REVIEW: Role of Cooperative H+/e- Linkage (Redox Bohr Effect) at Heme a/CuA and Heme a3/CuB in the Proton Pump of Cytochrome c Oxidase
S. Papa
Institute of Bioenergetics and Biomembranes, National Council of Research (CNR); Department of Medical Biochemistry and Biology, University of Bari, 70124 Bari, Italy; fax: +39-080-5478109; E-mail: papabchm@cimedoc.uniba.it
Received September 24, 2004
It is a pleasure to contribute to the special issue published in honor of Vladimir Skulachev, a distinguished scientist who greatly contributes to maintain a high standard of biochemical research in Russia. A more particular reason can be found in his work (Artzabanov, V. Y., Konstantinov, A. A., and Skulachev, V. P. (1978) FEBS Lett., 87, 180-185), where observations anticipating some ideas presented in my article were reported. Cytochrome c oxidase exhibits protonmotive, redox linked allosteric cooperativity. Experimental observations on soluble bovine cytochrome c oxidase are presented showing that oxido-reduction of heme a/CuA and heme a3/CuB is linked to deprotonation/protonation of two clusters of protolytic groups, A1 and A2, respectively. This cooperative linkage (redox Bohr effect) results in the translocation of 1 H+/oxidase molecule upon oxido-reduction of heme a/CuA and heme a3/CuB, respectively. Results on liposome-reconstituted oxidase show that upon oxidation of heme a/CuA and heme a3/CuB protons from A1 and A2 are released in the outer aqueous phase. A1 but not A2 appears to take up protons from the inner aqueous space upon reduction of the respective redox center. A cooperative model is presented in which the A1 and A2 clusters, operating in close sequence, constitute together the gate of the proton pump in cytochrome c oxidase.
KEY WORDS: cytochrome c oxidase, proton pump, cooperative coupling
Allosteric cooperativity is a fundamental property in different hemoproteins and enzymes [1, 2]. This property confers to the protein the capacity to feel and respond to the physiological state of cells and overall organisms. The prototype of an allosteric protein is provided by hemoglobin. In hemoglobin the binding of O2 to the heme iron pulls this by 0.5 Å in the plane of the porphyrin ring [3]. This small movement of the iron, which pulls after it the histidine coordinated at the fifth position, is amplified by a conformational wave that modifies the surface contacts of the globin chains. The result is a sequential increase in the binding affinity from the first to the fourth oxygen molecule [3]. Another important conformational change initiated by oxygen binding is the rupture at the surface of the beta globins of a salt bridge between aspartate 94 and histidine 146. As a consequence the pK of the histidine decreases with proton release in the aqueous phase. This cooperative interaction, known as the alkaline Bohr effect, since it takes place at pH above 6, makes the oxygen affinity of hemoglobin increase with pH [3, 4]. The Bohr effect results in an amplified physiological function in that it makes hemoglobin bind oxygen in the lung where the pH can be slightly higher than 7.4 and to release oxygen to tissues where pH is around pH 7.4 or lower [2].
As far as membrane bound hemoproteins are concerned, b and a type cytochromes of energy-transfer respiratory chains exhibit a pH dependence of the midpoint redox potential [5]. It means that electron transfer at the metal is linked to proton exchange by protolytic groups in the enzyme [6]. Starting from this property of cytochromes, I proposed in the seventies that cooperative linkage between the redox state of the metal and pK of protolytic group(s) in the protein could result in redox-driven proton pumping across the coupling membrane if electron delivery to the metal was associated with pK increase of a group with proton uptake from one of the two aqueous phases separated by the osmotic barrier of the membrane, and electron transfer to a downhill redox center to pK decrease of the same group, or another in proton connection with the first, with proton release in the opposite aqueous phase [7, 8]. This thermodynamic coupling between electron transfer and proton pumping, against a respiratory steady-state protonmotive force of some 250 mV in coupling membranes, will require an effective DeltapK of 4-5 units and a DeltaEh of 250-300 mV at least. It can be noted that this principle of vectorially organized redox-linked pK shifts of protolytic groups is now incorporated in some of the more recent models proposed for the proton pumping activity of cytochrome c oxidase [9-12]. It is feasible that cooperative protonmotive mechanisms may well operate in proton pumps in series or in parallel with direct primary protonmotive catalytic steps. An analogy can also be drawn, as anticipated some time ago [13], with the detailed models now put forward for the light driven proton pump of bacteriorhodopsin [14].
REDOX-LINKED CONFORMATIONAL CHANGES IN CYTOCHROME c
OXIDASE
Cytochrome c oxidase (Fig. 1) presents definite signs of redox-linked allosteric cooperativity. The Em of the four metal centers of the oxidase (CuA, heme a, heme a3, and CuB) is pH dependent [5, 15-17]. There are anticooperative redox interactions of the metal centers [15, 18]. X-Ray crystallographic analysis shows redox-coupled structural changes in subunit I of the oxidase [19]. Picosecond time resolved tryptophan fluorescence also indicates redox-linked conformational changes [20]. In both cases the conformational changes appear to take place in the environment of heme a.
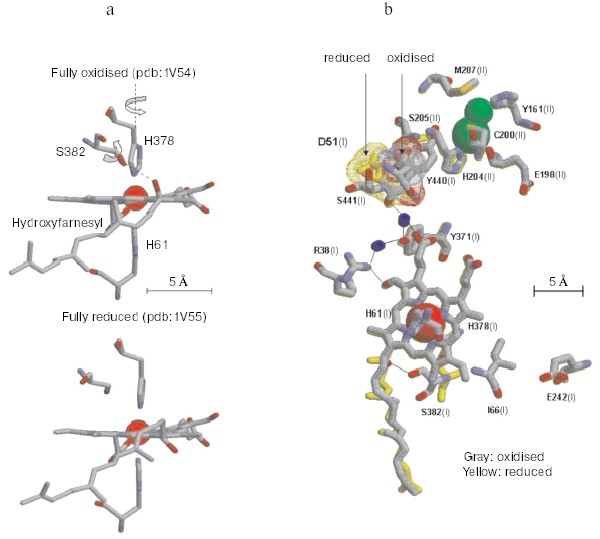
Non-resonant Raman spectroscopy has revealed in the oxidized bovine cytochrome c oxidase a peak at around 1645 cm-1 that disappears in the reduced state [21]. Comparison of the Raman spectra of the unliganded oxidase with those exhibited by the CN-ligated oxidase, quinol oxidase which lacks CuA, cytochrome c, and imidazole derivatives of the extracted hemochrome allowed to assign the redox-sensitive Raman peak to a transition in the stretching vibrational mode of the C=C or C=N bond in the imidazole ring of one of the two axial histidine ligands of the low spin heme a [21]. Inspection of the X-ray crystallographic structures of the oxidized and fully reduced bovine oxidase reveals that reduction results in a few degree rotation of the imidazole plane of I-His378 on the axis perpendicular to the porphyrin plane [19] (see color insert, Fig. 2a). This movement of the histidine ligand, in a sense reminiscent of the movement of the histidine ligand induced by oxygen binding in hemoglobin, although small appears to be the closest sign of a perturbation of the protein structure induced by a change in the redox state of the heme a iron. The X-ray crystallographic structures show that the reduction of the oxidase results also in more significant conformational changes at heme a. Reduction causes the rupture of a hydrogen bond between the OH group of the hydroxyfarnesyl substituent of heme a and I-Ser382, with concomitant movement of serine and the hydrocarbon chain of the hydroxyfarnesyl [19]. A conformational wave induced by reduction of the oxidase appears to reach the cytosolic surface of subunit I in contact with subunit II. A segment of subunit I from I-Gly49 to I-Asn55 moves towards the surface with the carboxylic group of Asp51 becoming exposed to the aqueous phase [12, 19] (see color insert, Fig. 2b). Again an analogy seems to exist between these redox linked local/global conformational changes in the oxidase and those underlying the Bohr effect in hemoglobin.Fig. 1. Sketch of cytochrome c oxidase with location of the redox centers relative to the plane of the coupling membrane as obtained from X-ray crystallographic structures (see [10] for review).
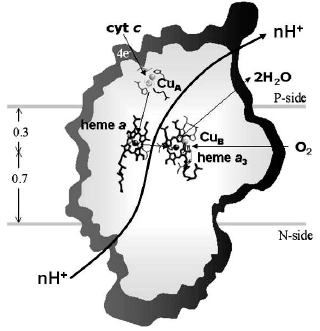
The X-ray crystallographic structures show also a hydrogen bond between I-Arg38 and the formyl substituent of heme a, which was anticipated by resonance Raman spectroscopy studies [22] (Fig. 2b). This approach also revealed the presence of a water molecule near the formyl [9].Fig. 2. Redox-linked conformational changes in bovine cytochrome c oxidase. The picture was elaborated with the Rasmol 26 program using the 1V54 atomic coordinates from the PDB bank of the monomer of cytochrome c oxidase, superimposing the 1V54 (1.8 Å resolution) and the 1V55 (1.9 Å resolution) atomic coordinates from the PDB data bank of the oxidized and reduced crystal structure of bovine cytochrome c oxidase, respectively (“What if” program). a) Details of the redox-linked movement of I-H378, I-S382, and hydroxyl-farnesyl at the low spin heme a [19, 21]. b) The redox-linked movement of I-Asp51 (see text and [12, 19]) is evidenced by dotting in color van der Waals atomic radii. The stick structures of other residues shown evidenced only minor position changes of their atomic coordinates. The thin line shows possible hydrogen bond networks [12, 19].
REDOX-LINKED PROTON TRANSLOCATION IN CYTOCHROME c
OXIDASE
Measurements of proton release and uptake, associated with the oxidation and reduction respectively of heme a and CuA in the soluble CO-inhibited bovine cytochrome c oxidase (COX) (a3 and CuB clamped in the reduced state) have shown that oxido-reduction of these centers is coupled to H+ transfer, with an H+/COX ratio varying between 0.7 and 0.9 in the pH range 6.0-8.5 [23]. These data have been recently confirmed by Forte et al. who showed that in the soluble bovine [24] and P. denitrificans cytochrome c oxidase [25] the reduction of heme a is associated with the uptake of 0.6-0.8 H+/COX.
Electron/proton coupling at heme a only, with a H+/COX coupling ratio of 0.7-0.9, was apparently inconsistent with previous measurements of the pH dependence of the Em of heme a, which in the CO-inhibited COX was reported to amount to not more than -20 mV per pH unit increase in the same pH range [17]. A solution to this was provided by the finding that CuA exhibits Em value and a pH dependence completely superimposed on that of heme a, the Em of both centers decreasing by around 20 mV/pH unit increase [23]. This shows that oxido-reduction of both heme a and CuA is linked to pK shifts of two or more common acid-base groups, whose overall balance results in the observed H+ release upon oxidation of the two centers and proton uptake upon their reduction [6]. The observed movement, upon reduction, of I-Asp51 from a hydrophobic interior to an aqueous environment, would result in proton dissociation from the carboxylic group of this residue [12, 19]. In the oxido-reduction of heme a this negative Bohr effect of I-Asp51 can however be associated with positive Bohr effects [26, 27]. Mathematical analysis of the pH dependence of proton transfer coupled to oxido-reduction of heme a and CuA and of the Em of the two redox centers in the CO-liganded soluble cytochrome c oxidase resulted in a best-fit of the experimental points by an equation representing the case in which both heme a and CuA shared coupling with a negative Bohr effect (I-Asp51, ?), pKox 7.3 - pKred 4.0, and a minimum of two positive Bohr effects (pKox 4.0 - pKred 6.9 and pKox 5.4 - pKred 9.0, respectively) [27]. It seems significant in this context that S205 of subunit II, which is not too distant from the binuclear CuA center, is in the crystal of the oxidized oxidase hydrogen bonded to I-Asp51 [12, 19]. In any event, it should be kept in mind that, in principle, the experimental points could be fitted by similar equations incorporating protolytic groups in the cluster with different pK shifts (cf. [28]).
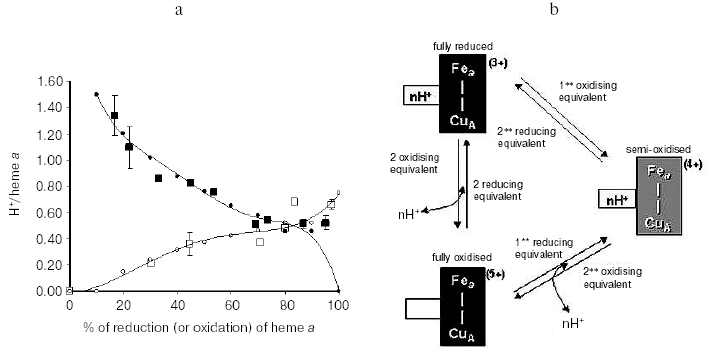
An important consequence of the interactive coupling of the oxido-reduction of both heme a and CuA with pK shifts in a common cluster of protolytic groups (A1) is that, while one electron reduction of the heme a/CuA center is sufficient to produce maximal protonation of the cluster, release of the proton bound to the cluster will take place only when both heme a and CuA are oxidized [29] (Fig. 3). The consequence is that at the steady state one electron at once has to pass through CuA and heme a to result in the translocation of around 1 H+ per electron. This rule of “one electron at once” might represent one of the causes of the slip, observed under certain conditions, in the proton pump of cytochrome c oxidase [27, 30, 31].
The vectorial nature of Bohr protons linked to oxido-reductions of heme a/CuA was investigated in the mixed valence CO-liganded bovine oxidase reconstituted in liposomes, in which heme a3 and CuB at the binuclear site were clamped in the reduced state [29]. The oxidation of heme a/CuA, reduced by an external reductant, was associated with H+ release in the external aqueous phase. The expected H+ uptake associated with re-reduction of heme a/CuA was considerably delayed with respect to completion of the reduction of these centers, unless CCCP was added [29]. These results thus showed that whilst oxidation of heme a/CuA results in the release in the external bulk phase of the Bohr protons linked to these centers, their reduction results in the uptake of the Bohr protons from the inner space. Consistent with this picture are previous results of Artzabanov et al. [32] who showed oxidation/reduction of heme a upon alkalinization/acidification, respectively, of the matrix space in intact rat liver mitochondria.Fig. 3. Proton release/uptake coupled with oxidation/reduction of heme a and CuA in CO-inhibited cytochrome c oxidase [29]. a) H+ uptake/release associated with stepwise reductive-oxidative titration of CuA and heme a in CO-liganded cytochrome c oxidase. The measured H+/heme a, CuA ratios for proton uptake-release associated with consecutive stepwise reductive-oxidative titrations with hexammineruthenium (II) and ferricyanide are shown by filled and empty squares, respectively. Simulation of the H+/heme a, CuA ratios is shown by small black circles for reductive titration and small empty circles for oxidation titration. The bars on the symbols (where given) indicate the standard errors of the mean value of three to four measurements. For experimental details see [29]. b) Model of interactive proton release/uptake coupled with oxidation/reduction of CuA and heme a in CO-inhibited cytochrome c oxidase. Reproduced with permission from [29].
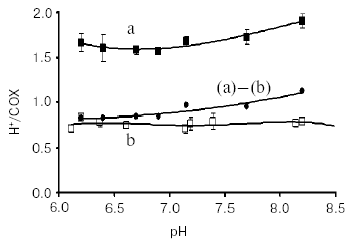
Redox Bohr protons have also been analyzed in the soluble unliganded bovine cytochrome c oxidase. In this case the fully oxidized oxidase was preincubated in the presence of 150 mM KCl, conditions under which the binuclear center is reported to be occupied by Cl-, thus preventing OH-/H2O exchange at this site [24]. Oxido-reduction of the four metal centers in the unliganded oxidase resulted in the release/uptake of 1.7-1.9 H+/COX. This H+ exchange is, at pH values around neutrality, around twice that observed upon oxido-reduction of heme a/CuA in the mixed-valence CO-liganded oxidase (Fig. 4) [31, 33]. Thus, oxido-reduction of heme a3/CuB at the binuclear center results in the exchange at a second cluster of protolytic groups, A2, of 0.8-0.9 Bohr protons.
The contribution of cooperative H+/e- linkage at heme a/CuA and a3/CuB (redox Bohr effects) in proton pumping was investigated in bovine cytochrome c oxidase reconstituted in liposomes. In a series of experiments the four metal centers in the unliganded oxidase were fully reduced in anaerobiosis by the photo activated EDTA-riboflavin system [34]. The four centers were then oxidized by a stoichiometric amount of ferricyanide. This resulted in a synchronous H+ release in the external bulk phase that amounted to an H+/COX ratio of 1.63 ± 0.37 (Fig. 5).Fig. 4. pH dependence of proton release/uptake associated with oxidation/reduction of metal centers induced by ferricyanide/ferrocyanide in anaerobic soluble bovine heart cytochrome c oxidase. Filled squares (a): H+/COX ratios associated with oxido-reduction of hemes a, a3, CuA, and CuB in the unliganded oxidase. Empty squares (b): H+/COX ratios associated with oxido-reduction of heme a and CuA in the CO-liganded COX, heme a3 and CuB clamped in the reduced state [23]. Soluble cytochrome c oxidase was preincubated in 150 mM KCl before induction of the redox transitions. For calculation of the H+/COX ratios in the unliganded oxidase presented in (a), molar extinction coefficient of 0.188 µM-1*cm-1 [35, 36] for heme a plus heme a3 was used. This is higher than molar extinction coefficient of 0.152 µM-1*cm-1 used in previous calculations [31, 33].
In another series of experiments the reconstituted, unliganded oxidase, fully reduced in anaerobiosis, was subjected to oxidation by a stoichiometric amount of oxygen, in the absence or presence of ferrocytochrome c. The aerobic oxidation of the oxidase resulted in fast H+ release in the external phase amounting, in a number of these experiments, to an H+/COX ratio of 1.9 ± 0.06 (Fig. 5). When the oxidation was immediately followed by a complete re-reduction of the metal centers of the oxidase by ferrocytochrome c, an additional H+ release was observed, which brought the overall H+/COX ratio to 2.66 ± 0.07 (Fig. 5). It should be noted that for similar experiments Verkhovsky et al. [36] reported an H+/COX ratio for the oxidation phase approaching 2, thus practically equal to that measured by us, but an overall H+/COX ratio approaching 4 when the oxidation phase was immediately followed by re-reduction of the metal centers. The reasons for this latter discrepancy are at present not clear (see also [37]).Fig. 5. Proton release associated with anaerobic ferricyanide oxidation of unliganded fully reduced soluble (a) and liposome-reconstituted bovine cytochrome c oxidase (b), aerobic oxidation (c) and aerobic oxidation-re-reduction (d) of liposome-reconstituted cytochrome c oxidase. a) Anaerobic reduction of cytochrome c oxidase (1 µM aa3 plus 0.5 µM cytochrome c) was attained by succinate oxidation in the presence of a trace of frozen-thawed broken beef-heart mitochondria. Oxidation was produced by the addition of an amount of ferricyanide stoichiometric with the sum of the reduced metal centers in the presence of antimycin plus myxothiazol [33]. b, c) Anaerobic full reduction of the oxidized oxidase, 0.8-1.0 µM aa3 plus 0.5 µM cytochrome c (b), 0.5-1.0 µM aa3 plus 0.1 µM cytochrome c (c) was attained by the photo-activated EDTA/riboflavin system [34]. d) Anaerobic full reduction of the oxidase, 1 µM aa3 plus 1 µM cytochrome c, was produced by 1 mM ascorbate and 20 µM hexammineruthenium. Oxidation was produced by the addition of an amount of ferricyanide stoichiometric with the sum of the reduced metal centers (b), or with an amount of O2 stoichiometric with that of aa3 (c and d). The concentration of aa3 was determined using molar extinction coefficient of 0.188 µM-1*cm-1 for heme a plus heme a3 [35, 36]. This is higher than molar extinction coefficient of 0.152 µM-1*cm-1 used in previous calculations [27]. (Capitanio, G., Martino, L., Capitanio, N., and Papa, S., unpublished results.)
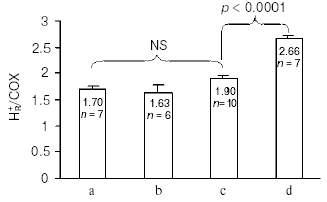
The H+/COX ratio for proton release in the external aqueous phase associated with the oxidation of the metal centers by ferricyanide in the reconstituted oxidase was practically equal to the H+/COX ratio for proton release induced by ferricyanide oxidation of the metal centers in the soluble oxidase. The H+/COX ratio for proton release induced by aerobic oxidation of the reconstituted oxidase was also very close, if not equal, to the above H+/COX ratios. It has to be concluded that the H+ release observed in the ferricyanide and the O2 oxidation of the fully reduced reconstituted oxidase derives from deprotonation of protolytic groups in the clusters A1 and A2 responsible for the redox Bohr effects linked to heme a/CuA and heme a3/CuB, respectively.
H+/COX ratios obtained from simulations based on different membrane orientation of the Bohr effect linked to oxido-reduction of heme a3/CuB and H+ pumping in the normal or reverse direction associated to reverse electron flow in the ferricyanide oxidation of reduced oxidase were found to be incompatible with the experimental H+/COX ratios.
DISCUSSION
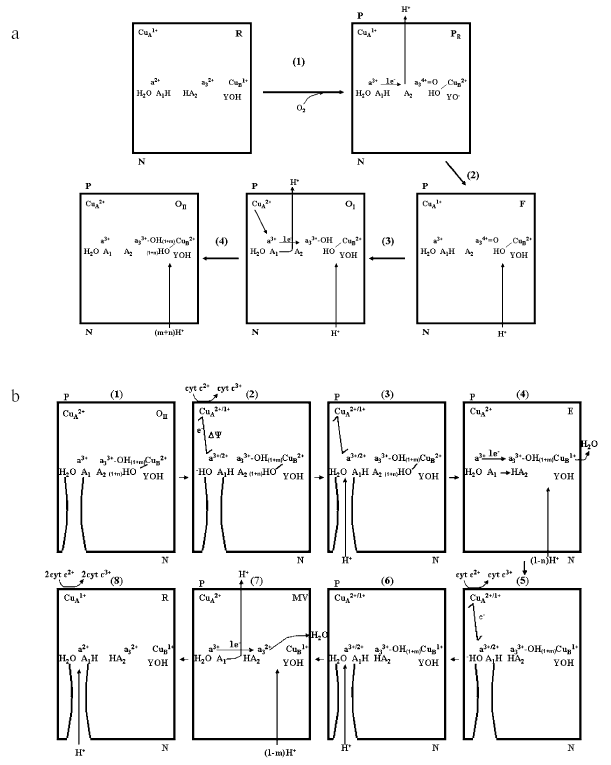
Schemes on Figs. 6a and 6b describe the sequence of electron and proton transfer steps in a single turnover, in which the fully reduced enzyme is oxidized by oxygen and then re-reduced by ferrocytochrome c2+. In the fully reduced oxidase both the clusters A1 and A2 associated to heme a/CuA and heme a3/CuB respectively are in the protonated state (Fig. 6a). A1 and A2 each bind approximately one H+. The number of these protons appears, however, to vary slightly with pH [23, 31, 33]. Upon binding of O2 (intermediate A is not shown) with oxidation of heme a, heme a3, and CuB in the reductive cleavage of O2 to form the PR compound the Bohr proton of the cluster A2 is released in the outer aqueous space (P). CuA (or heme a) being still reduced holds the Bohr proton on A1. In the conversion of R to PR no scalar proton is taken up from the inner aqueous space (N) [10, 38]. A tyrosine of subunit I, I-Tyr244 (bovine numbering) [19, 39, 40], or another tyrosine [41] connected with the binuclear center, donates the proton consumed in the formation of CuB2+-OH. In the PR --> F conversion, one proton is taken up from the inner space and reprotonates I-Tyr-O- [42]. F is converted to OI upon reduction by CuA1+ of a34+=O to a33+-OH, with simultaneous uptake of one scalar H+ from the inner space [10, 37]. The oxidation of both CuA and heme a results in the release in the outer space of the Bohr proton of cluster A1. OI is then converted to OII with partial protonation to H2O of the two OH- bound to a3 and CuB, the extent of this depending on the actual pH [31, 33]. In conclusion, in the oxidative phase of a single turnover two H+ deriving from the A1 and A2 clusters in the oxidase are released in the outer space. No proton pumping from the inner to the outer space would take place in this phase.
In the re-reduction of OII (Fig. 6b) the first electron delivered to the oxidase rapidly distributes itself between CuA and heme a, this is associated with transfer of a proton from an H2O near the formyl of heme a to the cluster A1 [9, 12]. In this phase the only electrogenic event is represented by the rapid 10-20 µsec electron transfer from CuA to heme a located some distance below the outer surface (cf. [43-45]) (Fig. 1). The proton movement from H2O to A1 might cover a short distance essentially parallel to the plane of the membrane, thus it will not result in significant charge translocation along the axis perpendicular to the plane of the membrane. The OH- generated near heme a is then released in the outer space or neutralized by an incoming proton from the inner space. This phase, too, might not result in the generation of a transmembrane potential, if the H2O/A1 site at heme a is exposed to a H2O channel continuous with the outer aqueous bulk phase [12]. The single electron now moves from heme a to CuB (intermediate E) with electrogenic uptake from the inner phase of (1 - n)H+ consumed in the protonation of the OH- to H2O at CuB. The electron transfer from heme a/CuA to CuB results in proton transfer from the A1 cluster (whose effective pK decreases) to the A2 cluster (whose pK simultaneously increases). No proton release in the outer bulk phase would take place in the transfer of the first electron from heme a/CuA to heme a3/CuB (cf. [43-45]). Delivery of a second electron to CuA/heme a results again in proton transfer from H2O to A1 at the heme a. OH- is reprotonated to H2O by H+ from the inner space. Transfer of the second e- to heme a3 is associated with protonation of the second OH- to H2O at the binuclear site by (1 - m)H+ from the inner space. Oxidation of both heme a and CuA results in deprotonation of A1. Since A2 is already protonated, an H+ is released in the bulk phase [46]. This situation might be mimicked by the CO-inhibited oxidase in which heme a3 and CuB are clamped in the reduced state. Finally, delivery of two additional electrons converts the oxidase in the fully reduced state in which both A1 and A2 are protonated. Thus, one H+ is pumped from the inner to the outer space in the reductive phase of the single turnover of the enzyme. The schemes described in Fig. 6 (a and b) result in an H+/COX ratio of ~3 (see Fig. 5) for the overall H+ release in the outer bulk phase in the R --> OII --> R transition. As mentioned above Verkhovsky et al. [36] have reported an overall H+/COX ratio of ~4 for this transition. A higher ratio would require additional H+ release at one of the steps described in the scheme of Fig. 6b and/or some aerobic re-oxidation of the metals at the binuclear site during the re-reduction phase.Fig. 6. Schemes describing the sequence of electron transfer and proton translocation steps in a single turnover transition of the membrane associated cytochrome c oxidase from the fully reduced (R) to the oxidized (OII) state (a) and from the OII to the R state (b). For details, see text.
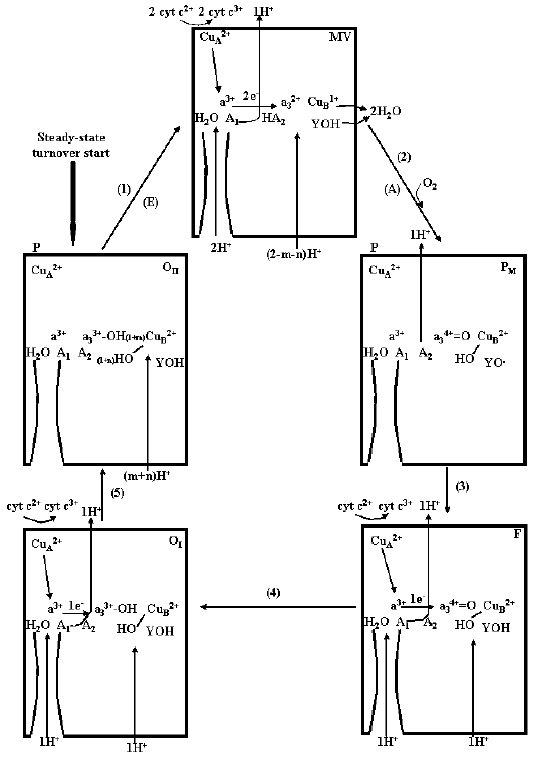
The scheme in Fig. 7 describes the sequence of electron transfer and proton translocation steps in the catalytic cycle of cytochrome c oxidase at the respiratory steady state, resulting in pumping of ~1 H+ from the inner to the outer aqueous phase per each electron being transferred from ferrocytochrome c to O2. It should be noted that, at difference of the single turnover R --> OII --> R transition, at the steady state one electron at once is transferred, through CuA/heme a, from ferrocytochrome c2+ to the binuclear site. Furthermore, the fully reduced R compound is not formed during the aerobic steady-state catalytic cycle (see also [37]). This can explain difference in the H+/COX ratios for overall proton release in the outer bulk phase in the R --> OII --> R transition with respect to that in the reduction of O2 to H2O in the steady-state catalytic cycle of the oxidase.
Under normal physiological conditions, a turnover of the oxidase starts with the fully oxidized enzyme. Reduction of the binuclear center by two electrons, delivered by ferrocytochrome c2+, one at once via CuA/heme a to a3/CuB, results in the uptake of 2H+ from the inner space. One H+ is held by the A2 cluster, whose effective pKa increases upon reduction of a3/CuB, the other H+ is pumped in the outer space. O2 binds to the reduced state (compound A is not shown) and upon reductive cleavage of O2 with oxidation of a3/CuB (state PM) the second proton, taken up from the inner space in the previous reductive step and held on A2 [46], is pumped in the outer space. Note that, in the respiratory catalytic cycle for the reductive cleavage of O2, three electrons derive from a3/CuB reduced in the previous step, and a fourth electron with a proton is provided by I-Tyr [10, 37, 41]. In the transitions from PM to F and F to OI one electron at once is transferred from cytochrome c, via CuA/a, to the binuclear site with pumping of 1 H+ from the inner to the outer space for each of the two steps. In the cycle the pumped protons follow a pathway whose critical elements are represented by the two clusters A1 and A2 both undergoing pK shift upon reduction/oxidation of the heme a/CuA and heme a3/CuB, respectively. These clusters do not contribute scalar protons consumed in the reduction of O2 to H2O at the binuclear site. These protons are taken up from the inner space by a separate pathway.Fig. 7. Catalytic cycle for reduction of O2 to H2O by ferrocytochrome c and proton pumping in the cytochrome c oxidase in the coupling membrane at the respiratory steady-state. For details, see text.
There is a wealth of structural and functional observations showing that oxido-reduction of the low spin heme a/CuA is associated with protonmotive local/global cooperative conformational changes. The H+/e- coupling at heme a/CuA appears to represent the primary step in the uptake of pumped protons from the inner aqueous space. This proton uptake involves a cluster of protolytic groups A1, whose effective pK is governed by the redox state of heme a/CuA. Protons taken up by cluster A1 upon reduction of CuA/heme a move, upon electron transfer from heme a to heme a3, to cluster A2, whose effective pK is governed by the redox state of a3/CuB. Upon oxidation of a3/CuB protons are released from A2 in the outer bulk phase. The cooperative H+/e- coupling at a3/CuB synchronizes the pumping process with the partial steps of O2 reduction to H2O and allows the energy, thus made available, to pump protons against a steady state protonmotive force of some 250 mV. The A1 and A2 clusters operating in close sequence constitute together the gate of the pump. The energy needed to pump protons from the inner to the outer space is distributed between pK shifts of A1 and A2 and is provided by the steps of O2 reduction to H2O at the binuclear site. The A1-A2 pathway for proton pumping has to be separated from the pathway along which the scalar protons consumed in the reduction of O2 to H2O are taken up from the inner space. This prevents pumped protons from being annihilated in the reduction of O2 to H2O.
A discussion of the specific residues contributing to the A1 and A2 clusters, as well as the proton conduction pathways used for proton uptake from the inner space and release in the outer space, is beyond the scope of this paper. It can, however, be argued that I-Arg38 and the propionates of heme a contribute to cluster A1, whilst the propionates of heme a3 and I-Arg438 and I-Arg439 contribute to cluster A2 [10, 37]. Development of the present working hypothesis is associated with clarification of these issues.
The author would like to thank Nazzareno Capitanio, Giuseppe Capitanio, Emanuele De Nitto, Luca Martino, Peter Nicholls, Luigi Palese, and Shinia Yoshikawa for their contribution to the development of ideas presented in this paper.
This work was supported by Grants from the National Project on Bioenergetics: genetic, biochemical and physiopathological aspects (2003, MIUR, Italy), and the Center of Excellence on “Comparative Genomics”, University of Bari.
REFERENCES
1.Monod, J., Wyman, J., and Changeux, I. P. (1965)
J. Mol. Biol., 12, 88-118.
2.Perutz, M. F. (1976) Br. Med. Bull.,
32, 193-194.
3.Perutz, M. F., Kilmartin, J. V., Nishikura, K.,
Fogg, J. H., Butler, P. J., and Rollema, H. S. (1980) J. Mol.
Biol., 138, 649-668.
4.Kilmartin, J. V., and Rossi Bernardi, L. (1973)
Physiol. Rev., 53, 836-890.
5.Dutton, P. L., and Wilson, D. F. (1974) Biochim.
Biophys. Acta, 346, 165-212.
6.Clark, W. M. (1960) Oxidation-Reduction
Potentials of Organic Systems, William and Wilkins Co, Baltimore,
USA.
7.Papa, S. (1976) Biochim. Biophys. Acta,
456, 39-84.
8.Papa, S., Guerrieri, F., Lorusso, M., Izzo, G.,
Boffoli, D., and Maida, I. (1981) in Vectorial Reactions in Electron
and Ion Transport (Palmieri, F., et al., eds.) Elsevier/North
Holland Biomedical Press, p. 57.
9.Rousseau, D., Sassaroli, M., Ching, Y., and
Dasgupta, S. (1988) Ann. N. Y. Acad. Sci., 550,
223-237.
10.Brzezinski, P., and Larsson, G. (2003)
Biochim. Biophys. Acta, 1605, 1-13.
11.Brzezinski, P. (2004) Trends Biochem.
Sci., 29, 380-387.
12.Tsukihara, T., Shimokata, K., Katayama, Y.,
Shimada, H., Muramoto, K., Aoyama, H., Mochizuki, M., Shinzawa-Itoh,
K., Yamashita, E., Yao, M., Ishimura, Y., and Yoshikawa, S. (2003)
PNAS, 100, 15304-15309.
13.Papa, S. (1989) Highlights in Modern
Biochemistry (Kotyk, A., et al., eds.), Vol. 1, pp. 781-796.
14.Heberle, J. (2000) Biochim. Biophys. Acta,
1458, 135-147.
15.Wikstrom, M., Krab, K., and Saraste, M. (1981)
Cytochrome c Oxidase. A Synthesis, Academic Press, London, pp.
111-115.
16.Van Gelder, B. F., van Rijin, J. L. M. L.,
Schilder, G. J. A., and Wilms, J. (1977) in Structure and Function
of Energy-Transduction Membranes (van Dam, K., and van Gelder, B.
F., eds.) Elsevier/North Holland, Amsterdam, pp. 61-68.
17.Ellis, W. R., Wang, H., Blair, D. F., Gray, H.
B., and Chan, S. I. (1986) Biochemistry, 25, 161-167.
18.Nicholls, P., and Petersen, L. C. (1974)
Biochim. Biophys. Acta, 357, 462-467.
19.Yoshikawa, S., Shinzawa-Itoh, K., Nakashima, R.,
Yaono, R., Yamashita, E., Inoue, N., Yao, M., Fei, M., Libeu, C. P.,
Mizushima, T., Yamaguchi, H., Tomizaki, T., and Tsukihara, T. (1998)
Science, 280, 1723-1729.
20.Kanti Das, T., and Mazundar, S. (2000)
Biopolymers (Biospectroscopy), 57, 316-322.
21.Capitanio, N., Piccoli, C., Capitanio, G., Perna,
G., Boffoli, D., Capozzi, V., and Papa, S. (2005) Physica
Scripta, in press.
22.Callahan, P. M., and Babcock, G. T. (1983)
Biochemistry, 22, 452-461.
23.Capitanio, N., Capitanio, G., Minuto, M., De
Nitto, E., Palese, L. L., Nicholls, P., and Papa, S. (2000)
Biochemistry, 39, 6373-6379.
24.Forte, E., Barone, M. C., Brunori, M., Sarti, P.,
and Giuffre, A. (2002) Biochemistry, 41, 13046-13052.
25.Forte, E., Scandurra, F. M., Richter, O. M. H.,
D'Itri, E., Sarti, P., Brunori, M., Ludvig, B., and Giuffre, A. (2004)
Biochemistry, 43, 2957-2963.
26.Louro, R. O., Catarino, T., Le Gall, J., Turner,
D. L., and Xavier, A. V. (2001) Chem. Biochem., 2,
22-28.
27.Papa, S., Capitanio, N., and Capitanio, G. (2004)
Biochim. Biophys. Acta, 1655, 353-364.
28.Xavier, A. V. (2004) Biochim. Biophys.
Acta, 1658, 23-30.
29.Capitanio, N., Capitanio, G., Boffoli, D., and
Papa, S. (2000) Biochemistry, 39, 15454-15461.
30.Capitanio, N., Capitanio, G., Demarinis, D. A.,
De Nitto, E., Massari, S., and Papa, S. (1996) Biochemistry,
35, 10800-10806.
31.Papa, S., Capitanio, N., Capitanio, G., and
Palese, L. L. (2004) Biochim. Biophys. Acta, 1658,
95-105.
32.Artzabanov, V. Y., Konstantinov, A. A., and
Skulachev, V. P. (1978) FEBS Lett., 87, 180-185.
33.Capitanio, N., Capitanio, G., De Nitto, E.,
Boffoli, D., and Papa, S. (2003) Biochemistry, 42,
4607-4612.
34.Traber, R., Kramer, H. E., and Hemmerich, P.
(1982) Biochemistry, 21, 1687-1693.
35.Verkhovsky, M. I., Morgan, J. E., and Wikstrom,
M. (1995) Biochemistry, 34, 7483-7491.
36.Verkhosvsky, M. I., Jasaitis, A., Verkhovskaya,
M. L., Morgan, J. E., and Wikstrom, M. (1999) Nature,
400, 480-483.
37.Michel, H. (1999) Biochemistry, 38,
15129-15140.
38.Mitchell, R., and Rich, P. R. (1994) Biochim.
Biophys. Acta, 1186, 19-26.
39.Ostermeier, C., Harrenga, A., Ermler, U., and
Michel, H. (1997) Proc. Natl. Acad. Sci. USA, 94,
10547-10553.
40.Proshlyakov, D. A., Pressler, M. A., and Babcock,
G. T. (1998) Proc. Natl. Acad. Sci. USA, 95,
8020-8025.
41.Budiman, K., Kannt, A., Lyubenova, S., Richter,
H. O. M., Ludwig, B., Michel, H., and MacMillan, F. (2004)
Biochemistry, 43, 11709-11716.
42.Van Eps, N., Szundi, I., and Einarsdottir, O.
(2003) Biochemistry, 42, 5065-5073.
43.Ruitenberg, M., Kannt, A., Bamberg, E., Ludwig,
B., Michel, H., and Fendler, K. (2000) PNAS, 97,
4632-4636.
44.Verkhosvsky, M. I., Tuukkanen, A., Backgren, C.,
Puustinen, A., and Wikstrom, M. (2001) Biochemistry, 40,
7077-7083.
45.Ruitenberg, M., Kannt, A., Bamberg, E., Fendler,
K., and Michel, H. (2002) Nature, 417, 99-102.
46.Rich, P. R. (1995) Aust. J. Plant.
Physiol., 22, 479-486.