Generation of Superoxide-Radical by the NADH:Ubiquinone Oxidoreductase of Heart Mitochondria
A. D. Vinogradov* and V. G. Grivennikova
Department of Biochemistry, Faculty of Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (7-095) 939-1376; E-mail: adv@biochem.bio.msu.su* To whom correspondence should be addressed.
Received September 30, 2004
Besides major NADH-, succinate-, and other substrate oxidase reactions resulting in four-electron reduction of oxygen to water, the mitochondrial respiratory chain catalyzes one-electron reduction of oxygen to superoxide radical O·2- followed by formation of hydrogen peroxide. In this paper the superoxide generation by Complex I in tightly coupled bovine heart submitochondrial particles is quantitatively characterized. The rate of superoxide formation during deltaµH+-controlled respiration with succinate depends linearly on oxygen concentration and contributes approximately 0.4% of the overall oxidase activity at saturating (0.25 mM) oxygen. The major part of one-electron oxygen reduction during succinate oxidation (~80%) proceeds via Complex I at the expense of its deltaµH+-dependent reduction (reverse electron transfer). At saturating NADH the rate of O·2- formation is substantially smaller than that with succinate as the substrate. In contrast to NADH oxidase, the rate-substrate concentration dependence for the superoxide production shows a maximum at low (~50 µM) concentrations of NADH. NAD+ and NADH inhibit the succinate-supported superoxide generation. Deactivation of Complex I results in almost complete loss of its NADH-ubiquinone reductase activity and in increase in NADH-dependent superoxide generation. A model is proposed according to which complex I has two redox active nucleotide binding sites. One site (F) serves as an entry for the NADH oxidation and the other one (R) serves as an exit during either the succinate-supported NAD+ reduction or superoxide generation or NADH-ferricyanide reductase reaction.
KEY WORDS: superoxide radical, respiratory chain, Complex I, mitochondria
Abbreviations: SMP) submitochondrial particles; SOD) superoxide dismutase; ClCCP) carbonyl cyanide-m-chlorophenylhydrazone; deltaµH+) difference of electrochemical potentials of protons across the coupling membrane; NEM) N-ethylmaleimide; O·2-) superoxide radical.
There exist a number of enzymes (substrate:oxygen oxidoreductases)
capable of oxygen reduction to H2O2 in different
cell organelles [1]. Up to 30% of hydrogen peroxide
formed in liver is produced by mitochondria [2].
Intact mitochondria (outer and inner membranes plus matrix) contain
several H2O2-producing enzymes, which are
tissue-specific. The “enzyme” that is common for the
mitochondria in any tissue is the respiratory chain. The rate of
H2O2 production by intact mitochondria strongly
depends on their functional state (state 3 or state 4) and also on the
nature of the substrate (NAD+-dependent substrates,
succinate) [1]. Submitochondrial particles
(inside-out closed vesicles formed by the inner mitochondrial membrane)
are also capable of hydrogen peroxide formation in the presence of NADH
or succinate [3-5]. It has been
demonstrated that superoxide radical is the immediate precursor of
H2O2 [6] that forms in
spontaneous or superoxide dismutase (SOD)-catalyzed [7] disproportionation:
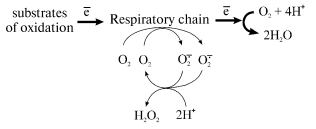
Any reduced component of the respiratory chain under aerobic conditions can serve as a potential source of one-electron oxygen reduction. [Footnote: Countless papers directly or indirectly related to superoxide generation by mitochondria have been published. The detailed analysis of their content could be the scope of special review. Here, only limited citations of the papers directly related to our studies are given. We also do not consider here some special cases, such as cyanide-resistant respiration of mitochondria from plants or some yeasts or prokaryotes where hydrogen peroxide can be the major product of oxygen reduction.] Standard inhibitory analysis has revealed that superoxide generation by the respiratory chain at significant rates occurs at Complex I and Complex III level; in the latter case the reaction proceeds only when the natural electron pathway in the Q-cycle is disturbed by specific inhibitors [8-10]. Thus the major superoxide-producing component of the mammalian respiratory chain is NADH:ubiquinone oxidoreductase (Complex I), a gigantic device composed of 46 different polypeptides bearing at least 8 individual redox components (FMN, 5-6 iron-sulfur clusters, tightly bound ubiquinone) [11-13]. What particular redox component(s) of Complex I serve as one-electron reductant of oxygen is not known.
Most of the current knowledge about hydrogen peroxide formation by mitochondria and other cell organelles has been accumulated in the late seventies and is summarized in a number of comprehensive reviews [1, 14, 15]. Recently interest in this problem has been greatly increased mostly because of numerous hypotheses (with different degrees of experimental support) on the extreme importance of the superoxide radical and hydrogen superoxide for the life and death of cells. It should be noted that only a few new findings on the subject have been published during the last 25 years.
Our interest in the problem originated more from the potential use of superoxide generation by Complex I as a tool to study the enzyme mechanism than from the possible physiological importance of O·2- generation. The tightly coupled inside-out submitochondrial particles seems to be the best choice for such studies because they allow the simulation of different functional states of the respiratory chain (state 3, state 4) and also escaping the permeability problems and possible involvement of other than the respiratory chain enzymes. In this paper, we present data on general properties of oxygen one-electron reduction by Complex I in submitochondrial particles. The results allow the development of a hypothesis on the mechanism and participants of the superoxide generation by Complex I in the presence of NADH and/or succinate as the substrates.
MATERIALS AND METHODS
Bovine heart submitochondrial particles (SMP) were prepared [16] and activated [17] as described. Deactivated SMP were obtained after incubation of the active preparations for 30 min at 37°C. Deactivation of SMP which were used for the measurements of their NADH oxidase activities was performed in the presence of NEM to prevent completely the D- to A-form transition [12] as follows. SMP (5 mg/ml) were suspended in the standard mixture comprising 0.25 M sucrose, 50 mM Tris-HCl, and 0.2 mM EDTA (pH 8.0). NEM (1 mM) was added and the mixture was incubated for 30 min at 37°C. The suspension was diluted 5-fold with cold standard mixture containing 10 mM cysteine and precipitated by centrifugation for 1 h at 30,000g. The pellets were rinsed with the standard mixture and resuspended (5 mg/ml). The mitochondrial matrix proteins were prepared from the supernatant obtained after the first precipitation of submitochondrial particles [16]. One molar K-phosphate (pH 8.0) was added to the supernatant (1/50 of the original volume), pH was adjusted to 6.0 with 1 M acetic acid, and the mixture was centrifuged for 1 h at 160,000g. The clear yellow supernatant was collected, pH was adjusted to 8.0 with 1 M KOH, and the material was stored at -20°C.
The NADH oxidase activity was measured photometrically at 340 nm (epsilon340mM = 6.22) or at 380 nm (epsilon380mM = 1.25), when NADH concentration was higher than 150 µM, in the presence or absence of 5 µM ClCCP (state 3 and state 4, respectively). The succinate oxidase activity was measured either with an oxygen-sensitive electrode or as an increase in absorption at 278 nm (fumarate formation, epsilon278mM = 0.3). The xanthine oxidase activity was assayed photometrically at 295 nm (uric acid formation, epsilon295mM = 10) using 80 µM hypoxanthine as the substrate. The superoxide generation was measured photometrically at 550 nm by tracing the SOD (3 units/ml)-sensitive acetylated cytochrome c (15 µM) reduction (epsilon550mM = 20) [18] in the standard mixture containing 0.1 M K-phosphate (pH 8.0) and the substrates of oxidation (NADH or potassium succinate). Other details of the experiments are indicated in the legends to the figures and table.
NADH, NAD+, succinic acid, EDTA, rotenone, ClCCP, buttermilk xanthine oxidase, bovine liver SOD, and catalase were from Sigma (USA). Hypoxanthine was obtained from Merck (Germany). Acetylated cytochrome c was prepared as described [19] or was obtained from Sigma. Other reagents were of the highest quality commercially available.
RESULTS
Quantitative measurements of the rate of one-electron oxygen reduction in general and particularly that for mitochondria or submitochondrial fragments are restricted by a number of serious limitations. First, the reaction product, superoxide radical O·2- is an extremely unstable specie. The bimolecular rate constant for its spontaneous dismutation in aqueous solutions at pH > 4.8 (pKa for O·2H) was estimated as 100 M-1*sec-1 [7]. This means that superoxide anion at the concentration of about 10-5 M disappears in the time scale of seconds and the standard biochemical assay procedure such as discontinuous or continuous tracing of product accumulation can hardly be applied. Moreover, the spontaneous dismutation is effectively catalyzed by transition metal complexes (Cu2+*EDTA) and it is expected that the mitochondrial preparations which are highly enriched in a number of redox components may have high unspecific superoxide dismutase activity in addition to the specific SOD [7], thus being able to mask their own superoxide generating activity. Two major approaches to quantitative assay of O·2- production are usually applied. One is based on the use of some oxidant (or reductant) rapidly reacting with O·2- and measurement of the reduced (or oxidized) form of the reagent. The mitochondrial preparations themselves are highly capable of oxidation or reduction of the redox active reagents with the rates that may be comparable with the rate of superoxide production. To exclude these interfering reactions the reagent production is usually measured in the presence and absence of a “kinetic excess” of added SOD, the difference between the two reaction rates corresponding to the true rate of superoxide production. Another way to overcome the problems is to add an excess of SOD and to measure the rate of hydroperoxide formation. The latter approach can also be complicated by the presence of catalase.
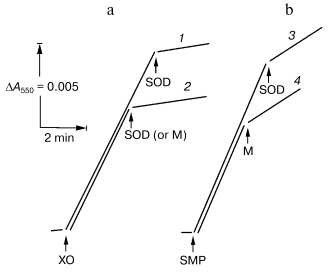
In this work, we used “direct” assay of superoxide production invented by Azzi and his collaborators [18], that is, tracing of the SOD-sensitive fraction of acetylated cytochrome c reduction. To work out this procedure as applied for SMP test experiments with the hypoxanthine-xanthine oxidase system as a source of O·2- [7] were performed. Figure 1 depicts the reduction of acetylated cytochrome c by hypoxanthine-xanthine oxidase or by SMP oxidizing NADH. The residual SOD-insensitive NADH:cytochrome c reductase activity catalyzed by SMP was evidently due to the direct, superoxide-nonmediated reduction of acetylated cytochrome c by Complex I and/or by NADH. SMP showed no SOD activity (Fig. 1a) in contrast to the mitochondrial matrix proteins, which significantly inhibited both cytochrome c reduction by hypoxanthine-xanthine oxidase or by NADH-SMP (Fig. 1, a and b).
The table shows quantitative characteristics of superoxide radical formation by SMP upon oxidation of either NADH (1 mM, a concentration that is much higher than Km for NADH oxidase [20]) or succinate. The rates of O·2- generation under any conditions with NADH or succinate as the substrates were not more than 0.4% of the respective oxidase activities resulting in four-electron reduction of oxygen to water. Only weak sensitivity of the NADH-supported generation to uncoupler was seen and the reaction was slightly stimulated by rotenone (or piericidin), i.e., under the conditions where reoxidation of all Complex I redox components was prevented. These findings are in accord with the data showing only small changes of the iron-sulfur clusters reduction level upon transition from the deltaµH+-controlled NADH oxidation (state 4) to uncoupled respiration (state 3) [21, 22]. Presumably, both in state 3 and state 4 the rate of respiration with NADH is limited by the iron-sulfur center N-2-ubiquinone oxidoreduction. It worth noting that the steady-state N-2-associated ubisemiquinone level (SQNf) is drastically decreased upon uncoupling [23]. The succinate-supported O·2- generation was substantially higher than that supported by NADH oxidation and the former was almost completely inhibited by rotenone or uncoupler. Thus, it is safe to conclude that the succinate-supported superoxide generation in state 4 is due to reverse electron transfer (O·2--supported reduction of Complex I by ubiquinol). The succinate-induced O·2- production was substantially decreased in the presence of NAD+, the substrate of the reverse electron transfer. When NADH and succinate were oxidized simultaneously the overall rate of oxygen consumption was the same as that seen with NADH alone, whereas partial electron flows from either substrate were diminished by about 30% (the table, sample 3). This behavior agrees with independent functioning of the individual respiratory complexes and can hardly be explained in terms of “supercomplex” formation [24]. Interestingly, the rate of superoxide generation with succinate and NADH (the table, sample 3) was decreased down to the activity observed with NADH alone. The residual superoxide formation observed upon uncoupled succinate oxidation most likely resulted from one-electron reduction of oxygen by Complex III (presumably by the Q·- species participating in the Q-cycle or by Rieske iron-sulfur protein) [8, 9].Fig. 1. Generation of superoxide by xanthine-hypoxanthine oxidase and NADH-SMP systems. a) The reaction was started by the addition of xanthine oxidase (XO, 10 µg/ml). Superoxide generation was followed in the presence (curve 1) or absence (curve 2) of SMP (100 µg/ml). b) Generation of superoxide by SMP (100 µg/ml) oxidizing NADH (20 µM) in the presence of rotenone (10 µM). Superoxide dismutase (SOD, 3 units/ml) or mitochondrial matrix proteins (M, 40 µg/ml) were added where indicated. The amount of the mitochondrial matrix proteins approximately corresponds to the relative content of the membrane-bound and matrix proteins in the original preparation of bovine heart mitochondria (1 : 0.4). The specific SOD activity of matrix was 125 units/mg. The addition of matrix proteins did not alter the rate of hypoxanthine oxidation.
Oxidase activities and superoxide generation by coupled submitochondrial
particles (0.25 M sucrose, 50 mM Tris-HCl, 0.1 M
potassium phosphate, pH 8.0, 30°C)
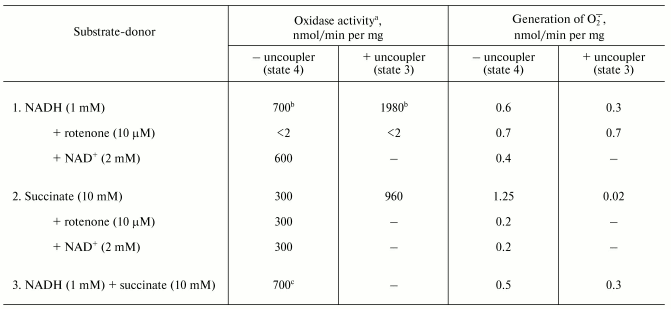
a Oxidase activities were measured with an oxygen-sensitive
electrode as described in the “Materials and Methods”
section.
b The presence of 0.1 M potassium phosphate (or
0.1 M potassium chloride) caused partial uncoupling and decreased
the respiratory control ratio from 5.5 to 2.8 (the rates of NADH
oxidation in state 4 and state 3 in the absence of potassium phosphate
were 290 and 1450 nmol/min per mg of protein, respectively).
Potassium phosphate did not affect the respiratory control ratio with
succinate as the substrate. Potassium phosphate was added to decrease
the SOD-insensitive cytochrome c reduction.
c The oxidase activity measured as a decrease of absorption
at 380 nm (oxidation of NADH) was 500 nmol/min per mg of
protein, the rate of succinate oxidation thus was 200 nmol/min
per mg of protein when both NADH and succinate were present.
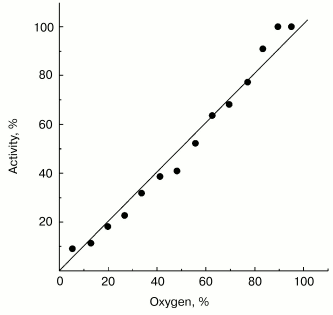
Possible functional significance of superoxide generation by the mitochondrial respiratory chain is widely discussed in the current literature. It might be expected that a specific binding site for one-electron oxygen reduction exists if superoxide generation by respiratory chain, indeed, plays an important role in the cell physiology and the affinity of oxygen to this hypothetical “site” could be under fine control as it is generally established for enzymatic catalysis. Note should be made that this assumption is not obligatory: several oxidoreductases (such as SOD [7] or catalase [25]) are known to catalyze first-order reactions (with respect of the substrate concentrations) within concentration ranges much higher than those conceivable for physiologically relevant conditions. The data presented in Fig. 2 show that this is also the case for succinate-supported, reverse electron transfer-mediated superoxide generation. Linear dependence of O·2- generation on oxygen concentration was found within the range of 0-0.25 mM (oxygen concentration in aqueous solutions under normal air-saturation condition).
In light of widely discussed toxicity of O·2- and H2O2 it was of interest to check whether these species have any inhibitory effect on the respiratory activity of SMP. Incubation of SMP (respiratory control ratio of 5.5 with NADH as the substrate) in the presence of hypoxanthine and xanthine oxidase for 20 min at 30°C, i.e., under the conditions where superoxide radical was actively generated (see Fig. 1) changed neither respiratory control ratio nor the rate of the reverse electron transfer. Prior thermal deactivation of Complex I [16] also did not reveal any toxic effect of superoxide radical or hydrogen peroxide on SMP. The standard parameters of NADH oxidation by SMP were also not changed when assayed in the presence of 10 mM H2O2 (results are not shown).
Non-additivity of the succinate- and NADH-supported superoxide generation (in fact, NADH inhibits the succinate-supported reaction; the table) seemed quite puzzling. The arguments have been presented in our previous papers for different reaction pathways in forward and reverse electron transfer catalyzed by Complex I. Particularly, evidence for the presence of at least two different nucleotide binding sites for NADH and NAD+ have been published [26, 27]. The results presented below are in agreement with the two-nucleotide binding site model. Figure 3 shows the dependence of NADH oxidase and the rate of the NADH-supported O·2- generation (state 4) on the substrate concentration. An increase in NADH over optimal concentration resulted in inhibition of superoxide production (Fig. 3a), whereas only very slight substrate inhibition was seen for the NADH oxidase activity (Fig. 3b). It has been shown that NAD+ is a very weak competitive inhibitor of NADH oxidation both in state 3 and state 4 (Ki = 1.25 mM [20, 26]). We found that NAD+ strongly decreased the rate of superoxide production if succinate or NADH were used as the substrates, whereas NAD+ did not significantly affect the NADH oxidase activity (see the table). We believe that it is difficult if not impossible to explain the data shown in the table and Fig. 3 by a model other than that with two nucleotide specific binding sites.Fig. 2. Generation of superoxide as a function of oxygen concentration. To calibrate the oxygen concentration scale the zero-order oxidation of succinate (10 mM) by SMP (100 µg/ml) was traced in a separate sample (not shown) by following fumarate formation (see “Materials and Methods”) up to “anaerobiosis” which was seen as sharp decrease in absorption change at 280 nm. The univalent oxygen consumption in calibration samples was neglected. Superoxide formation was measured as the continuous first-order curve of SOD-sensitive cytochrome c reduction (see “Materials and Methods”) under exactly the same conditions as were used for calibration. Saturating concentration of acetylated cytochrome c (15 µM) was present. One hundred percent activity corresponds to the rate of superoxide production of 1.2 nmol/min per mg of protein.
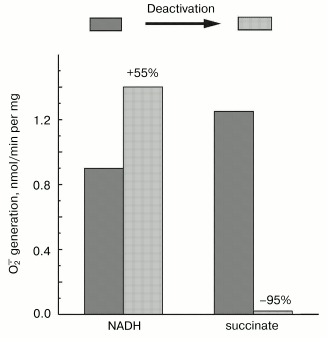
Mitochondrial Complex I exists as two distinct slowly interconvertible forms: A-form which is active proton-translocating NADH:ubiquinone oxidoreductase and D-form which is inactive, slowly oxido-reduction activated enzyme [20, 28]. A/D-transition of Complex I has been demonstrated for purified enzyme [29], submitochondrial particles [16], and intact mitochondria [30] and in ex vivo experiments in perfused rat hearts [31]. In terms of its catalytic activities, the D-form is similar to the rotenone- or piericidin-inhibited Complex I. Since rotenone does not prevent NADH-supported superoxide generation (in fact it slightly stimulates the reaction, see the table) we proposed that the deactivation of Complex I would also result in increase in NADH-supported superoxide generation. According to this hypothesis [28], deactivation of Complex I under anoxia followed by oxygenation after reperfusion may result in physiologically dangerous increase in H2O2 concentration. The results presented in Fig. 4 support this hypothesis, at least its enzymological part: deactivation of Complex I was accompanied by loss of the NADH oxidase activity and an increase in the NADH-supported superoxide formation.Fig. 3. Dependence of NADH-induced superoxide production (a) and NADH oxidase activity (b) on NADH concentration. a) Generation of superoxide by SMP (100 µg/ml) was measured in the presence of rotenone (10 µM). b) Coupled NADH oxidase activity (state 4) of SMP (25 µg/ml).
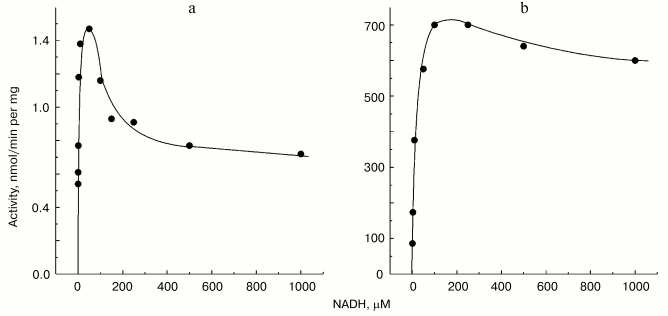
Fig. 4. Effect of deactivation of Complex I on superoxide generation. The reaction rate was measured with 50 µM NADH as the substrate in the standard reaction mixture containing 30 µg/ml of active or deactivated (treated with NEM, see “Materials and Methods”) SMP. Superoxide generation in the presence of 10 mM succinate was measured for active or thermally deactivated SMP (100 µg/ml, no NEM treatment). The reactions were started by the addition of the substrate (NADH or succinate).
DISCUSSION
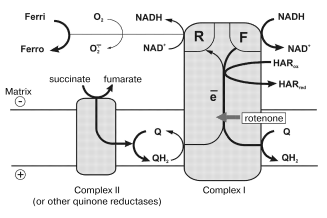
In this paper, the quantitative parameters of superoxide generation by heart mitochondrial respiratory chain are reported. Using tightly coupled submitochondrial particles we were able to characterize this reaction, to our knowledge for the first time, under physiologically relevant conditions, i.e., corresponding to the deltaµH+-controlled (state 4) and uncoupled (state 3) oxidation of NADH, the major feeding source for the respiratory chain. Succinate oxidation as the example of other flavin-dependent electron donors was also studied. The results of our studies explain why the rate of succinate-supported, reverse electron transfer-mediated superoxide generation is greater than the NADH-dependent reaction and they also support our hypothesis [12, 32] on different electron transfer pathways in the forward and reverse reactions catalyzed by Complex I. The model, which explains the findings reported here, is shown in Fig. 5. Complex I bears at least two NADH (NAD+) specific sites. One site (F) has high affinity to NADH (KmNADH ~ 7 µM) and low affinity to NAD+ (KiNAD+ for competitive inhibition of NADH oxidation is of the order of >10-3 M) [20]. ADP-ribose, a competitive inhibitor of all NADH dehydrogenase activities of Complex I, binds exclusively to the F-site, thus showing no inhibition of succinate-supported NAD+ reduction [26]. Site F serves as the entry point for NADH oxidation by ubiquinone and coupled translocation of 4 H+ per molecule of NADH oxidized [33].
Site R has relatively high affinity to NAD+ (KmNAD+ in the reverse electron transfer reaction is 7 µM [20]), and also binds NADH (KiNADH for competitive inhibition of the reverse electron transfer reaction is 40 µM [20]). The existence of site R is evident from the unidirectional effect of ADP-ribose on NADH oxidation [26] and from the ternary complex (NADH·E·NAD+) kinetic mechanism of transhydrogenating reactions catalyzed by Complex I or by its low molecular mass fragments [27]. It seems likely that ferricyanide anion previously widely used as an artificial electron acceptor for Complex I [34], but not hexaammineruthenium cation [35], interacts with the enzyme at site R. Indeed, NADH:ferricyanide reductase is strongly inhibited by high concentrations of NADH [36] just as is superoxide generation (Fig. 3a), whereas NADH:hexaammineruthenium reductase is not susceptible to the substrate inhibition [35]. The results reported here suggest that site R is specifically involved in univalent reduction of oxygen (see Fig. 5).
What particular redox component (flavin, iron-sulfur centers, ubisemiquinone) of Complex I is an immediate donor for one-electron reduction of oxygen? We believe that FMNH2 or flavin semiquinone is the univalent reductant for O·2- formation. First, it is well known that free FADH2 and FMNH2 (mid-point redox potential is -280 mV [37]) are rapidly oxidized by O2 [38]. This property of flavins is used in a number of flavin-dependent oxidases. Second, the specific Complex I inhibitors, rotenone and piericidin prevent the formation of iron-sulfur center N-2 associated ubisemiquinone during coupled (state 4) NADH oxidation [22] whereas they do not prevent (in fact, stimulate) the superoxide generation (the table). Third, the transhydrogenase reaction which proceeds with ternary complex formation (both F- and R-sites are involved) is effectively catalyzed by a small fragment of Complex I, so-called flavoprotein, FP [39], which contains FMN and only a single strongly modified iron-sulfur cluster (most likely N-3). Interestingly, the presence of two FMN per mole of Complex I has been proposed by Albracht [40] based on a number of circumstantial evidences.Fig. 5. A model for the reaction sites in Complex I. Ferri, HARox and Ferro, HARred are oxidized and reduced forms of ferricyanide anion and hexaammineruthenium cation, respectively. Relative thickness of the arrows corresponds arbitrarily to relative rates of the particular reaction pathway. See text for further explanations.
The presence of the specific site R participating in the deltaµH+-dependent ubiquinol:NAD+ reductase reaction (reverse electron transfer) and in superoxide generation by Complex I raises the question of the physiological significance of these reactions. Space does not permit discussing the possible physiological function(s) of the reverse electron transfer (-dependent NAD+ reduction). The only note pertinent for the present discussion is that succinate, the most widely used substrate, is not the only and, most likely, not the major electron donor at ubiquinone level. Each first dehydrogenation in the fatty acid beta-oxidation cycle, oxidation of alpha-glycerophosphate, choline, sarcosine, and dihydroorotic acid (in eukaryotes) are coupled with direct ubiquinone reduction, and site R may participate in “universalization” of the reducing equivalents in the form of NADH via reverse electron transfer. Another aspect of the problem is the control of intramitochondrial NADH oxidation. No natural ligands affecting affinity of NADH to its “classical” binding site (site F) are known. On the other hand, it is hard to believe that the rate of NADH oxidation is only regulated by the respiratory control mechanism (directly, by inhibition of electrogenic steps within Complex I, or indirectly, by Q/QH2 ratio). The structural (and functional) separation of the sites created for NADH oxidation and/or NAD+ reduction provides a “futile” cycle at any NADH/NAD+ ratio in the mitochondrial matrix. Such a cycle would serve as a fine tool for the “kinetic” control of the major entry point to the respiratory chain by the “thermodynamic” redox potential of the matrix.
Physiological significance of O·2- generation by the respiratory chain (mainly by Complex I) seems to us of minor importance, at least under normal mitochondrial functioning. The rate of superoxide formation is quite low and combined operation of SOD (Fig. 1), catalase [41], and glutathione peroxidase [42] in matrix is expected to diminish or neglect completely the potential ability of the respiratory chain to create a significant amount of superoxide. This, by no mean, makes superoxide production by mitochondria unimportant (see review [1]). First, this process may become of great importance under some extreme conditions such as reoxygenation after anoxia when Complex I is converted into its de-activated form [31] (see Fig. 4). Second, the relative “leakage” of the respiratory chain for univalent oxygen reduction may be tissue specific; comparative quantitative analysis of mitochondria from different tissues and species would be most interesting. At last, it should be emphasized that well-known hydrogen peroxide formation by mitochondria is not necessarily due to unspecific interactions of oxygen with the respiratory chain. Mitochondria contain several enzymes potentially capable of superoxide and/or hydrogen peroxide formation, those are monoamine oxidase of the outer mitochondrial membrane and flavin containing enzymes in matrix such as dehydrogenases of alpha-keto acids and D,T-diaphorase. NAD(P)H can serve as the electron donor for the latter enzymes and the steady-state level of the reduced nucleotides certainly depends on the activities of the respiratory chain and the energy-linked transhydrogenase. At present, the analysis of relative contribution of different enzymes into overall superoxide and hydrogen peroxide production by mitochondria remains strongly dependent on reliability of the methods for quantitative determination of these analytically “difficult” compounds.
This study was supported by the Russian Foundation for Basic Research (grant 02-04-48679 and grant 03-04-48202), by the program Leading Schools in Science (grant 00-15-97798), and by USA NIH Fogarty International Research grant 1R03TW006041.
REFERENCES
1.Chance, B., Sies, H., and Boveris, A. (1979)
Physiol Rev., 59, 527-605.
2.Boveris, A., Oshino, N., and Chance, B. (1972)
Biochem. J., 128, 617-630.
3.Boveris, A., and Chance, B. (1973) Biochem.
J., 134, 707-716.
4.Hinkle, P. C., Butow, R. A., and Racker, E. (1967)
J. Biol. Chem., 242, 5162-5173.
5.Loschen, G., Flohe, L., and Chance, B. (1971)
FEBS Lett., 18, 261-264.
6.Loschen, G., Azzi, A., Richten, C., and Flohe, L.
(1974) FEBS Lett., 42, 68-72.
7.Fridovich, I. (1974) Adv. Enzymol.,
41, 35-97.
8.Ksenzenko, M. Y., Konstantinov, A. A., Khomutov, G.
B., Tikhonov, A. N., and Ruuge, E. K. (1988) FEBS Lett.,
155, 19-24.
9.Sun, J., and Trumpower, B. (2003) Arch. Biochem.
Biophys., 419, 198-206.
10.Boveris, A., and Cadenas, E. (2000) IUBMB
Life, 50, 245-250.
11.Walker, J. E. (1992) Q. Rev. Biophys.,
25, 253-324.
12.Grivennikova, V. G., and Vinogradov, A. D. (2003)
Uspekhi Biol. Chim., 43, 19-58.
13.Vinogradov, A. D. (2001) Biochemistry
(Moscow), 66, 1086-1097.
14.Azzi, A., Davies, K. J. A., and Kelly, F. (2004)
FEBS Lett., 558, 3-6.
15.Skulachev, V. P. (1996) Q. Rev. Biophys.,
29, 169-202.
16.Kotlyar, A. B., and Vinogradov, A. D. (1990)
Biochim. Biophys. Acta, 1019, 151-158.
17.Burbaev, D. Sh., Moroz, I. A., Kotlyar, A. B.,
Sled, V. D., and Vinogradov, A. D. (1989) FEBS Lett.,
254, 47-51.
18.Azzi, A., Montecucco, C., and Richter, C. (1975)
Biochem. Biophys. Res. Commun., 65, 597-603.
19.Minakami, S., Titani, K., and Ishikura, H. (1958)
J. Biochem. (Tokyo), 64, 341-352.
20.Vinogradov, A. D. (1993) J. Bioenerg.
Biomembr., 25, 367-375.
21.Krishnamoorthy, A. I. G., and Hinkle, P. C.
(1988) J. Biol. Chem., 263, 17566-17575.
22.Kotlyar, A. B., Sled, V. D., Burbev, D. Sh.,
Moroz, I. A., and Vinogradov, A. D. (1990) FEBS Lett.,
264, 17-20.
23.Magnitsky, S., Toulokhonova, L., Yano, T., Sled,
V. D., Hägerhäll, C., Grivennikova, V. G., Burbaev, D. S.,
Vinogradov, A. D., and Ohnishi, T. (2002) J. Bioenerg.
Biomembr., 34, 193-208.
24.Schägger, H., and Pfeiffer, K. (2000)
EMBO J., 19, 1777-1783.
25.Nicholls, P., and Schonbaum, G. R. (1963) in
The Enzymes (Boyer, P., Lardy, H. A., and Myrbäck, K.,
eds.) Vol. 8, Academic Press, N. Y., pp. 147-225.
26.Zharova, T. V., and Vinogradov, A. D. (1997)
Biochim. Biophys. Acta, 130, 256-264.
27.Zakharova, N. V., Zharova, T. V., and Vinogradov,
A. D. (1999) FEBS Lett., 444, 211-216.
28.Vinogradov, A. D., and Grivennikova, V. G. (2001)
IUBMB Life, 52, 129-134.
29.Maklashina, E. O., Sled, V. D., and Vinogradov,
A. D. (1994) Biochemistry (Moscow), 59, 707-713.
30.Grivennikova, V. G., Kapustin, A. N., and
Vinogradov, A. D. (2001) J. Biol. Chem., 276,
9038-9044.
31.Maklashina, E., Sher, Y., Zong-Zhou, H., Gray, M.
O., Karliner, J. S., and Cecchini, G. (2002) Biochim. Biophys.
Acta, 1556, 6-12.
32.Vinogradov, A. D., Gavrikova, E. V.,
Grivennikova, V. G., Zharova, T. V., and Zakharova, N. V. (1999)
Biochemistry (Moscow), 64, 136-152.
33.Galkin, A. S., Grivennikova, V. G., and
Vinogradov, A. D. (2001) Biochemistry (Moscow), 66,
435-443.
34.Singer, T. P. (1974) Meth. Biochem. Anal.,
22, 123-175.
35.Sled, V. D., and Vinogradov, A. D. (1993)
Biochim. Biophys. Acta, 1141, 262-268.
36.Dooijewaard, G., and Slater, E. C. (1976)
Biochim. Biophys. Acta, 440, 1-15.
37.Clark, W. M. (1960) Oxidation-Reduction
Potentials of Organic Systems, Williams & Wilkins
Coo, Baltimore.
38.Dixon, M. (1971) Biochim. Biophys. Acta,
226, 269-284.
39.Hatefi, Y., and Galante, Y. M. (1977) Proc.
Natl. Acad. Sci. USA, 74, 846-850.
40.Albracht, S. P. J., and Hedderich, R. (2000)
FEBS Lett., 485, 1-6.
41.Nohl, H., and Hegner, D. (1978) FEBS
Lett., 89, 126-130.
42.Flohe, L., Günzler, W. A., and Ladenstein,
R. (1976) in Glutathione: Metabolism and Function (Arias, I. M.,
and Jakoby, W. B., eds.) Raven, N. Y., pp. 115-135.