The Adenine Nucleotide Translocase--a Carrier Protein Potentially Required for Mitochondrial Generation of NAD
M. Ziegler
Universitetet i Bergen, Molekylarbiologisk institutt, Thormohlensgt. 55, 5020 Bergen, Norway; E-mail: Mathias.Ziegler@mbi.uib.noReceived September 23, 2004
Just about 20 years ago I learned what bioenergetics is and “what it is eaten with” (“S chem eio ed'at”--one of V. P. Skulachev's favorite expressions that I remember). I took a one-semester course on the subject at Moscow State University. Being a student at the Biochemistry division of the then 2nd Moscow Medical Institute I was encouraged by our professors to attend that course. They must have known that, at least at that time, no textbook would have even nearly covered the broad range of topics addressed in V. P. Skulachev's lectures. And in the textbooks one would also not find the “Songs on bioenergetics” which were an integral part of the lectures. At that time I didn't have the faintest idea that four years later I would spend a few months in the Belozersky laboratory, where these lectures were generated.
During my Ph.D. work in Berlin, I was, among other things, involved in studying the kinetics of the adenine nucleotide translocase. Initially, these measurements were done by using radioactive substrate, spinning the mitochondria through a silicone oil layer to remove excess substrate and radioactivity before HPLC analysis of nucleotides. For the mess with the silicone oil I soon started to hate the translocase. To make things worse, it was summer and the density of the oil changed just as did the temperature, precluding any day-to-day reproducibility of the experiments. Perhaps it was good luck that the permeability transition pore was not known at that time. Therefore, the function of the translocase was suspected just to exchange ATP against ADP. Almost. There was the idea that fatty acids might also be a transport substrate and thus the translocase would be able to “catalyze” uncoupling [1]. Although it was again the translocase, I was very excited to go to the Bioenergetics group at the Belozersky laboratory to be involved in studying this possibility. It was amazing how much I could learn in just three months. For me, the most spectacular method was the time-resolved measurement of proton translocation across artificial membranes that was developed in Skulachev's laboratory [2]. Apart from the scientific methods, it was also impressive how intense the exchange of ideas in this department was. I could talk to virtually anybody about my project and would then walk away with a bunch of new ideas how to approach the problem. This fantastic atmosphere diffused immediately the frustration that one often gets after an unsuccessful experiment. In fact, I had to agree that there are no unsuccessful experiments. First, you learn how the things do not work. Second, you have a lot of new things to discuss again with the people in the department. Despite of all these wonderful impressions, my personal relationship to the translocase did not really improve.
A year later, in May 1989, I attended a FEBS advanced course on bioenergetics in Budapest. Of course, I met V. P. Skulachev there again and learned about another of his faculties, besides science. He then predicted the fall of the Berlin Wall to happen in the near future. Living in East Berlin at that time, I had but a smile for this fortune-telling. At that course, I also met Harvey Penefsky who invited me to do a post-doc with him. Thanks to history, Skulachev's prediction came true and I could indeed go to Syracuse, New York State, a year later.
In Penefsky's laboratory, I was introduced into the field of ATP synthase and received excellent training, particularly in the use of fluorescent probes. The intention was to study conformational changes in the mitochondrial ATP synthase using flash photolysis of a caged proton. The synthesis of the caged proton was set up by Harvey himself and the only pleasant thing I remember about it is the scent of vanillin, the starting material. Fortunately, Harvey had an excellent technician who then could make a lot of the probe. In the meantime, I had started setting up the fluorescent measurement of protons translocated into submitochondrial particles (SMPs) or phospholipid vesicles (see color insert, Fig. 1) [3]. Measuring proton movements was, of course, somewhat reminiscent of the time in Moscow. In addition, it turned out that there were people that I had met in Skulachev's laboratory (Boris Chernyak and Yannik Milgrom) now working next door in the department in Syracuse. It's a small world! What I didn't expect either was that I would again get involved with the translocase! Not in Penefsky's laboratory! Well, as described below, this is apparently unavoidable for me.
Using the fluorescent dye pyranine, entrapped in SMPs, I could readily measure accumulation of protons following addition of ATP or NADH, as indicated by a decrease in the fluorescence. Even entrapping the caged proton, I could measure instantaneous internal acidification following illumination. One day I made a fascinating observation and Harvey was just as excited about it as I was: following addition of oligomycin, pyranine-loaded SMPs responded with a fluorescence signal indicating acidification, even in the presence of uncoupler! Clearly, this had to mean that oligomycin caused a release of protons when binding to Fo. Here it was, the “proton well”! It took quite a long time, not to mention the frustration, until it turned out that under this condition pyranine was not reporting any pH-dependent signal, but was specifically sequestered within the membranes and its fluorescence quenched. The effect had an unusual dependence on the presence of adenine nucleotides on both sides of the SMPs.Fig. 1. Principle of the measurement of proton translocation into submitochondrial particles using the fluorescence response of entrapped pyranine. Upon protonation of pyranine, the fluorescence is quenched, but only in the pH-sensitive part of the pyranine excitation spectrum.
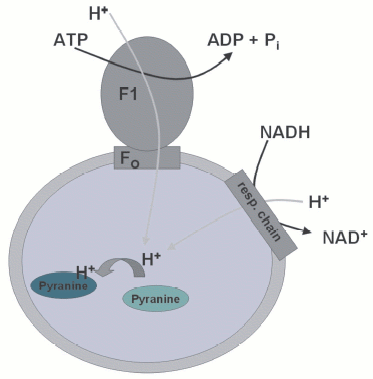
The use of specific inhibitors (carboxyatractyloside and bongkrekic acid) removed all doubt--it was the translocase, again. As far as we understood, what happened was that oligomycin caused the formation of a highly specific binding site for pyranine, but this, in turn, was dependent on the conformation of the translocase! The conclusion, therefore, was that Fo and the translocase interact highly specifically within the membrane of the SMPs [3]. Although this was a very interesting observation, there was no time to look into it in more detail. As a quite useful byproduct, I had developed an elegant method to measure nucleotide binding to the translocase. I could in no time reproduce the nucleotide specificity known for the translocase, confirmed that pyrophosphate is also a substrate, etc. Pyranine entrapped in SMPs would faithfully report binding to the translocase as long as oligomycin was present. For those who might share my destiny in not being able to get away from the translocase, I provide the recipe (Box 1). It is indeed very easy. Perhaps, it may also help understanding the role of the translocase in the formation of the PTP. Since part of my current work is related to apoptosis, specifically the mitochondrial pathway, this is a somewhat scary thought for me.
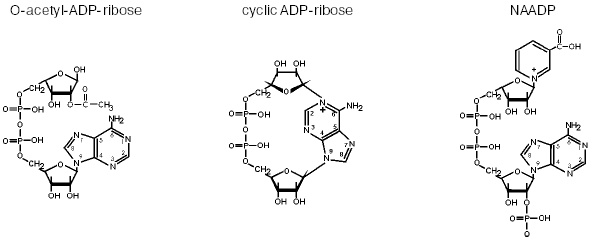
For about ten years, I have been studying another very interesting part of bioenergetics--the functions and biosynthesis of pyridine nucleotides. In the classical view of bioenergetics, there would be no question that these molecules have the sole function of electron transfer. Therefore, their synthesis was not supposed to be of major interest, because in the redox reactions there is no consumption or degradation of NAD(P). However, just like ATP, the pyridine nucleotides have been demonstrated to carry a variety of signaling functions [4]. Thus, NAD(P) is used to form second messengers with intracellular calcium mobilizing activity. Among them, cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) are known best (Fig. 2). Moreover, NAD also serves as substrate for the posttranslational modification of proteins. These protein modifications include mono- and poly-ADP-ribosylation. Only very recently, a further mode of NAD-mediated protein modification was detected, the NAD-dependent deacetylation of proteins [5]. In these reactions, the nicotinamide moiety of NAD is replaced by the acetyl group previously attached to the protein, thereby forming a novel molecule, O-acetyl ADP-ribose (Fig. 2). It is likely that this NAD derivative exerts regulatory influence on important cellular events [5, 6].
All known NAD(P)-mediated signaling reactions include the cleavage of nicotinamide from the molecule and a further reaction of the remaining ADP-ribose moiety. Since the resynthesis of NAD from ADP-ribose and nicotinamide requires at least four molecules of ATP, these reactions have to be tightly controlled. Moreover, depending on the physiological state of the cell, NAD-mediated regulatory reactions may consume considerable amounts of the pyridine nucleotide. Consequently, there is a constant need for the biosynthesis of NAD to supply these reactions and to replenish the cellular pool of pyridine nucleotides for bioenergetic purposes as well. This realization has boosted the interest in the biosynthetic pathways for the generation of NAD and NADP. Although the principal reactions have been known for a long time, the molecular characterization of the enzymes involved has largely been achieved only in the past few years [4]. A hitherto unknown route of NAD biosynthesis, in which nicotinamide riboside serves as the initial substrate, has been established recently [7].Fig. 2. Messenger molecules derived from NAD(P). Cyclic ADP-ribose and NAADP are potent intracellular calcium-releasing agents. The mode of action of cyclic ADP-ribose is similar to and functionally redundant with that of inositol-1,4,5-trisphosphate (InsP3). NAADP is active on a calcium store distinct from the endoplasmic reticulum and may serve as the initiator of ordered intracellular calcium waves. O-Acetyl-ADP-ribose represents a “byproduct” of NAD-dependent protein deacetylation catalyzed by enzymes of the Sir2 family. Its biological role has not been revealed, but it is likely to exert regulatory functions [6].
In mammals, the major route of NAD de novo synthesis proceeds via reaction of nicotinamide (or nicotinic acid) with phosphoribosyl pyrophosphate yielding the corresponding mononucleotide, nicotinamide mononucleotide (NMN) (or nicotinic acid mononucleotide (NaMN)). The precursors, nicotinamide and nicotinic acid, are known as niacin or vitamin B6. Only now, being in Norway, I actually became aware that this was discovered by a Norwegian (Conrad A. Elvehjem) who was born and lived in the United States and for several years (1958-1962) served as president of the University of Wisconsin. In his seminal works on the “black tongue disease”, he established that niacin was the missing constituent in the diet causing pellagra and that adding niacin was sufficient to cure the symptoms [8].
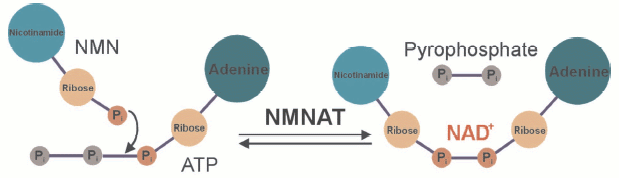
The mononucleotides NMN and NaMN are reacted with ATP to give rise to NAD or NaAD under the release of pyrophosphate (see color insert, Fig. 3). The nicotinic acid containing form, NaAD, is then amidated and thereby converted to NAD. As indicated above, NMN can also be generated from nicotinamide riboside by a specific kinase [7]. It is obvious then that the final step, the formation of NAD from NMN and ATP represents a key reaction, because all pathways, de novo and salvage, require the activity of the corresponding enzyme, nicotinamide mononucleotide adenylyl transferase, NMNAT (Fig. 3). Indeed, this enzyme has become of major interest owing to its critical role not only for the cellular energy supply. A major link between NAD biosynthesis and transcriptional control was originally established in the yeast Saccharomyces cerevisiae. The NAD-dependent protein deacetylase Sir2 (silent information regulator 2) was demonstrated to catalyze histone deacetylation and thereby specific silencing of transcription [5, 6]. Enhanced catalytic activity of Sir2 (e.g., by overexpressing it) resulted in a substantial extension of the replicative lifespan. Interestingly, Sir2 activation was also found to be the critical event for the lifespan extension under conditions of calorie restriction, a well-known biological phenomenon. Moreover, the activation of Sir2 could be ascribed to an enhanced reutilization of nicotinamide, a byproduct of the Sir2-mediated histone deacetylation. The reason for this effect is that nicotinamide is a strong endogenous inhibitor of Sir2. Therefore, its accumulation would inhibit Sir2 and thereby suppress its silencing activity. Indeed, overexpression of individual enzymes of the nicotinamide salvage pathway, including NMNAT, results in a similar extension of the lifespan as found for Sir2 overexpression. In the meantime, similar observations have been made in higher organisms including C. elegans and human cells [5].
Even the degeneration of nerve cells after injury is governed by a similar mechanism. Usually, transection of neurites results in a rapid degeneration (a few days) of the nerve ends, a process called Wallerian degeneration. A mouse strain is known which exhibits a largely delayed Wallerian degeneration, the Wlds (Wallerian degeneration slow) mice. It turned out that these mice carry a mutation that leads to a triplication of the NMNAT gene and thereby to a substantially higher degree of its expression [9]. The cellular level of NAD is unchanged suggesting that it is not the level of NAD, but its rapid resynthesis that is responsible for the longer survival of the nerve termini. It turned out that this effect is indeed mediated by the mouse homolog of Sir2, that is, slowed Wallerian degeneration in the Wlds mice is a phenomenon based on the same mechanism as lifespan extension under conditions of calorie restriction and governed by NAD biosynthetic enzymes.Fig. 3. The key step of NAD biosynthesis. Nicotinamide mononucleotide adenylyl transferase (NMNAT) joins two nucleotides, NMN and AMP, to form the dinucleotide. In humans, primarily the nicotinamide derivative is thought to be used. However, the nicotinic acid derivative of NAD can be formed from nicotinic acid mononucleotide (instead of NMN). In this case, the nicotinic acid adenine dinucleotide is then amidated by NAD synthase to yield NAD. Three human isoforms of NMNAT are known, which appear to have different subcellular localizations.
Not only for these reasons has NMNAT become of major interest. In human cells, the enzyme originally identified is located within the nucleus [10]. Suspected to be just a metabolic enzyme, this was, of course, quite a surprise. What could NAD synthesis be good for in the nucleus? It has to be considered, however, that besides the Sir2 homolog (SIRT1), the major consumer of cellular NAD, poly-ADP-ribose polymerase 1 (PARP1), is also located within the nucleus. PARP1 has a high affinity to DNA strand breaks and upon binding to them becomes catalytically activated. It then synthesizes polymers of ADP-ribose (from NAD) and attaches them to specific target proteins. This protein modification, in turn, appears to be of utmost importance for the recruitment of DNA repair complexes, and also for the regulation of other major cellular events including transcription, cell cycle regulation, and apoptosis. Experimentally, PARP1 can be activated, for example, by treating cultured cells with agents damaging DNA such as hydrogen peroxide. Depending on the extent of DNA damage the consumption of NAD for the poly-ADP-ribosylation reaction will vary. Accordingly, the cells may repair the damage without further problems, the polymers may initiate the mitochondrial pathway of apoptosis [11], or the cells cannot cope with the stress and die by necrosis at least in part due to energy depletion. Indeed, if damage is severe, almost the entire store of cellular NAD may become depleted [12]. Therefore, the presence of NMNAT within the nucleus may represent a means to supply PARP1 directly with NAD under these conditions. We have now found that the nuclear NMNAT actually activates PARP1 and that this activation can be abolished by phosphorylation of NMNAT (Berger et al., unpublished observations). This observation establishes a quite interesting link between NAD biosynthesis and consumption. In addition, the functional link with SIRT1 places NMNAT at a key position of cellular survival: lifespan extension via SIRT1 activation or cell death via over-activation of PARP1.
Another important question relates to the subcellular compartmentation of NAD and its biosynthesis. It is certainly a quite unexpected situation that to this day it is totally unknown how the mitochondrial pyridine nucleotide pool is generated (except, perhaps, in plants, whose mitochondria are permeable to NAD). There is no doubt that a major proportion of NAD(P) is localized within the matrix of these organelles [13]. On the other hand, since nuclear poly-ADP-ribosylation may consume virtually all cellular NAD, there has to be a pathway to provide access to the mitochondrial NAD pool under these conditions. While the latter could potentially be mediated by the permeability transition pore (PTP), the precursors and their intramitochondrial conversions to generate NAD remain unknown. Since the concentration of NAD(P) in mitochondria is high relative to that in the cytosol, it is rather unlikely that pyridine nucleotides are physiologically taken up via the PTP. It is interesting in this regard, that two other human isoforms of NMNAT have been discovered [14]. When fused to the green fluorescent protein, they appear to localize primarily to the cytoplasm. However, the third isoform may also, at least in part, localize to mitochondria [14]. This is an intriguing possibility that we also investigate now. Unfortunately, there is a rather dangerous aspect to it: a substrate for NMNAT is ATP. This could be transported into the organelles, but is likely to be generated from ADP by the ATP synthase. In any case, the substrate would have to enter the matrix via the translocase!
It is also thanks to Vladimir Petrovich Skulachev that there are at least a few quite pleasant memories about this protein! My most cordial congratulations on his birthday and best wishes for his future scientific and personal life! From a scientific point of view, I would suggest a low calorie diet, supplemented with sufficient niacin and a glass of wine every now and then--it contains resveratrol, a potent activator of Sir2. In other words, I hope to enjoy more of his exciting scientific ideas for many years to come.
REFERENCES
1.Andreyev, A. Yu., Bondareva, T. O., Dedukhova, V.
I., Mokhova, E. N., Skulachev, V. P., and Volkov, N. I. (1988) FEBS
Lett., 226, 265-269.
2.Drachev, L. A., Jasaitis, A. A., Kaulen, A. D.,
Kondrashin, A. A., Liberman, E. A., Nemecek, I. B., Ostroumov, S. A.,
Semenov, A. Yu., and Skulachev, V. P. (1974) Nature, 249,
321-324.
3.Ziegler, M., and Penefsky, H. S. (1993) J. Biol.
Chem., 268, 25320-25328.
4.Berger, F., Ramirez-Hernandez, M. H., and Ziegler,
M. (2004) Trends Biochem. Sci., 29, 111-118.
5.Blander, G., and Guarente, L. (2004) Annu. Rev.
Biochem., 73, 417-435.
6.Denu, J. M. (2003) Trends Biochem. Sci.,
28, 41-48.
7.Bieganowski, P., and Brenner, C. (2004)
Cell, 117, 495-502.
8.Elvehjem, C. A., Madden, R. J., Strong, F. M., and
Woolley, D. W. (1938) J. Biol. Chem., 123, 137-149.
9.Mack, T. G., Reiner, M., Beirowski, B., Mi, W.,
Emanuelli, M., Wagner, D., Thomson, D., Gillingwater, T., Court, F.,
Conforti, L., Fernando, F. S., Tarlton, A., Andressen, C., Addicks, K.,
Magni, G., Ribchester, R. R., Perry, V. H., and Coleman, M. P. (2001)
Nat. Neurosci., 4, 1199-1206.
10.Schweiger, M., Hennig, K., Lerner, F., Niere, M.,
Hirsch-Kauffman, M., Specht, T., Wiese, C., Oei, S. L., and Ziegler, M.
(2001) FEBS Lett., 492, 95-100.
11.Yu, S. W., Wang, H., Poitras, M. F., Coombs, C.,
Bowers, W. J., Federoff, H. J., Poirier, G. G., Dawson, T. M., and
Dawson, V. L. (2002) Science, 297, 259-263.
12.Chiarugi, A. (2002) Trends Pharmacol.
Sci., 23, 122-129.
13.Di Lisa, F., and Ziegler, M. (2001) FEBS
Lett., 492, 4-8.
14.Zhang, X., Kurnasov, O. V., Karthikeyan, S.,
Grishin, N. V., Osterman, A. L., and Zhang, H. (2003) J. Biol.
Chem., 278, 13503-13511.
BOX 1. PROTOCOL FOR THE PYRANINE FLUORESCENCE-BASED MEASUREMENT
OF SUBSTRATE BINDING TO THE ADENINE NUCLEOTIDE TRANSLOCASE [3]
Pyranine and ATP loaded SMPs report specifically binding of transport substrates at the matrix side (external side of SMPs). For unknown reasons the quenching of pyranine fluorescence by external translocase substrates occurs only if oligomycin has been added to the particles. Presumably, there is a specific interaction between the ATP synthase (Fo) and the translocase, which is “fixed” by oligomycin, and this state is sensed by the fluorescent dye.
Preparation of Pyranine-SMPs
1. Sonicate a mitochondrial suspension (~15-20 mg/ml) in 60 mM K2SO4, 10 mM MgSO4, 100 mM sucrose, 1 mM Tricine, pH 8.0, and (that's the most important part) 0.5 mM pyranine + 1 mM ATP.
2. Thoroughly wash the pyranine-SMPs by centrifugation. They can then be stored frozen.
3. Thawed pyranine-SMPs can be separated from free pyranine by gel filtration (e.g., using Sephadex G-75).
Fluorescence Measurements
4. Excitation wavelength, 470 nm; emission wavelength, 511 nm. These settings permit to measure both internal acidification (following NADH or ATP addition to the particles) or translocase-dependent fluorescence quenching (following oligomycin addition). Ideally, a pyranine quencher (e.g., butane bis 2,2´-bipyridinium, 12 mM) should be added to the medium to minimize fluorescence of pyranine adsorbed on the external surface of the particles. This used to be available from Molecular Probes (USA).
5. Titration of translocase substrates (for the matrix side, which is the external surface of the pyranine-SMPs). 1) Use about 0.1-0.2 mg/ml pyranine-SMPs in a solution consisting of 100 mM MOPS-KOH, pH 7.5, 60 mM K2SO4, 10 mM MgSO4. To avoid interference with pH effects, the particles are uncoupled using FCCP and valinomycin. 2) Oligomycin (4 µg/ml) is added to make the system responsive to external nucleotides (this is the mystical part). 3) When a stable baseline is obtained, add the desired nucleotide which is supposed to be a substrate of the translocase (as a control, ADP or ATP are used). Depending on the concentration and affinity to the translocase the pyranine fluorescence will decrease and reach a stable level within a few minutes. 4) Using several concentrations the decrease in pyranine fluorescence can be used as a measure for binding of the compound to the translocase. Relate the fluorescence decrease to the maximal decrease observed (saturation of the translocase binding sites by excess of substrate). 5) The obtained values can then be used to calculate the affinity (Km) of the compound in question to the translocase. In analogy to the Michaelis-Menten kinetics, use the relative fluorescence decreases in place of the reaction rate (v). Vmax should be about 1, because all values were related to the maximal quenching. Since no activity is actually measured, this value is meaningless anyway. In principle, of course, this value should correspond to the amount of available binding sites. However, it is not possible to quantify this properly.
6. Good luck!