodies favor folding not only through stabilization of the native protein, but also of the folding intermediates. In fact, antibodies can bind to conformational determinants formed at early stages of folding [17, 20-22, 26]. In situations when folding is hampered by a high activation barrier, stabilization of the transition state by antibody can lower the free energy of its formation.
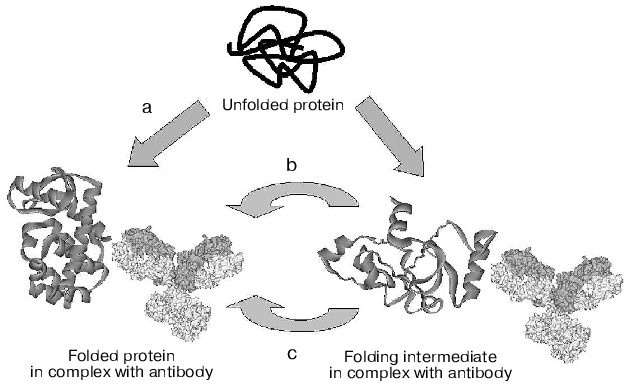
Alternative mechanisms of chaperone-like activity of antibodies, discussed above, are presented in Fig. 2. These are: stabilization of the native protein conformation with a shift of equilibrium between unfolded and native conformations of the protein antigen towards the native form (mechanism (a)); inhibition of folding intermediates aggregation through screening of hydrophobic surfaces by antibody (mechanism (b)), and, lastly, lowering of activation barrier through stabilization of the folding transition state (mechanism (c)). Today, only the hypothesis of folding due to stabilization of the protein native conformation has serious experimental foundation [48]. Which of the mechanisms is responsible for the chaperone-like activity of antibodies in most of the cases remains to be determined.
Fig. 2. Schematic representation of the three alternative mechanisms of chaperon-like activity of antibodies (see details in text).
PERSPECTIVES OF STUDIES OF THE INFLUENCE OF ANTIBODIES ON PROTEIN FOLDING
Despite various data on the ability of antibodies to influence folding and inhibit polypeptide aggregation, the phenomenon of chaperone-like activity of antibodies is still little studied. Except for the antibody hampering aggregation of the mutant form of lysozyme [48] and several antibodies inhibiting aggregation of beta-amyloid peptide [39, 50] and prion protein (PrP) [39, 46], epitopes of antibodies with chaperone-like activity have not been characterized.
Antibody dependent folding can serve as a convenient model for studying folding coupled to binding, as well as to help in determining the general mechanisms of folding. The understanding of circumstances under which antibodies influence folding is necessary for correct interpretation of experiments in which folding is detected based on restoration of binding of conformational antibodies with the protein.
Antibodies can serve as an effective tool for renaturation of recombinant proteins from inclusion bodies. Disulfide isomerase, peptidyl-prolyl-cis-trans-isomerase, and chaperonine GroEL immobilized on agarose were successfully used for “refolding chromatography” on renaturation of MHC class I-like protein from inclusion bodies [56]. Analogous to this system, it would be logical to assume that immobilized antibodies might be used for simultaneous affinity purification and renaturation of recombinant proteins from inclusion bodies.
Lastly, immunotherapy in the near future may become one of the promising methods of treatment of human disorders associated with protein and peptide aggregation. Recent studies have shown the possibility of using antibodies for preventing aggregation of beta-amyloid peptide in Alzheimer disease [38, 39] and prion protein (PrP) [46, 47].
REFERENCES
1.Fink, A. L. (1998) Fold. Des., 3,
R9-R23.
2.Markossian, K. A., and Kurganov, B. I. (2004)
Biochemistry (Moscow), 69, 971-984.
3.Dobson, C. M. (2003) Nature, 426,
884-890.
4.Hartl, F. U., and Hayer-Hartl, M. (2002)
Science, 295, 1852-1858.
5.Wright, P. E., and Dyson, H. J. (1999) J. Mol.
Biol., 293, 321-331.
6.Dyson, H. J., and Wright, P. E. (2002) Curr.
Opin. Struct. Biol., 12, 54-60.
7.Williams, D. C., Jr., Benjamin, D. C., Poljak, R.
J., and Rule, G. S. (1996) J. Mol. Biol., 257,
866-876.
8.Rizzo, P., Tinello, C., Punturieri, A., and
Taniuchi, H. (1992) Biochim. Biophys. Acta, 1159,
169-178.
9.Davies, D. R., and Cohen, G. H. (1996) Proc.
Natl. Acad. Sci. USA, 93, 7-12.
10.Snyder, P. D., Jr., Wold, F., Bernlohr, R. W.,
Dullum, C., Desnick, R. J., Krivit, W., and Condie, R. M. (1974)
Biochim. Biophys. Acta, 350, 432-436.
11.Melchers, F., and Messer, W. (1970) Biochem.
Biophys. Res. Commun., 40, 570-575.
12.Feinstein, R. N., Jaroslow, B. N., Howard, J. B.,
and Faulhaber, J. T. (1971) J. Immunol., 106,
1316-1322.
13.Ben-Yoseph, Y., Geiger, B., and Arnon, R. (1975)
Immunochemistry, 12, 221-226.
14.Solomon, B., and Balas, N. (1991) Biotechnol.
Appl. Biochem., 14, 202-211.
15.Michaeli, D., Pinto, J. D., Benjamini, E., and de
Buren, F. P. (1969) Immunochemistry, 6, 101-109.
16.Goldberg, M. E. (1991) Trends Biochem.
Sci., 16, 358-362.
17.Blond, S., and Goldberg, M. (1987) Proc. Natl.
Acad. Sci. USA, 84, 1147-1151.
18.Blond-Elguindi, S., and Goldberg, M. E. (1990)
Biochemistry, 29, 2409-2417.
19.Goldberg, M. E., Semisotnov, G. V., Friguet, B.,
Kuwajima, K., Ptitsyn, O. B., and Sugai, S. (1990) FEBS Lett.,
263, 51-56.
20.Jarrett, N. M., Djavadi-Ohaniance, L., Willson,
R. C., Tachibana, H., and Goldberg, M. E. (2002) Protein Sci.,
11, 2584-2595.
21.Raman, C. S., Jemmerson, R., and Nall, B. T.
(2000) Protein Sci., 9, 129-137.
22.Allen, M. J., Jemmerson, R., and Nall, B. T.
(1994) Biochemistry, 33, 3967-3973.
23.Fedorov, A. N., Friguet, B., Djavadi-Ohaniance,
L., Alakhov, Y. B., and Goldberg, M. E. (1992) J. Mol. Biol.,
228, 351-358.
24.Hamlin, J., and Zabin, I. (1972) Proc. Natl.
Acad. Sci. USA, 69, 412-416.
25.Braakman, I., Hoover-Litty, H., Wagner, K. R.,
and Helenius, A. (1991) J. Cell. Biol., 114, 401-411.
26.Friguet, B., Djavadi-Ohaniance, L., King, J., and
Goldberg, M. E. (1994) J. Biol. Chem., 269,
15945-15949.
27.Michaeli, D., Pinto, J. D., and Benjamini, E.
(1969) Immunochemistry, 6, 371-378.
28.Chavez, L. G., Jr., and Benjamin, D. C. (1978)
J. Biol. Chem., 253, 8081-8086.
29.Carlson, J. D., and Yarmush, M. L. (1992)
Biotechnology, 10, 86-91.
30.Solomon, B., and Schwartz, F. (1995) J. Mol.
Recognit., 8, 72-76.
31.Katzav-Gozansky, T., Hanan, E., and Solomon, B.
(1996) Biotechnol. Appl. Biochem., 23, 227-230.
32.Ermolenko, D. N., Zherdev, A. V., Dzantiev, B.
B., and Popov, V. O. (2002) Biochem. Biophys. Res. Commun.,
291, 959-965.
33.Bezsudnova, E. D., Zherdev, A. V., Ermolenko, D.
N., Yakovleva, I. V., Sviridov, V. V., Popov, V. O., and Dzantiev, B.
B. (2003) Appl. Biochem. Microbiol., 39, 509-517.
34.Xu, Q., Xie, Z., Ding, J., Lin, S.-X., and Xu, G.
(2004) Protein Sci., 13, 1851-1858.
35.Grigorieva, J. A., Dainiak, M. B., Katrukha, A.
G., and Muronetz, V. I. (1999) Arch. Biochem. Biophys.,
369, 252-260.
36.Morris, G. E., Frost, L. C., Newport, P. A., and
Hudson, N. (1987) Biochem. J., 248, 53-59.
37.Speed, M. A., Morshead, T., Wang, D. I., and
King, J. (1997) Protein Sci., 6, 99-108.
38.Schenk, D. (2002) Nat. Rev. Neurosci.,
3, 824-828.
39.Solomon, B. (2002) Curr. Med. Chem.,
9, 1737-1749.
40.Solomon, B. (2003) J. Mol. Neurosci.,
20, 283-286.
41.Solomon, B., Koppel, R., Hanan, E., and Katzav,
T. (1996) Proc. Natl. Acad. Sci. USA, 93, 452-455.
42.Solomon, B., Koppel, R., Frankel, D., and
Hanan-Aharon, E. (1997) Proc. Natl. Acad. Sci. USA, 94,
4109-4112.
43.Schenk, D., Barbour, R., Dunn, W., Gordon, G.,
Grajeda, H., Guido, T., Hu, K., Huang, J., Johnson-Wood, K., Khan, K.,
Kholodenko, D., Lee, M., Liao, Z., Lieberburg, I., Motter, R., Mutter,
L., Soriano, F., Shopp, G., Vasquez, N., Vandevert, C., Walker, S.,
Wogulis, M., Yednock, T., Games, D., and Seubert, P. (1999)
Nature, 400, 173-177.
44.Janus, C., Pearson, J., McLaurin, J., Mathews, P.
M., Jiang, Y., Schmidt, S. D., Chishti, M. A., Horne, P., Heslin, D.,
French, J., Mount, H. T., Nixon, R. A., Mercken, M., Bergeron, C.,
Fraser, P. E., St. George-Hyslop, P., and Westaway, D. (2000)
Nature, 408, 979-982.
45.Bacskai, B. J., Kajdasz, S. T., McLellan, M. E.,
Games, D., Seubert, P., Schenk, D., and Hyman, B. T. (2002) J.
Neurosci., 22, 7873-7878.
46.Peretz, D., Williamson, R. A., Kaneko, K.,
Vergara, J., Leclerc, E., Schmitt-Ulms, G., Mehlhorn, I. R., Legname,
G., Wormald, M. R., Rudd, P. M., Dwek, R. A., Burton, D. R., and
Prusiner, S. B. (2001) Nature, 412, 739-743.
47.White, A. R., Enever, P., Tayebi, M., Mushens,
R., Linehan, J., Brandner, S., Anstee, D., Collinge, J., and Hawke, S.
(2003) Nature, 422, 80-83.
48.Dumoulin, M., Last, A. M., Desmyter, A.,
Decanniere, K., Canet, D., Larsson, G., Spencer, A., Archer, D. B.,
Sasse, J., Muyldermans, S., Wyns, L., Redfield, C., Matagne, A.,
Robinson, C. V., and Dobson, C. M. (2003) Nature, 424,
783-788.
49.Ma, L., Hsieh-Wilson, L. C., and Schultz, P. G.
(1998) Proc. Natl. Acad. Sci. USA, 95, 7251-7256.
50.Legleiter, J., Czilli, D. L., Gitter, B.,
DeMattos, R. B., Holtzman, D. M., and Kowalewski, T. (2004) J. Mol.
Biol., 335, 997-1006.
51.Booth, D. R., Sunde, M., Bellotti, V., Robinson,
C. V., Hutchinson, W. L., Fraser, P. E., Hawkins, P. N., Dobson, C. M.,
Radford, S. E., Blake, C. C., and Pepys, M. B. (1997) Nature,
385, 787-793.
52.Canet, D., Last, A. M., Tito, P., Sunde, M.,
Spencer, A., Archer, D. B., Redfield, C., Robinson, C. V., and Dobson,
C. M. (2002) Nat. Struct. Biol., 9, 308-315.
53.Rotman, M. B., and Celada, F. (1968) Proc.
Natl. Acad. Sci. USA, 60, 660-667.
54.Melchers, F., and Messer, W. (1970) Eur. J.
Biochem., 17, 267-272.
55.Crumpton, M. J. (1966) Biochem. J.,
100, 223-232.
56.Altamirano, M. M., Woolfson, A., Donda, A.,
Shamshiev, A., Briseno-Roa, L., Foster, N. W., Veprintsev, D. B., de
Libero, G., Fersht, A. R., and Milstein, C. (2001) Proc. Natl. Acad.
Sci. USA, 98, 3288-3293.