REVIEW: Mitochondrial Metabolism of Reactive Oxygen Species
A. Yu. Andreyev1,2, Yu. E. Kushnareva1,3, and A. A. Starkov1,4*
1Alumni of Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia2University of California, San Diego, La Jolla, California 92093, USA
3The Burnham Institute, Del E. Webb Center for Neuroscience and Aging, 10901 N. Torrey Pines Road, La Jolla, California 92037, USA
4Weill Medical College Cornell University, 585 E. 68th St., New York, NY 10021, USA; E-mail: ans2024@med.cornell.edu
* To whom correspondence should be addressed.
Received September 28, 2004
Oxidative stress is considered a major contributor to etiology of both “normal” senescence and severe pathologies with serious public health implications. Mitochondria generate reactive oxygen species (ROS) that are thought to augment intracellular oxidative stress. Mitochondria possess at least nine known sites that are capable of generating superoxide anion, a progenitor ROS. Mitochondria also possess numerous ROS defense systems that are much less studied. Studies of the last three decades shed light on many important mechanistic details of mitochondrial ROS production, but the bigger picture remains obscure. This review summarizes the current knowledge about major components involved in mitochondrial ROS metabolism and factors that regulate ROS generation and removal. An integrative, systemic approach is applied to analysis of mitochondrial ROS metabolism, which is now dissected into mitochondrial ROS production, mitochondrial ROS removal, and mitochondrial ROS emission. It is suggested that mitochondria augment intracellular oxidative stress due primarily to failure of their ROS removal systems, whereas the role of mitochondrial ROS emission is yet to be determined and a net increase in mitochondrial ROS production in situ remains to be demonstrated.
KEY WORDS: mitochondria, reactive oxygen species, superoxide, antioxidants, oxidative stress
“Living with the risk of oxidative stress is a price that aerobic organisms must pay for more efficient bioenergetics” (quoted from V. P. Skulachev [1]). Oxidative stress has been generating much recent interest primarily because of its accepted role as a major contributor to etiology of both “normal” senescence and severe pathologies with serious public health implications [2-8]. The term is used loosely to define a cluster of interrelated phenomena that includes elevated generation of reactive oxygen species (ROS) and oxidative damage to cell constituents. Causative links within the cluster are incompletely understood but are likely to include a positive feedback loop (“vicious cycle”) wherein ROS-mediated oxidative damage to cell favors elevated ROS production, and so on. The triggering factors for the oxidative stress may be diverse, ranging from hereditary or acquired genetic defects (mutations) or environmental factors (radiation or toxins) to pure stochastic events such as metabolic fluctuations.
Mitochondria, which harbor the bulk of oxidative pathways, are packed with various redox carriers and centers that can potentially leak single electrons to oxygen and convert it into superoxide anion, a progenitor ROS. The initial observations of ROS production in mitochondrial fragments came as early as 1966 [9] but passed almost unnoticed until 1971 when Loschen, Flohe, and Chance [10] demonstrated for the first time succinate supported H2O2 production by intact pigeon heart mitochondria. The following studies by Britton Chance's group systematically demonstrated that metabolically competent mitochondria generate ROS [11]. The ensuing 30 years of studies have revealed several important mechanistic details of mitochondrial ROS production, but the bigger picture still remains obscure.
Intracellular generation of ROS per se is an inevitable (and sometimes physiologically important) process [1]. To counter it, mitochondria, and cells in general, possess numerous ROS defense systems. It should have been implicit that the true source of oxidative stress is not the ROS generation per se but spatiotemporal imbalance of ROS production and detoxification, and, yet, until recently even the capacity of mitochondrial ROS defense was unknown. Furthermore, a contribution of specifically mitochondrially derived ROS to oxidative damage to cell constituents remains to be demonstrated, especially in pathology. Therefore, lack of factual data hampers the development of a comprehensive view on the role of mitochondria in oxidative stress. In a more limited attempt, this review aims at summarizing the current knowledge on mitochondrial ROS metabolism within the concept of a balance between ROS producing and detoxifying systems in mitochondria.
MULTIPLICITY OF ROS-PRODUCING SOURCES IN MITOCHONDRIA
Given a moderate redox potential of the superoxide/dioxygen couple (E1/2 = -0.16 V [12]), the reaction of one-electron reduction of oxygen is thermodynamically favorable for numerous mitochondrial oxidoreductases [13]. Taking into account that superoxide is effectively removed from the reaction (see “Mitochondrial ROS detoxifying systems” below) and the possibility of highly reduced state of many redox carriers (see discussion under “ROS production at Complex I”), the reaction becomes virtually irreversible. Therefore, which of the carriers do become the sites of ROS production is kinetically controlled. So far, a measurable ROS production by at least nine of the mammalian mitochondrial enzymes has been reported. The nine (they are “Nazgul” [14]) enzymes are ubiquitously present in mammalian mitochondria but their capacity in producing ROS, as well as their expression, varies greatly among tissues and species. Therefore, defining a single “main” source of ROS in vivo may prove difficult if possible at all.
The submitochondrial localization of the nine ROS generating sites (marked by asterisks) is shown in Fig. 1. Seven of these sites are briefly discussed here; Complex I (C-I) and Complex III (C-III) will be discussed in separate sections.
1) Cytochrome b5 reductase is located in the outer mitochondrial membrane. This protein is widely distributed in mammalian tissues [15]. It oxidizes cytoplasmic NAD(P)H and reduces cytochrome b5 in the outer membrane. It may also reduce ascorbyl free radical and, therefore, be involved in regeneration of ascorbate in mammalian liver [15]. The enzyme is upregulated in the patients suffering from schizophrenia, thus implying a role in the etiology of the disease [16, 17]. There is a single report that mitochondrial cytochrome b5 reductase may produce superoxide with a high rate of ~300 nmol/min per mg protein [17].Fig. 1. Known sources of ROS and ROS-detoxifying systems in mitochondria. Selected ROS-producing enzymes and ROS-detoxifying systems are shown in the context of their location within mitochondria. See text for further detail. Abbreviations: COX) cytochrome c oxidase; c) cytochrome c; C-III) Complex III; MnSOD) mitochondrial manganese superoxide dismutase; Cat) catalase; SDH) succinate dehydrogenase; ACO) aconitase; Prx3red) peroxiredoxin reduced; Prx3ox) peroxiredoxin oxidized; Q) coenzyme Q; DHOH) dihydroorotate dehydrogenase; KGDHC) alpha-ketoglutarate dehydrogenase complex; alphaGDH) alpha-glycerophosphate dehydrogenase; PDHC) pyruvate dehydrogenase complex; IDH) isocitric dehydrogenase, NAD+-dependent; Trx2red) thioredoxin-2 reduced; Trx2ox) thioredoxin-2 oxidized; Grx2red) glutaredoxin-2 reduced; Grx2ox) glutaredoxin-2 oxidized; TrxR2) thioredoxin-2 reductase; MDH) malate dehydrogenase; IDH1) isocitric dehydrogenase, NADP+-dependent; ME) malic enzyme, NADP+-dependent; GR) glutathione reductase; GSH) reduced glutathione; GS-SG) oxidized glutathione dipeptide; GPx) glutathione peroxidase; PGPx) phospholipid hydroperoxide glutathione peroxidase; C-I) Complex I; TH) transhydrogenase; Cyt. b5 reductase) cytochrome b5 reductase; MAOs) monoamine oxidases A and B; OM) outer mitochondrial membrane; IM) inner mitochondrial membrane. ROS species that are detoxified by the corresponding systems are shown enclosed in a square frame; asterisks indicate sources of ROS.
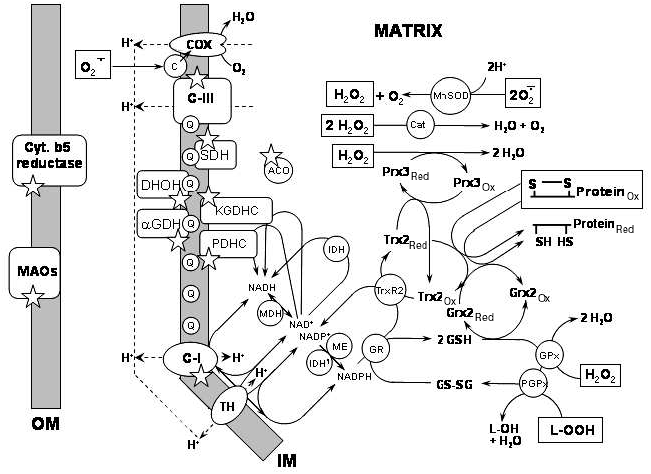
2) Monoamine oxidases (MAO-A and MAO-B, EC 1.4.3.4) are also located in the outer membrane and are ubiquitously expressed in various mammalian tissues. These enzymes catalyze oxidation of biogenic amines accompanied by release of H2O2. MAOs of brain mitochondria play a central role in the turnover of monoamine neurotransmitters. Their potential for H2O2 generation may far exceed that of other mitochondrial sources. For example, tyramine oxidation by rat brain mitochondria produces H2O2 at a rate ~50 times higher than Complex III inhibited with antimycin A [18]. MAOs may be a major source of H2O2 in tissues in ischemia [19, 20], aging [21], and during oxidation of exogenous amines [22]. Some authors suggested that an upregulation of MAO and the resulting elevated H2O2 production might be responsible for the mitochondrial damage in Parkinson's disease [23].
3) Dihydroorotate dehydrogenase (DHOH, EC 1.3.3.1 or EC 1.3.99.11) is located at the outer surface of the inner membrane and is ubiquitously distributed in mammalian tissues [24]. It catalyzes conversion of dihydroorotate to orotate, a step in the synthesis of pyrimidine nucleotides. In the absence of its natural electron acceptor, coenzyme Q of the inner mitochondrial membrane, reduced DHOH can produce H2O2 in vitro [24]. Superoxide production by DHOH has also been suggested [25, 26] but later re-ascribed to Complex III [27], thus rendering the issue controversial.
4) Dehydrogenase of alpha-glycerophosphate (aka glycerol-3-phosphate dehydrogenase, aka mGPDH, EC 1.1.99.5) is also located at the outer surface of the inner membrane and is ubiquitously expressed in mouse tissues with brown fat, muscle, and brain possessing the highest activity [28]. It is a FAD-containing enzyme catalyzing oxidation of glycerol-3-phosphate to dihydroxyacetone phosphate and utilizing mitochondrial coenzyme Q as electron acceptor. This reaction is involved in lipid metabolism and in the glycerol phosphate shuttle that regenerates cytosolic NAD+ from the NADH formed in glycolysis. The enzyme expression is upregulated in hyperthyroid animals [29, 30]. Mouse [31] and Drosophila [32] mitochondria oxidizing sn-glycerol-3-phosphate produce H2O2. Mechanistically, in this reaction at least in Drosophila the bulk of H2O2 was produced at the enzyme per se, whereas about 30% was produced at Complex I because of reverse electron transfer [32] as discussed in the following section.
5) Succinate dehydrogenase (SDH, aka succinate:ubiquinone oxidoreductase, Complex II, EC 1.3.5.1) is a flavoprotein located at the inner surface of the inner membrane. The enzyme oxidizes succinate to fumarate using coenzyme Q as electron acceptor. Isolated SDH incorporated in liposomes can produce ROS, most likely via its FAD, reduced in the absence of electron acceptor [33]. In submitochondrial particles oxidizing succinate, ROS production can be inhibited by carboxin, an inhibitor of SDH [34]. However, the same inhibitor also suppressed antimycin-induced ROS production and ROS production supported by NADH oxidation, two reactions thought to be independent of SDH. Therefore, it is unclear whether SDH as part of intact respiratory chain is capable of ROS generation.
6) Aconitase (mitochondrial (m-) aconitase, EC 4.2.1.3) is localized in the mitochondrial matrix. It catalyzes conversion of citrate to isocitrate as part of the tricarboxylic acid cycle. The enzyme is inactivated upon oxidation of its iron-sulfur cluster by superoxide [35]. Upon inactivation, isolated aconitase induces production of hydroxyl radical, most likely mediated by released Fe2+ [36].
7) alpha-Ketoglutarate dehydrogenase complex (KGDHC, aka 2-oxoglutarate dehydrogenase) is tightly associated with the matrix side of the inner membrane [37]. It catalyzes oxidation of alpha-ketoglutarate to succinyl-CoA using NAD+ as electron acceptor. KGDHC is composed of multiple copies of three enzymes: alpha-ketoglutarate dehydrogenase (E1k component, EC 1.2.4.2), dihydrolipoamide succinyltransferase (E2k component, EC 2.3.1.12), and lipoamide dehydrogenase (E3 component, EC 1.6.4.3). The E3 component of KGDHC is a flavin-containing enzyme; it is identical to the E3 component of another mitochondrial enzyme, pyruvate dehydrogenase (PDHC). This component is also known as dihydrolipoamide dehydrogenase (Dld) and is ubiquitously present in mammalian mitochondria. Two recent studies demonstrated that both PDHC and KGDHC can generate superoxide and hydrogen peroxide; ROS production was shown with isolated purified enzymes from bovine heart [38, 39] and in isolated mouse brain mitochondria [39]. The source of ROS in KGDHC appears to be the dihydrolipoamide dehydrogenase component [39]; earlier, isolated dihydrolipoamide dehydrogenase was shown to produce ROS [40]. The ROS production from KGDHC is stimulated by low availability of its natural electron acceptor, NAD+ [38, 39].
Although these seven sources were shown to produce ROS with appreciable rates in experiments with either isolated enzymes or mitochondria, their contribution to mitochondrial ROS production under physiological conditions is not yet known.
ROS PRODUCTION AT COMPLEX I
Complex I (aka NADH-ubiquinone oxidoreductase, C-I) is an integral inner membrane multi-protein complex exposed to both matrix and intermembrane space. It oxidizes NADH using coenzyme Q as electron acceptor in a reversible reaction coupled with proton pump generating transmembrane potential [41]. This represents one of the two major entry points into the respiratory chain for reducing equivalents derived from tricarboxylic acid cycle substrates (the other being SDH). One of the earliest studies [42] demonstrated that isolated Complex I can generate superoxide in the presence of NADH. The reaction apparently required tightly bound ubiquinone because it was inhibited by rotenone, an inhibitor that blocks electron transfer in close proximity to the ubiquinone binding site. This quinone-dependence suggests that the mechanism of ROS production observed in [42] somewhat resembled that of Complex III mediated superoxide formation reaction (involving formation of reactive ubisemiquinone, discussed in the next section). In other studies cited in this chapter, rotenone either enhanced ROS production by NADH-reduced Complex I or had no effect on it.
Mechanistic studies reported so far have not yielded a consensus about the site of ROS production in Complex I [8, 43, 44]. Studies with both isolated Complex I and submitochondrial particles demonstrated that a ROS producing site is located between flavin and the rotenone-binding site (Fig. 2) [45-48] and that there may be more than one superoxide producing site in that region [49]. Others suggested that the ROS producing site in Complex I may be the flavin [44, 50] or a complex of half-reduced NAD* radical bound to the flavin [51]. In our assessment, the data overall favors the opinion that ROS is most likely produced by one of the iron-sulfur centers [47, 48], not by a flavin per se. Exogenous quinones enhance ROS production in Complex I [42], possibly acting as a redox shuttle between the iron-sulfur centers and oxygen [52].
There are three major experimental paradigms employed in studies on ROS production attributed to Complex I: reverse electron transfer (RET), rotenone-induced ROS production, and ROS production in normally functioning respiratory chain.Fig. 2. Forward and reverse electron transfer in the respiratory chain. The scheme shows the sequence of electron transfer reactions in forward (indicated by arrow “F.E.T.”) and reverse (“R.E.T.”) directions. Coupling of electron transfer to proton translocation steps/membrane potential is omitted for clarity. Abbreviations: IM) inner mitochondrial membrane; TCA) tricarboxylic acid cycle; SDH) succinate dehydrogenase; C-III) complex III; c) cytochrome c; COX) cytochrome c oxidase; FMN) flavin mononucleotide; N-1a, N-1b, N-2, N-3, N-4, N-5) iron-sulfur centers of complex I; CoQ) coenzyme Q.
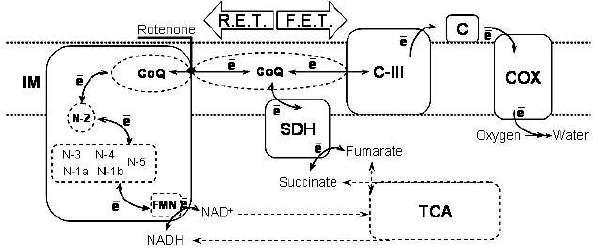
RET, initially discovered in experiments with submitochondrial particles, became one of the first reported mitochondrial reactions supporting ROS production [46]. RET is a set of reactions in the respiratory chain that allow electrons to be transferred against the gradient of redox potentials of electron carriers, from reduced coenzyme Q to NAD+ instead of oxygen. To proceed, this thermodynamically unfavorable process has to be coupled to utilization of the energy of membrane potential. The reduction of coenzyme Q for this reaction requires FADH2-linked oxidizable substrate (e.g., succinate or alpha-glycerophosphate).
In tightly coupled submitochondrial particles treated with antimycin A, RET from succinate to added NAD+ is accompanied by massive production of H2O2 [13, 46]. Both the NAD+ reduction and ROS production require high membrane potential (provided by ATP hydrolysis) and are prevented by an uncoupler [13, 46]. The ROS production is prevented by Complex I inhibitors acting at the rotenone-binding site, indicating the following sequence of electron transfer reactions: reduced Coenzyme Q --> rotenone-binding site --> ROS-generating site --> NAD+ in the matrix of mitochondria (Fig. 2). NAD+ is not required but substantially enhances the ROS production [51].
In intact mitochondria oxidizing succinate or alpha-glycerophosphate, the requirement for high membrane potential is met under non-phosphorylating (Britton Chance's “State 4” [53]) conditions (e.g., [32, 43, 54]). RET supports very high rates of ROS production, ranging from 0.5 to 3 nmol H2O2/min per mg mitochondrial protein [54-56], an equivalent of 5-20% of total oxygen consumption under these conditions. Consistent with high energy requirement for RET, this ROS production is sharply regulated by the amplitude of transmembrane potential [54-56] so that a 10% decrease in the membrane potential inhibits 90% of ROS production. Therefore, it is inhibited by any energy-utilizing process, e.g., ATP synthesis [10, 11], Ca2+ uptake [57], or uncoupling [10, 11, 54-56]. RET-supported ROS production is also suppressed by acidification of the matrix [58], lending additional indirect evidence that it originates from Complex I. It is known that ROS production attributed to Complex I in submitochondrial particles is higher at more alkaline pH [45, 59]. RET-supported ROS production in intact mitochondria is inhibited by rotenone because it blocks the flow of electrons from coenzyme Q to Complex I.
The second experimental paradigm is rotenone-induced ROS production; it is observed in submitochondrial particles (SMPs) or intact mitochondria oxidizing NAD+-linked substrates such as pyruvate or glutamate plus malate [31, 51, 56, 59]. In intact mitochondria, rotenone-induced ROS production requires very high degree of reduction of redox carriers upstream of the rotenone binding site (as measured via redox state of matrix pyridine nucleotides) [43]. With NAD+-linked substrates, it is apparently not regulated by the membrane potential because inhibiting Complex I ultimately results in dissipation of the membrane potential. Rotenone-induced ROS production can be “mimicked” by inhibiting the respiratory chain downstream of the rotenone-binding site, e.g., by Complex III inhibitors or cytochrome c depletion. These treatments induce the highly reduced state of mitochondrial redox centers and carriers that is required for ROS production [43]. The redox properties of the ROS producing site are consistent with the N-1a iron-sulfur center of Complex I [43].
In the absence of Complex I inhibitors (a third, most recent experimental paradigm), ROS production supported by NAD+-linked substrates is stimulated by high membrane potential [55]. These conditions also favor high degree of reduction of redox carriers proximal to the proton pump of Complex I. The dependence of ROS production rate on the amplitude of membrane potential is not as steep as in case of RET, consistent with a more thermodynamically favorable process [55].
Summarizing, in all three experimental paradigms ROS generation requires highly reduced status of the involved redox carriers/centers, at least, more electronegative than the standard redox potential of the NADH/NAD+ couple. Whether the absolute values of these redox potentials are similar or different is still an open question. The rates of ROS production in these paradigms are substantially different, but as there was no quantitative comparison of redox status, it is not known whether one or more sites are involved in ROS production.
Although Complex I of the mitochondrial electron transport chain has been widely accepted as a major site of mitochondrial ROS production [8, 60-62], there are still many unresolved issues. Numerous publications report ROS production in mitochondria or intact cell attributable to Complex I; however, their mechanistically correct interpretation is difficult. For example, a typical premise is that all ROS production caused by Complex I inhibitors occurs at a Complex I site. This might have been a fair universal assumption until recent emergence of mitochondrial lipoamide dehydrogenase as a source of ROS (Dld, reviewed under 7 in previous section) but, obviously, this literature requires a reassessment. Additionally, there may be other sources of ROS within mitochondrial matrix that are in equilibrium with the NAD(P)H/NAD(P)+ couple. In intact mitochondria, Complex I inhibition by any means inevitably results in over-reduction of many if not all NAD+-linked matrix enzymes. Therefore, interpreting intact mitochondria data would be correct in terms of “ROS-generating site proximal to the rotenone binding site” [43] rather than “Complex I”.
The situation in intact cells is even more difficult to analyze; it is however quite likely that mitochondrial metabolite shuttles (e.g., malate/aspartate shuttle) would spread redox imbalance induced by Complex I inhibition into the cytoplasmic NAD(P) pool, with an implicit possibility of ROS production at cytochrome b5 reductase (reviewed in the previous section) and/or other cytoplasmic sites.
Strictly speaking, an unequivocal assessment of the ROS-producing ability of Complex I is possible only for isolated enzyme, provided it is not damaged by the isolation procedure and that it is sufficiently pure of other enzymes linked to the shared electron donors and acceptors. Given the complexity of mammalian Complex I, this is a Herculean undertaking that is yet to be accomplished.
Even in submitochondrial particles, the inhibition of Complex I activity by rotenone and other inhibitors might not correlate with the production of ROS, which may indicate the existence of a superoxide-producing rotenone-binding site other than Complex I [63]. Stimulatory effects of ADP [61] and Ca2+ [64-68] on mitochondrial ROS production are puzzling because both Ca2+ uptake/retention and ADP-induced oxidative phosphorylation decrease membrane potential and the level of reduction of Complex I. Some of these controversies will be addressed below (section “Balance of ROS production and removal”), while others require more research.
Q-CYCLE AND THE MECHANISM OF ROS PRODUCTION AT COMPLEX III
Unveiling of the mechanism of ROS generation in Complex III represents a remarkable example of deductive investigation in biochemical sciences. Complex III (aka bc1 complex, ubiquinone:cytochrome c reductase, C-III) of the mitochondrial respiratory chain is an enzyme complex oxidizing coenzyme Q (QH2) using cytochrome c as electron acceptor. The oxidation of coenzyme Q proceeds in a set of reaction known as the “Q-cycle” (see legend to Fig. 3) coupled to a vectorial translocation of protons, thereby generating transmembrane potential (reviewed in [69]). Complex III is capable of robust production of superoxide [59, 70-73], which then rapidly dismutates to form H2O2 [12]. The underlying mechanism has been a focus of thorough investigation [11, 42, 70, 73-75] resulting in a consensus view that an unstable semiquinone (Q·-) formed in the Qo center (Fig. 3A) is responsible for the superoxide formation, as was first suggested by Rich and Bonner [76]. However, this semiquinone has never been detected [13, 77, 78]. The effects of specific Complex III inhibitors played therefore the most important role in deducing both the site and the source of superoxide production.
Figure 3B shows the sites of action of three most frequently used inhibitors of Complex III. Myxothiazol prevents the binding of QH2 at the Qo-site, stigmatellin prevents the transfer of the first electron to iron-sulfur protein (ISP), and antimycin A interrupts the transfer of the second electron to the Qi-site. The specific inhibitors of the Complex III affect the production of superoxide in remarkable agreement with their supposed effect on the formation of the putative semiquinone Q·- at the center Qo. According to the classical Q-cycle hypothesis (reviewed in [69]), inhibitors acting at the quinone-reducing center (Qi), e.g., antimycin A, prevent the transfer of the second electron to the Qi-site thereby causing accumulation of unstable semiquinone at Qo-site; whereas the inhibitors of the Qo site inhibit semiquinone formation [13, 69, 78, 79] either by displacing quinol QH2 at center Qo (myxothiazol) or by specifically blocking the electron transfer reaction from quinol to ISP (stigmatellin) [13, 69, 78, 79].
Fig. 3. Q-cycle model of coenzyme Q oxidation. Scheme A illustrates the mechanism of superoxide formation in Complex III (see the text). The reaction starts from the oxidation of the CoQ quinol (QH2) in a bifurcated electron transfer reaction at the Qo-site of the complex III. The first electron is transferred to a high reduction potential chain consisting of the iron-sulfur protein (ISP, aka Rieske protein), cytochrome c1 (Cyt. c1), and cytochrome c (Cyt. c) and further to cytochrome c oxidase (not shown). The remaining semiquinone (Qo·-) is unstable. It donates the second electron to the low reduction potential chain consisting of two cytochromes b, cyt bl and cyt bh, which serve as a pathway conducting electrons to the Qi-site. There, these electrons reduce another CoQ molecule. To provide two electrons required for the complete reduction of CoQ quinone at the Qi-site, the Qo-site oxidizes two QH2 molecules in two successive turnovers. The first electron at the Qi-site generates a stable semiquinone (Qi·-) that is reduced to a quinol (QH2) by the second electron [69, 79, 176]. Scheme B illustrates the mechanism of ubiquinone oxidation known as the “Q-cycle” and indicates the sites of action of most frequently used inhibitors of Complex III.
Therefore, antimycin A should stimulate superoxide production as it was demonstrated (reviewed in [13, 78]), whereas myxothiazol and stigmatellin should prevent and/or inhibit the effect of antimycin A. Indeed, both myxothiazol [54, 74, 75, 80, 81] and stigmatellin [82, 83] inhibited superoxide production in mammalian mitochondria.
Superoxide formation at the Complex III site was directly demonstrated by Dr. Konstantinov and colleagues in Skulachev's laboratory. Using the EPR superoxide probe Tiron (1,2-dihydroxybenzo-3,5-disulfonate) they detected superoxide production by inside-out submitochondrial particles oxidizing succinate [70]. These particles produced superoxide when inhibited with antimycin A but not when inhibited with cyanide (cytochrome c oxidase inhibitor) alone or with antimycin + cyanide, exactly as it would be expected if the superoxide was produced by the semiquinone Q·- at the center Qo [70]. Cyanide indirectly inhibits electron transport from ubiquinol to ISP by blocking downstream Complex IV and causing “backup” of electron flow [70]. Further studies by Konstantinov's group demonstrated that the effects of center Qo inhibitors mucidin, 2,3-dimercaptopropanol, and myxothiazol on the superoxide production were also exactly as expected, that is, inhibitory [74, 84].
Superoxide production by antimycin-inhibited Complex III is relatively independent of membrane potential. However, it was found that ROS production by antimycin-inhibited submitochondrial particles exerts a bell-shaped dependence on the redox poise of the respiratory chain [74, 83] rather than a sigmoidal dependence that would be expected for an unstable semiquinone Q·- residing in the Qo-site [74]. Such a redox behavior characterizes a stable semiquinone formed at equilibrium via a reversible dismutation of a quinone and a quinol, which is hardly compatible with an unstable semiquinone species Q·- at center Qo as a source of superoxide [74]. The mechanism of this phenomenon is not yet resolved.
PERMEABILITY OF INNER MITOCHONDRIAL MEMBRANE TO SUPEROXIDE AND
SIDEDNESS OF SUPEROXIDE PRODUCTION IN MITOCHONDRIA
Another unresolved issue is the sidedness of superoxide production. Early studies demonstrated that antimycin-induced superoxide production could be detected with SMPs but not with intact mitochondria ([70, 82] and references therein). The superoxide-generating Qo-site is located closer to the outer surface of the inner membrane of mitochondria (that is, to the inner surface of the SMPs), whereas a superoxide-detecting probe (negatively charged Tiron [70, 82] or cytochrome c) is outside of the particles. Several other studies demonstrated that mitochondria did release detectable, apparently Complex III-generated, superoxide into the external space [85-87]. This discrepancy raises a question about the permeability of mitochondrial inner membrane to superoxide. The data on the issue are contradictory; several studies demonstrated that superoxide could easily penetrate the plasma membrane of erythrocytes or even liposomes by means of an anion channel [88-90], whereas other studies found that the penetration of superoxide through the membranes of thylakoids and phospholipid liposomes is too slow or otherwise insignificant to be of any importance [88, 91, 92]. An attempt to demonstrate that mitochondrial membranes are permeable to superoxide has been made recently. A report by Martin Brand's group demonstrates that matrix aconitase can be readily inactivated by superoxide generated extramitochondrially by xanthine oxidase and xanthine; extramitochondrial superoxide dismutase prevented this inactivation [93]. The authors argue that superoxide may be diffusing through the inner mitochondrial membrane as hydroperoxyl radical (O2H*) to inactivate aconitase ([93] and references therein). However, mitochondrial aconitase is readily inactivated by hydrogen peroxide [94]. The inner mitochondrial membrane is no barrier for H2O2 whereas added superoxide dismutase would only augment the formation of H2O2. Therefore, it is not clear how experiments described in [93] can prove that superoxide permeates mitochondrial membranes. Several other lines of arguments against the idea that superoxide can permeate mitochondrial inner membrane are presented in [95]; however, the issue is still far from being ultimately resolved.
A recent report addresses the sidedness of superoxide production by mitochondrial Complex I and Complex III; the authors argue that Complex I produces superoxide exclusively into the mitochondrial matrix, whereas Complex III produces superoxide to both matrix side and cytosolic side [95].
It should be noted that understanding of membrane permeability for superoxide and sidedness of its production are important both theoretically and practically. Not only would it allow us to better interpret the accumulated literature data, it may also predict where to expect the most damage caused by mitochondrial ROS with broad implications for targeted development of protective strategies.
MITOCHONDRIAL ROS DETOXIFYING SYSTEMS
Mammalian mitochondria possess a multi-leveled ROS defense network of enzymes and non-enzymatic antioxidants. The complexity of this network is confounded by tissue specificity of the defense mechanism and by ongoing discovery of its new elements. The enormity of the subject precludes a comprehensive review; this chapter describes only selected, primarily enzymatic subsystems that are thought to represent mainstream mitochondrial ROS detoxifying pathways.
Membrane lipid peroxide removal systems. The “perimeter” layer of ROS defenses is formed by the systems protecting membrane lipids from peroxidation. These are chiefly alpha-tocopherol (TP) and phospholipid hydroperoxide glutathione peroxidase (where present). The TP is a ubiquitous lipid-soluble free radical scavenging antioxidant present in mitochondrial membranes. It reduces lipid radicals and can be regenerated by reduced coenzyme Q within the membrane or by water-soluble ascorbic acid at the water/membrane interface. A physiological role, redox chemistry, tissue-specific distribution in mitochondria, etc. have been comprehensively reviewed elsewhere (e.g., see [96] for a recent review and [97] for distribution and content of TP in mitochondria of rodents).
Phospholipid hydroperoxide glutathione peroxidase. Phospholipid hydroperoxide glutathione peroxidase (PHGPx, aka GPx4, EC 1.11.1.12) is a selenoenzyme that belongs to the glutathione peroxidase family and utilizes glutathione as source of reducing equivalents. Broad selectivity allows it to reduce phospholipid hydroperoxides, H2O2, cholesterol peroxides [98, 99], and even thymine peroxide [100]. It is the only enzyme known to reduce peroxidized phospholipids within membranes and it is thought to play an important role in cellular ROS defense system [101]. Homozygous knockout mice completely lacking GPx4 die ab utero, but heterozygous mice are viable and fertile [102]. GPx4 is expressed in two isoforms; a longer protein (L-form) is distributed to mitochondria [103]. Detailed information on tissue distribution of mitochondrial (L-form) GPx4 is not available, except that it is absent in mouse liver [104]. L-form RNA transcript is present only in testis in mice [105]; in rat tissues it is also found in testis with some traces in kidney, intestine, and cortex [106]. Such narrow tissue specificity raises some doubts whether GPx4 is of general importance for mitochondria.
MnSOD. The second layer of ROS defenses is formed by enzymes dealing with the primary ROS generated in mitochondria, i.e., superoxide radical and H2O2. The former is a substrate for mitochondrial manganese-containing superoxide dismutase (MnSOD, aka SOD2, EC 1.15.1.1). This enzyme is located exclusively inside the mitochondrial matrix; its only known function is to facilitate dismutation of superoxide radical to H2O2, thereby protecting mitochondrial iron-sulfur cluster containing enzymes from superoxide attack [107].
Homozygous MnSOD knockout mice do not survive longer than a few days after birth [108, 109]. However, heterozygous mutant mice possessing only 50% of MnSOD activity are viable and fertile and appear normal [108, 109]. The animals are normally resistant to oxidative stress-promoting hyperoxia [110] even when exposed to lethal levels of oxygen [111]. Their life span and rate of aging are similar to wild type despite more accumulated DNA damage and cancer occurrence later in life [112]. However, heart mitochondria isolated from these animals show severe dysfunction, i.e., inhibition of mitochondrial Complex I and respiration with NAD+-linked substrates, inhibition of aconitase, and increased sensitivity to Ca2+ [113]. Liver mitochondria show a similar pattern [114].
MnSOD does not require any cofactors so the efficiency of this system is determined solely by the amount of enzyme present. The MnSOD activity is unevenly distributed among tissues; in mice, the activity in liver and kidneys is highest followed by brain and heart, muscle, and spleen with the lowest activity in lungs, where it is about 20 times lower than in liver [115].
Surprisingly, an overexpression of MnSOD to 6-10 times above the normal level results in developmental abnormalities and decreased fertility of mice [116].
Cytochrome c. The intermembrane space of mitochondria contains ~0.7 mM cytochrome c [117] that is capable of superoxide removal. Cytochrome c can be alternatively reduced by the respiratory chain or superoxide [118]. The reduced cytochrome c is regenerated (oxidized) by its natural electron acceptor, cytochrome c oxidase.
In Skulachev's laboratory, the antioxidant properties of cytochrome c were confirmed in experiments with isolated mitochondria [119], but the physiological role and in vivo efficiency of this superoxide-scavenging system remain to be explored. A fascinating ability of this ROS-defense system to generate useful metabolic energy while detoxifying potentially harmful superoxide has earned it the title “the ideal antioxidant” [120]. Indeed, the oxidation of superoxide-reduced cytochrome c by cytochrome c oxidase generates proton-motive force that mitochondria can use to produce ATP [121].
Catalase. Superoxide dismutation leads to formation of another ROS, H2O2 that per se can be quite toxic and has to be detoxified by other enzymes. One such enzyme is catalase (EC 1.11.1.6), which converts H2O2 into O2 and H2O. In murine tissues, catalase activity is highest in liver followed by kidneys, lungs, heart, and brain [122]. It cardiac tissue, it is thought to be present only in mitochondria, where it comprises up to 0.025% of all protein [123].
The role of catalase in the ROS-defense network is not well understood. Even in heart, the contribution of catalase to H2O2 removal is thought to be insignificant compared to that of glutathione peroxidase, another H2O2-detoxifying enzyme [124].
Glutathione. Glutathione (GSH, L-gamma-glutamyl-L-cysteinylglycine) is a tripeptide featuring the thiol (-SH) of cysteine as its active group. Various aspects of GSH metabolism, biochemistry, functions, and analysis have recently been extensively reviewed [125, 126]. Mitochondria contain ~10-12% of total GSH amount in a cell, but due to their relatively small matrix volume the concentration of GSH in mitochondrial matrix is somewhat higher than that in the cytoplasm [127]. Mitochondria lack enzymes needed for GSH biosynthesis; the intramitochondrial pool of GSH is replenished by rapid net uptake of GSH from the cytoplasm [128-130]. There are several systems capable of transporting GSH into mitochondria, including specialized low and high affinity GSH-transporters [129] and dicarboxylate and 2-oxoglutarate carriers [130]. The concentration of glutathione within mitochondria is in the range from 2 to 14 mM [127, 128, 131]; about ~90% of glutathione is in its reduced form, GSH [127, 131, 132]. Actual concentrations of total (reduced + oxidized) glutathione in mitochondria vary depending on the metabolic state, age, and tissue [131]. However, given the low micromolar estimates for steady-state levels of H2O2 in the matrix of mitochondria [133], it is likely that even a significant decrease in GSH levels may not have an impact on H2O2 detoxification by GSH-dependent enzymes. For rat heart mitochondria, the threshold level of GSH depletion was determined experimentally to be ~50% [134]; lesser changes in GSH level did not affect mitochondrial H2O2 emission, whereas a linear increase in H2O2 production was observed concomitant with GSH depletion exceeding ~50% [134].
Glutathione-S-transferase. Mitochondria utilize GSH in two major ways: as a recyclable electron donor and as a consumable in conjugation reactions [126]. The latter are catalyzed by glutathione-S-transferase (GST, EC 2.5.1.18) several isoforms of which are present in mitochondria [135]. These enzymes protect mitochondria from various toxins including products of lipid peroxidation such as 4-hydroxynonenal by adding a GSH molecule to the toxin; GSH is consumed and has to be replenished by uptake from the cytosol [128-130]. A sufficiently large intramitochondrial pool of GSH ensures an efficient operation of the GST-based detoxifying system.
Glutathione reductase. Reduced glutathione can either scavenge superoxide and hydroxyl radical non-enzymatically or by serving as an electron-donating substrate to several enzymes involved in ROS-detoxification (reviewed in [126]). In either case, GSH is oxidized to GSSG that cannot be exported to cytosol [136] and has to be reduced back to GSH in the mitochondrial matrix. The reduction is catalyzed by a specific enzyme, glutathione reductase (GR, aka GSSG reductase, aka GSR, EC 1.8.1.7, formerly EC 1.6.4.2), which is present in the matrix of mitochondria [137-140]. This enzyme utilizes intramitochondrial NADPH as a source of reducing equivalents.
In turn, mitochondrial NADPH can be regenerated by two major pathways, which are the substrate-dependent reduction by dehydrogenases of mitochondrial matrix and protonmotive force-dependent hydride ion transfer reaction utilizing intramitochondrial NADH to reduce NADP+. The former pathway is catalyzed primarily by NADP+-dependent isocitrate dehydrogenase (mNADP-IDH, aka IDPm, EC 1.1.1.42) and by malic enzyme (NADP-ME, EC 1.1.1.40) [132]; the latter is catalyzed by a protein of the inner mitochondrial membrane, nicotinamide nucleotide transhydrogenase (TH, EC 1.6.1.2) [141].
These NADPH regeneration pathways link mitochondrial ability to defend themselves against exogenously or endogenously generated ROS and their bioenergetic prowess and oxidative capacity. As a result, ROS detoxification dissipates energy derived from oxidizable substrates either directly, by oxidizing malate and isocitrate, or indirectly, by consuming protonmotive force generated by substrate oxidation (including malate and isocitrate). In either case, energy is spent to detoxify ROS via NADPH and GSH instead of being used for other functions like, e.g., ATP synthesis. The enzymes involved in NADPH reduction are differently expressed in various tissues, thereby defining which pathway of GSH-regeneration in mitochondria would dominate in a specific mammalian tissue. It is conceivable that tissue specificity of GSH-regenerating pathways results in tissue-specific mitochondrial resistance to ROS or ROS-related toxin challenges.
Hypothetical antioxidant function of NAD(P)H. Some authors hypothesize that NAD(P)H per se can serve as a directly operating non-enzymatic antioxidant [142]. Mammalian mitochondria contain high concentrations of NADH and NADPH (~3-5 mM of each, e.g., see [143]), which can react with and scavenge oxygen-centered radicals such as trioxocarbonate and nitrogen dioxide, thereby preventing damage to mitochondrial proteins and DNA. Because such reactions usually result in formation of superoxide and H2O2, the authors further hypothesize that mitochondrial MnSOD and glutathione peroxidase are sufficient to prevent ROS buildup [142]. However, this mechanism apparently requires too many ROS defense lines acting in concordance with each other for an efficient ROS detoxification.
In contrast, a degradation of mitochondrial NAD(P)+ can, under some special circumstances, be viewed as an efficient strategy against ROS production, as suggested by Skulachev [144]. A ROS-activated enzyme NADase (NAD+ glycohydrolase) is localized in the outer mitochondrial membrane, where it catalyzes the decomposition of NAD+ and NADP+ released from the mitochondrial matrix as a result of mitochondrial permeability transition. According to Skulachev, the exhaustion of the cellular NAD(P)+ pool must eventually stop all mitochondrial ROS production and also suppress some other intracellular ROS-producing systems. Therefore, mitochondrial ROS-activated NADase can be considered as an additional antioxidant system [144].
Glutathione peroxidase. Classical glutathione peroxidase (GPx1, aka cGPx, EC 1.11.1.9) is probably the best studied mitochondrial enzyme that utilizes GSH for the reduction of H2O2 to H2O.
This selenoenzyme is ubiquitously expressed in mammalian tissues [145] and can be detected in various cellular compartments including the mitochondrial matrix [137, 139, 146, 147] and intermembrane space [139]; the same gene encodes both the mitochondrial and extramitochondrial GPx1 [148]. The glutathione peroxidase activity is high in liver, kidney, and heart mitochondria and somewhat lower in brain and skeletal muscle mitochondria; however, detailed information on the expression and activity of GPx1 in mitochondria from different mammalian tissues is not available [104].
GPx1 has long been viewed the most important part of the cellular and mitochondrial ROS-defense network. However, this concept was shaken by data showing that homozygous knockout mice possessing no GPx1 activity are healthy, fertile, develop normally, and do not show any signs of tissue damage and oxidative stress [149-152]. On the other hand, GPx1 knockout mice are hypersensitive to a number of toxins known to induce oxidative stress, including paraquat, N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, and 3-nitropropionic acid [153-155]. Apparently, GPx1 is involved in protecting tissues and mitochondria against acute oxidative stress rather than in providing a major defense against low-level endogenous mitochondrial ROS production.
Peroxiredoxins and other oxins. Peroxiredoxins, or thioredoxin-dependent peroxide reductases, are recently discovered peroxidases that reduce H2O2 and lipid hydroperoxides (reviewed in [156, 157]).
Two isoforms of peroxiredoxins (Prx3 and Prx5) were found in mammalian mitochondria. Prx3 (aka SP-22) is ubiquitously present in various rat tissues, with the highest amount found in heart and adrenal gland, followed by liver and brain [158]. Similar Prx3 gene expression pattern was observed in bovine tissues except that it was highest in adrenal gland [159]. Prx3 gene expression is induced by oxidative stress; the enzyme apparently functions as an antioxidant in cardiac [160] and neuronal mitochondria [161], protecting them in vivo against oxidative damage. However, the capacity and efficiency of Prx3 in H2O2 removal comparing to other mitochondrial systems are not yet known.
Prx5 is the newest mitochondrial member of the peroxiredoxins family. The Prx5 gene is also ubiquitously expressed in bovine tissues, with the highest level found in testis [159]. Overexpression of human Prx5 in mitochondria of hamster ovary cells protected them from H2O2-induced oxidative damage, thereby suggesting a role for this protein in the mitochondrial ROS defense network [162].
Both Prx3 and Prx5 are regenerated in their active form by disulfide oxidoreductase thioredoxin (Trx2), a part of the mitochondrial thioredoxin system. Trx2 is reduced by thioredoxin reductase (TrxR2) that utilizes intramitochondrial NADPH as the source of reducing equivalents. Therefore, the efficient operation of Prx3 and Prx5 is dependent on efficient mitochondrial bioenergetics, similar to the GSH-linked systems described above.
Another member of this family, glutaredoxin (Grx2) can also catalyze Trx-disulfide oxidoreduction reactions. It can reduce both protein disulfides and mixed disulfides with GSH [163], while thioredoxins reduce efficiently only protein disulfides.
Thioredoxin, thioredoxin reductase (TrxR, EC 1.8.1.9, formerly EC 1.6.4.5), and glutaredoxin are ubiquitous proteins present in many if not all tissues and performing a multitude of functions aside from their role in cellular antioxidant defenses. A wealth of information on tissue distribution, genetics, functions, reaction mechanism, and other aspects of these proteins is available [164, 165]. However, not much is known about mitochondrial isoforms of these proteins, Trx2, TrxR2, and Grx2, and even less is known about their specific role in mitochondrial ROS defenses. The mitochondrial thioredoxin system may be essential for mammalian development because disruption of Trx2 gene in the mouse resulted in massive apoptosis during early embryogenesis and embryonic lethality [166]. However, overexpression of Trx2 or TrxR2, or both, does not necessarily improve cell survival or resistance to ROS-promoting factors, indicating that perhaps an unidentified variable controls the effect of these proteins [167].
Summarizing, recent research has uncovered a plethora of mitochondrial ROS-defense systems, but their functioning as a system is not well understood, especially with regard to their interaction with mitochondrial bioenergetic function and endogenous mitochondrial ROS.
BALANCE OF ROS PRODUCTION AND REMOVAL
Surprisingly, almost all published studies on the mitochondria-derived oxidative stress focus primarily on the ROS generation while ignoring ROS detoxifying capabilities of mitochondria. In a rare attempt to address this issue, Alexandre's group reported that brain mitochondria were capable of removal of H2O2 at a very high rate of ~6.5 nmol/min per mg protein [138]. This rate is 3 to 12 times faster that maximal reported rates of ROS production by mitochondria that could only be attained under non-physiological conditions such as succinate-supported RET or in the presence of antimycin A. However, this ROS-removal rate was measured by following the disappearance of an exogenously added H2O2 bolus and therefore might not reflect a true capability of mitochondria to detoxify endogenously produced ROS. Figure 4 presents the results of a simple experiment directly probing the balance of ROS production and removal in mitochondria. It illustrates that undermining the ROS defenses is a prerequisite for net mitochondrial ROS emission.
For these experiments, we utilized Complex III-mediated production of ROS induced by antimycin A (Ant A) in the presence of an uncoupler and succinate as electron donating substrate. Additionally, we timed the addition of the H2O2 detecting enzyme, horseradish peroxidase (HRP), to either prior to mitochondria (traces a and c) or 5 min after the induction of ROS production by Ant A (traces b and d). The block-scheme of the reactions in the experiment is shown by Eq. (1) and includes ROS production (1), ROS removal (2), and, when HRP is present, ROS detection (3). In the presence of HRP, both intact (trace c) and alamethicin-permeabilized (trace a) mitochondria demonstrated a typical response to Ant A by increasing their H2O2 production. Characteristically, permeabilized mitochondria incubated either in the absence (trace b) or in the presence (trace a) of HRP accumulated practically identical amounts of H2O2 as the signal was completely recovered upon addition of the enzyme (trace b). In contrast, intact mitochondria incubated in the absence of HRP did not accumulate any noticeable amount of H2O2 (trace d, compare to trace c).Fig. 4. Intact mitochondria do not accumulate ROS. Rat heart mitochondria (RHM) were prepared using classic differential centrifugation protocol with modifications as reported elsewhere [43]. The assay medium contained 250 mM sucrose, 2 mM potassium phosphate, 10 mM HEPES, 0.5 mM succinate, 1 µM FCCP, and 20 µM Amplex Red (Molecular Probes, USA). Where indicated, the medium was supplemented with 0.8 unit/ml of horseradish peroxidase (HRP). To obtain permeabilized mitochondria, an aliquot of the intact mitochondrial preparation was diluted to 0.25 mg/ml, treated for 2 min with 20 µg/ml alamethicin, pelleted at 14,000g (2 min), and resuspended in the assay medium. Additions: RHM, 0.25 mg/ml intact (c, d) or an equivalent amount of permeabilized (a, b) mitochondria; Ant A, 0.2 µM antimycin A; HRP, 0.8 unit/ml horseradish peroxidase. Trace a, permeabilized mitochondria incubated in the presence of HRP; trace b, permeabilized mitochondria; trace c, intact mitochondria incubated in the presence of HRP; trace d, intact mitochondria. See text for further details.
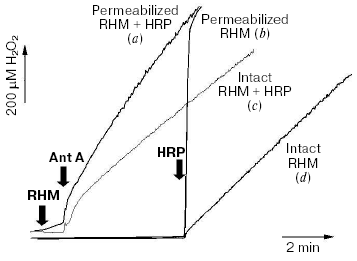
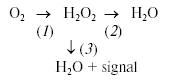
This allows us to draw three important conclusions. First, in intact mitochondria ROS removal systems (reaction 2) may be sufficient to cope with even the highest intramitochondrial rate of ROS production (reaction 1; see trace d). This implies that functionally and structurally intact mitochondria serve as a net sink rather than a net source of ROS. This conclusion is in agreement with the literature reporting high activity of ROS removal [138] and lack of ROS accumulation in post-treatment supernatants of mitochondria [168].
Second, ROS-defenses are severely undermined in structurally compromised mitochondria (trace b), presumably because of the loss of low molecular weight solutes from the matrix, and that turns mitochondria into net producers of ROS. According to [138], the glutathione peroxidase/glutathione reductase cycle serves as the main ROS removal system. Therefore, a loss of mitochondrial glutathione either via alamethicin (Fig. 4) or via the permeability transition pore [169] would inevitably result in an oxidative stress due to net ROS production by mitochondria.
The third conclusion is that depending on the concentrations of both the enzyme and mitochondria, a “trap-based” detection system (reaction 3) may outpace the capacity of mitochondrial ROS removal systems (reaction 2). If this is the case, the detection system (e.g., HRP-based, or EPR spin-traps) will report a rate of ROS production (reaction 1; see trace c) rather than the rate of ROS emission (the production (1) rate minus the removal (2) rate). It would not reflect a true level of ROS either, as it would always be nearly zero (trace d). Therefore, a proper choice of ROS detection system is crucial as it may severely interfere with the measured process.
The implications of these conclusions may pertain to a controversy surrounding ROS generation during Ca2+ overload and/or mitochondrial permeability transition. There are numerous reports implying that a massive mitochondrial accumulation of Ca2+ somehow promotes ROS production [64, 65, 68, 170, 171, 177] either per se or via the permeability transition [67, 172, 173]. However, the Ca2+ uptake per se should suppress ROS production because it dissipates the DeltaPsi and decreases the level of NAD(P)H reduction in mitochondria (see “ROS production at Complex I”). This reasoning was proved experimentally [57]. On the other hand, Ca2+ overload leads to inhibition of the major, GPx1-GR ROS removal system and thus may increase ROS emission [138]. Furthermore, if Ca2+ overload progresses into permeability transition the most active ROS defense systems are expected to rapidly become dysfunctional. In this case, direct loss of glutathione [174] into the cytoplasm down the concentration gradient [127] is exacerbated by loss of NAD(P)H and membrane potential, the two key components of GR-catalyzed GSH regeneration (see “Mitochondrial ROS detoxifying systems”). Therefore, although ROS production (which was measured in [57]) is initially suppressed, we hypothesize that mitochondrial ROS emission should eventually increase due to inactivation of ROS removal. This may explain the literature data on increased oxidative stress (ROS emission) under the conditions of Ca2+ overload and/or permeability transition [64, 65, 68, 170, 171].
CONCLUDING REMARKS
In the good old days, a picture of the universe was simple. Good mitochondria were producing ATP; evil mitochondria were producing ROS. There was a major site of ROS production (Complex I, or II, or III, etc.), and two enzymes to detoxify ROS--MnSOD and glutathione peroxidase. Mitochondrial ROS were, of course, the absolute evil; they augmented diseases, damaged genes, and made us age miserably. These good old days of simple biology are long gone in the wake of new findings, and emerging concept of mitochondrial ROS production is becoming more and more complex as new elements are discovered.
This review demonstrates that multiple sources of ROS are present in mitochondria, and that there are multiple systems involved in ROS detoxification. Which of them are the most important is to be determined on a case-by-case basis, as this is expected to depend on the species and tissue specificity and on metabolic state of mitochondria. Nevertheless, a few novel elements can be introduced to the concept of mitochondrially produced ROS in oxidative stress.
First, one should distinguish between ROS emission, ROS production, ROS removal, and a steady-state ROS concentration in and around mitochondria. The ROS production is the amount of ROS primarily generated by mitochondrial sites; the ROS removal is the amount of ROS mitochondria detoxify. Note that ROS removal capacity of mitochondria could be different toward exogenously or endogenously generated ROS. The ROS production minus ROS removal is the net ROS emission as can be measured outside of mitochondria. The steady state ROS concentration depends on both the ROS emission rate and the ROS production by extramitochondrial sources. The steady-state concentration of ROS calculated as H2O2 in a typical cell culture was reported to be in the low micromolar range [133].
It is tempting to view ROS emission as the only parameter of importance regarding the role of mitochondria in oxidative stress. Indeed, as it follows from our own data (Fig. 4) and the literature [138] undermining the ROS defense mechanisms is a critical event precipitating the ROS emission by mitochondria. Otherwise, very little (if any) ROS is emitted even with the most robust intra-mitochondrial ROS generators being fully active. With so many mitochondrial ROS defense systems, it seems likely that “defense failure” most adequately describes the role of mitochondria in the onset of oxidative stress. As reviewed above, various mechanisms leading to increased mitochondrial ROS production may further exacerbate the oxidative stress.
However, it should be noted that this concept is based on and directly applicable only to the behavior of isolated mitochondria in suspension. The situation in situ is severely complicated by spatiotemporal heterogeneity of the intracellular environment that likely results in the local and transient conditions favoring ROS emission because of uneven distribution of antioxidant enzymes and highly variable metabolic load of various parts of the mitochondrial network. Further uncertainties arise from differences in lifetimes and diffusion rates of various ROS species. It is conceivable that the same mitochondrion (or any part of it) may act alternatively as either a sink or a source of ROS. This spatiotemporal heterogeneity should be considered when interpreting experiments with intact cells.
The mitochondrial ROS emission may be greatly exacerbated by ROS-inflicted damage that results in further activation of mitochondrial ROS generating sites (“vicious cycle”). For example, mitochondrial enzyme aconitase can be damaged and inactivated by ROS in a way that leads to elevated hydroxyl radical production, thereby adding an autocatalytic, positive feedback loop to the oxidative stress cascade [36]. Therefore, preventing mitochondrial ROS emission rather than scavenging emitted ROS may perhaps be the most efficient strategy to minimize the contribution of mitochondria to oxidative stress.
Although reviewing the protective therapeutic strategies is beyond the scope of this manuscript, it would be incomplete without at least brief evaluation of basic principles behind them. The spatiotemporal heterogeneity renders unlikely the possibility of entirely preventing the mitochondrial ROS emission. An ideal strategy would be to boost the ROS defense systems using nontoxic catalytic antioxidants that are either delivered tissue-specifically or produced where needed from an inactive precursor; several biomedical research companies are working toward this goal, but the publicly available results are not satisfactory yet. Decreasing the primary ROS generation by preventing the over-reduction of intramitochondrial NAD(P)H [43] should also help to alleviate oxidative stress. In certain cases, this can be achieved by mild uncouplers [1, 175]. Another strategy could be to pharmacologically stimulate the expression of endogenous mitochondrial and intracellular antioxidant systems. The latter however requires a level of understanding of intracellular ROS physiology that is not yet achieved, despite a wave of interest rising internationally. Knowledge is at the roots of interest; we therefore hope this review will contribute to more scientists becoming involved in studies of this interesting yet poorly understood subject.
REFERENCES
1.Skulachev, V. P. (1996) Q. Rev. Biophys.,
29, 169-202.
2.Fiskum, G., Murphy, A. N., and Beal, M. F. (1999)
J. Cerebr. Blood Flow Metab., 19, 351-369.
3.Murphy, A. N., Fiskum, G., and Beal, M. F. (1999)
J. Cerebr. Blood Flow Metab., 19, 231-245.
4.Beal, M. F. (1996) Curr. Opin. Neurobiol.,
6, 661-666.
5.Bossy-Wetzel, E., Schwarzenbacher, R., and Lipton,
S. A. (2004) Nat. Med., 10 (Suppl.), S2-9.
6.Dawson, T. M., and Dawson, V. L. (2003)
Science, 302, 819-822.
7.Shen, J., and Cookson, M. R. (2004) Neuron,
43, 301-304.
8.Lenaz, G. (2001) IUBMB Life, 52,
159-164.
9.Jensen, P. K. (1966) Biochim. Biophys. Acta,
122, 157-166.
10.Loschen, G., Flohe, L., and Chance, B. (1971)
FEBS Lett., 18, 261-264.
11.Boveris, A., and Chance, B. (1973) Biochem.
J., 134, 707-716.
12.Sawyer, D. T., and Valentine, J. S. (1981)
Acc. Chem. Res., 14, 393-400.
13.Turrens, J. F. (2003) J. Physiol.,
552, 335-344.
14.Tolkien, J. R. R. (1954) The Lord of the
Rings, George Allen & Unwin.
15.Nishino, H., and Ito, A. (1986) J. Biochem.
(Tokyo), 100, 1523-1531.
16.Whatley, S. A., Curti, D., and Marchbanks, R. M.
(1996) Neurochem. Res., 21, 995-1004.
17.Whatley, S. A., Curti, D., Das Gupta, F.,
Ferrier, I. N., Jones, S., Taylor, C., and Marchbanks, R. M. (1998)
Mol. Psychiatry, 3, 227-237.
18.Hauptmann, N., Grimsby, J., Shih, J. C., and
Cadenas, E. (1996) Arch. Biochem. Biophys., 335,
295-304.
19.Simonson, S. G., Zhang, J., Canada, A. T., Jr.,
Su, Y. F., Benveniste, H., and Piantadosi, C. A. (1993) J. Cerebr.
Blood Flow Metab., 13, 125-134.
20.Kunduzova, O. R., Bianchi, P., Parini, A., and
Cambon, C. (2002) Eur. J. Pharmacol., 448, 225-230.
21.Maurel, A., Hernandez, C., Kunduzova, O.,
Bompart, G., Cambon, C., Parini, A., and Frances, B. (2003) Am. J.
Physiol. Heart Circ. Physiol., 284, H1460-1467.
22.Carvalho, F., Duarte, J. A., Neuparth, M. J.,
Carmo, H., Fernandes, E., Remiao, F., and Bastos, M. L. (2001) Arch.
Toxicol., 75, 465-469.
23.Kumar, M. J., Nicholls, D. G., and Andersen, J.
K. (2003) J. Biol. Chem., 278, 46432-46439.
24.Loffler, M., Becker, C., Wegerle, E., and
Schuster, G. (1996) Histochem. Cell Biol., 105,
119-128.
25.Forman, J. H., and Kennedy, J. (1975) J. Biol.
Chem., 250, 4322-4326.
26.Forman, H. J., and Kennedy, J. (1976) Arch.
Biochem. Biophys., 173, 219-224.
27.Dileepan, K. N., and Kennedy, J. (1985)
Biochem. J., 225, 189-194.
28.Koza, R. A., Kozak, U. C., Brown, L. J., Leiter,
E. H., MacDonald, M. J., and Kozak, L. P. (1996) Arch. Biochem.
Biophys., 336, 97-104.
29.Lee, Y. P., and Lardy, H. A. (1965) J. Biol.
Chem., 240, 1427-1436.
30.Dummler, K., Muller, S., and Seitz, H. J. (1996)
Biochem. J., 317 (Pt. 3), 913-918.
31.Kwong, L. K., and Sohal, R. S. (1998) Arch.
Biochem. Biophys., 350, 118-126.
32.Miwa, S., St-Pierre, J., Partridge, L., and
Brand, M. D. (2003) Free Rad. Biol. Med., 35,
938-948.
33.Zhang, L., Yu, L., and Yu, C. A. (1998) J.
Biol. Chem., 273, 33972-33976.
34.McLennan, H. R., and Degli Esposti, M. (2000)
J. Bioenerg. Biomembr., 32, 153-162.
35.Gardner, P. R. (2002) Meth. Enzymol.,
349, 9-23.
36.Vasquez-Vivar, J., Kalyanaraman, B., and Kennedy,
M. C. (2000) J. Biol. Chem., 275, 14064-14069.
37.Maas, E., and Bisswanger, H. (1990) FEBS
Lett., 277, 189-190.
38.Tretter, L., and Adam-Vizi, V. (2004) J.
Neurosci., 24, 7771-7778.
39.Starkov, A. A., Fiskum, G., Chinopoulos, C.,
Lorenzo, B. J., Browne, S. E., Patel, M. S., and Beal, M. F. (2004)
J. Neurosci., 24, 7779-7788.
40.Bunik, V. I., and Sievers, C. (2002) Eur. J.
Biochem., 269, 5004-5015.
41.Skulachev, V. P. (1988) Membrane
Bioenergetics, Springer-Verlag, New York.
42.Cadenas, E., Boveris, A., Ragan, C. I., and
Stoppani, A. O. (1977) Arch. Biochem. Biophys., 180,
248-257.
43.Kushnareva, Y., Murphy, A. N., and Andreyev, A.
(2002) Biochem. J., 368, 545-553.
44.Liu, Y., Fiskum, G., and Schubert, D. (2002)
J. Neurochem., 80, 780-787.
45.Takeshige, K., and Minakami, S. (1979)
Biochem. J., 180, 129-135.
46.Hinkle, P. C., Butow, R. A., Racker, E., and
Chance, B. (1967) J. Biol. Chem., 242, 5169-5173.
47.Herrero, A., and Barja, G. (2000) J. Bioenerg.
Biomembr., 32, 609-615.
48.Genova, M. L., Ventura, B., Giuliano, G., Bovina,
C., Formiggini, G., Parenti Castelli, G., and Lenaz, G. (2001) FEBS
Lett., 505, 364-368.
49.Kang, D., Narabayashi, H., Sata, T., and
Takeshige, K. (1983) J. Biochem. (Tokyo), 94,
1301-1306.
50.Kudin, A. P., Bimpong-Buta, N. Y., Vielhaber, S.,
Elger, C. E., and Kunz, W. S. (2004) J. Biol. Chem., 279,
4127-4135.
51.Krishnamoorthy, G., and Hinkle, P. C. (1988)
J. Biol. Chem.., 263, 17566-17575.
52.Genova, M. L., Pich, M. M., Biondi, A.,
Bernacchia, A., Falasca, A., Bovina, C., Formiggini, G., Parenti
Castelli, G., and Lenaz, G. (2003) Exp. Biol. Med. (Maywood),
228, 506-513.
53.Chance, B., and Williams, G. R. (1956) Adv.
Enzymol. Relat. Subj. Biochem., 17, 65-134.
54.Korshunov, S. S., Skulachev, V. P., and Starkov,
A. A. (1997) FEBS Lett., 416, 15-18.
55.Starkov, A. A., and Fiskum, G. (2003) J.
Neurochem., 86, 1101-1107.
56.Hansford, R. G., Hogue, B. A., and Mildaziene, V.
(1997) J. Bioenerg. Biomembr., 29, 89-95.
57.Starkov, A. A., Polster, B. M., and Fiskum, G.
(2002) J. Neurochem., 83, 220-228.
58.Lambert, A. J., and Brand, M. D. (2004)
Biochem. J., 382, 511-517.
59.Turrens, J. F., and Boveris, A. (1980)
Biochem. J., 191, 421-427.
60.Sipos, I., Tretter, L., and Adam-Vizi, V. (2003)
J. Neurochem., 84, 112-118.
61.Barja, G. (1999) J. Bioenerg. Biomembr.,
31, 347-366.
62.Herrero, A., and Barja, G. (2000) J. Bioenerg.
Biomembr., 32, 609-615.
63.Ramsay, R. R., and Singer, T. P. (1992)
Biochem. Biophys. Res. Commun., 189, 47-52.
64.Dykens, J. A. (1994) J. Neurochem.,
63, 584-591.
65.Kowaltowski, A. J., Castilho, R. F., and Vercesi,
A. E. (1995) Am. J. Physiol., 269, C141-147.
66.Kowaltowski, A. J., Castilho, R. F., and Vercesi,
A. E. (1996) FEBS Lett., 378, 150-152.
67.Kowaltowski, A. J., Naia-da-Silva, E. S.,
Castilho, R. F., and Vercesi, A. E. (1998) Arch. Biochem.
Biophys., 359, 77-81.
68.Kowaltowski, A. J., Netto, L. E., and Vercesi, A.
E. (1998) J. Biol. Chem., 273, 12766-12769.
69.Trumpower, B. L. (1990) J. Biol. Chem.,
265, 11409-11412.
70.Grigolava, I. V., Ksenzenko, M., Konstantinov, A.
A., Tikhonov, A. N., and Kerimov, T. M. (1980) Biokhimiya,
45, 75-82.
71.Loschen, G., Azzi, A., Richter, C., and Flohe, L.
(1974) FEBS Lett., 42, 68-72.
72.Dionisi, O., Galeotti, T., Terranova, T., and
Azzi, A. (1975) Biochim. Biophys. Acta, 403, 292-300.
73.Boveris, A., Cadenas, E., and Stoppani, A. O.
(1976) Biochem. J., 156, 435-444.
74.Ksenzenko, M., Konstantinov, A. A., Khomutov, G.
B., Tikhonov, A. N., and Ruuge, E. K. (1983) FEBS Lett.,
155, 19-24.
75.Turrens, J. F., Alexandre, A., and Lehninger, A.
L. (1985) Arch. Biochem. Biophys., 237, 408-414.
76.Rich, P. R., and Bonner, W. D. (1978) Arch.
Biochem. Biophys., 188, 206-213.
77.Junemann, S., Heathcote, P., and Rich, P. R.
(1998) J. Biol. Chem., 273, 21603-21607.
78.Turrens, J. F. (1997) Biosci. Rep.,
17, 3-8.
79.Crofts, A. R., Barquera, B., Gennis, R. B.,
Kuras, R., Guergova-Kuras, M., and Berry, E. A. (1999)
Biochemistry, 38, 15807-15826.
80.Zoccarato, F., Cavallini, L., Deana, R., and
Alexandre, A. (1988) Biochem. Biophys. Res. Commun., 154,
727-734.
81.Konstantinov, A. A., Peskin, A. V., Popova, E.,
Khomutov, G. B., and Ruuge, E. K. (1987) Biochim. Biophys. Acta,
894, 1-10.
82.Raha, S., McEachern, G. E., Myint, A. T., and
Robinson, B. H. (2000) Free Rad. Biol. Med., 29,
170-180.
83.Starkov, A. A., and Fiskum, G. (2001) Biochem.
Biophys. Res. Commun., 281, 645-650.
84.Ksenzenko, M., Konstantinov, A. A., Khomutov, G.
B., Tikhonov, A. N., and Ruuge, E. K. (1984) FEBS Lett.,
175, 105-108.
85.Nohl, H., and Hegner, D. (1978) Eur. J.
Biochem., 82, 563-567.
86.St-Pierre, J., Buckingham, J. A., Roebuck, S. J.,
and Brand, M. D. (2002) J. Biol. Chem., 277,
44784-44790.
87.Han, D., Williams, E., and Cadenas, E. (2001)
Biochem. J., 353, 411-416.
88.Mao, G. D., and Poznansky, M. J. (1992) FEBS
Lett., 305, 233-236.
89.Gus'kova, R. A., Ivanov, II, Kol'tover, V. K.,
Akhobadze, V. V., and Rubin, A. B. (1984) Biochim. Biophys.
Acta, 778, 579-585.
90.Lynch, R. E., and Fridovich, I. (1978) J.
Biol. Chem., 253, 4697-4699.
91.Frimer, A. A., Strul, G., Buch, J., and Gottlieb,
H. E. (1996) Free Rad. Biol. Med., 20, 843-852.
92.Takahashi, M. A., and Asada, K. (1983) Arch.
Biochem. Biophys., 226, 558-566.
93.Echtay, K. S., Murphy, M. P., Smith, R. A.,
Talbot, D. A., and Brand, M. D. (2002) J. Biol. Chem.,
277, 47129-47135.
94.Bulteau, A. L., Ikeda-Saito, M., and Szweda, L.
I. (2003) Biochemistry, 42, 14846-14855.
95.Muller, F. L., Liu, Y., and van Remmen, H. (2004)
J. Biol. Chem., 279, 49064-49073.
96.Packer, L., Weber, S. U., and Rimbach, G. (2001)
J. Nutr., 131, 369S-373S.
97.Lass, A., Forster, M. J., and Sohal, R. S. (1999)
Free Rad. Biol. Med., 26, 1375-1382.
98.Maiorino, M., Thomas, J. P., Girotti, A. W., and
Ursini, F. (1991) Free Rad. Res. Commun., 12/13,
131-135.
99.Thomas, J. P., Maiorino, M., Ursini, F., and
Girotti, A. W. (1990) J. Biol. Chem., 265, 454-461.
100.Bao, Y., Jemth, P., Mannervik, B., and
Williamson, G. (1997) FEBS Lett., 410, 210-212.
101.Imai, H., and Nakagawa, Y. (2003) Free Rad.
Biol. Med., 34, 145-169.
102.Yant, L. J., Ran, Q., Rao, L., van Remmen, H.,
Shibatani, T., Belter, J. G., Motta, L., Richardson, A., and Prolla, T.
A. (2003) Free Rad. Biol. Med., 34, 496-502.
103.Arai, M., Imai, H., Koumura, T., Yoshida, M.,
Emoto, K., Umeda, M., Chiba, N., and Nakagawa, Y. (1999) J. Biol.
Chem., 274, 4924-4933.
104.Esposito, L. A., Kokoszka, J. E., Waymire, K.
G., Cottrell, B., MacGregor, G. R., and Wallace, D. C. (2000) Free
Rad. Biol. Med., 28, 754-766.
105.Knopp, E. A., Arndt, T. L., Eng, K. L.,
Caldwell, M., LeBoeuf, R. C., Deeb, S. S., and O'Brien, K. D. (1999)
Mamm. Genome, 10, 601-605.
106.Pushpa-Rekha, T. R., Burdsall, A. L., Oleksa,
L. M., Chisolm, G. M., and Driscoll, D. M. (1995) J. Biol.
Chem., 270, 26993-26999.
107.Gardner, P. R., Raineri, I., Epstein, L. B.,
and White, C. W. (1995) J. Biol. Chem., 270,
13399-13405.
108.Li, Y., Huang, T. T., Carlson, E. J., Melov,
S., Ursell, P. C., Olson, J. L., Noble, L. J., Yoshimura, M. P.,
Berger, C., Chan, P. H., et al. (1995) Nat. Genet., 11,
376-381.
109.Lebovitz, R. M., Zhang, H., Vogel, H.,
Cartwright, J., Jr., Dionne, L., Lu, N., Huang, S., and Matzuk, M. M.
(1996) Proc. Natl. Acad. Sci. USA, 93, 9782-9787.
110.Tsan, M. F., White, J. E., Caska, B., Epstein,
C. J., and Lee, C. Y. (1998) Am. J. Respir. Cell Mol. Biol.,
19, 114-120.
111.Jackson, R. M., Helton, E. S., Viera, L., and
Ohman, T. (1999) Exp. Lung Res., 25, 631-646.
112.Van Remmen, H., Ikeno, Y., Hamilton, M.,
Pahlavani, M., Wolf, N., Thorpe, S. R., Alderson, N. L., Baynes, J. W.,
Epstein, C. J., Huang, T. T., Nelson, J., Strong, R., and Richardson,
A. (2003) Physiol. Genomics, 16, 29-37.
113.Van Remmen, H., Williams, M. D., Guo, Z.,
Estlack, L., Yang, H., Carlson, E. J., Epstein, C. J., Huang, T. T.,
and Richardson, A. (2001) Am. J. Physiol. Heart Circ. Physiol.,
281, H1422-1432.
114.Williams, M. D., van Remmen, H., Conrad, C. C.,
Huang, T. T., Epstein, C. J., and Richardson, A. (1998) J. Biol.
Chem., 273, 28510-28515.
115.Van Remmen, H., Salvador, C., Yang, H., Huang,
T. T., Epstein, C. J., and Richardson, A. (1999) Arch. Biochem.
Biophys., 363, 91-97.
116.Raineri, I., Carlson, E. J., Gacayan, R.,
Carra, S., Oberley, T. D., Huang, T. T., and Epstein, C. J. (2001)
Free Rad. Biol. Med., 31, 1018-1030.
117.Hackenbrock, C. R., Chazotte, B., and Gupte, S.
S. (1986) J. Bioenerg. Biomembr., 18, 331-368.
118.McCord, J. M., and Fridovich, I. (1970) J.
Biol. Chem., 245, 1374-1377.
119.Korshunov, S. S., Krasnikov, B. F., Pereverzev,
M. O., and Skulachev, V. P. (1999) FEBS Lett., 462,
192-198.
120.Pereverzev, M. O., Vygodina, T. V.,
Konstantinov, A. A., and Skulachev, V. P. (2003) Biochem. Soc.
Trans., 31, 1312-1315.
121.Mailer, K. (1990) Biochem. Biophys. Res.
Commun., 170, 59-64.
122.Ho, Y. S., Xiong, Y., Ma, W., Spector, A., and
Ho, D. S. (2004) J. Biol. Chem., 279, 32804-32812.
123.Radi, R., Turrens, J. F., Chang, L. Y., Bush,
K. M., Crapo, J. D., and Freeman, B. A. (1991) J. Biol. Chem.,
266, 22028-22034.
124.Antunes, F., Han, D., and Cadenas, E. (2002)
Free Rad. Biol. Med., 33, 1260-1267.
125.Pastore, A., Federici, G., Bertini, E., and
Piemonte, F. (2003) Clin. Chim. Acta, 333, 19-39.
126.Dringen, R. (2000) Progr. Neurobiol.,
62, 649-671.
127.Wahllander, A., Soboll, S., Sies, H., Linke,
I., and Muller, M. (1979) FEBS Lett., 97, 138-140.
128.Griffith, O. W., and Meister, A. (1985)
Proc. Natl. Acad. Sci. USA, 82, 4668-4672.
129.Martensson, J., Lai, J. C., and Meister, A.
(1990) Proc. Natl. Acad. Sci. USA, 87, 7185-7189.
130.Chen, Z., and Lash, L. H. (1998) J.
Pharmacol. Exp. Ther., 285, 608-618.
131.Rebrin, I., Kamzalov, S., and Sohal, R. S.
(2003) Free Rad. Biol. Med., 35, 626-635.
132.Vogel, R., Wiesinger, H., Hamprecht, B., and
Dringen, R. (1999) Neurosci. Lett., 275, 97-100.
133.Boveris, A., and Cadenas, E. (1997) Cellular
Sources and Steady-State Levels of Reactive Oxygen Species, Marcel
Dekker.
134.Han, D., Canali, R., Rettori, D., and
Kaplowitz, N. (2003) Mol. Pharmacol., 64, 1136-1144.
135.Raza, H., Robin, M. A., Fang, J. K., and
Avadhani, N. G. (2002) Biochem. J., 366, 45-55.
136.Olafsdottir, K., and Reed, D. J. (1988)
Biochim. Biophys. Acta, 964, 377-382.
137.Kozhemiakin, L. A., Bulavin, D. V., Udintsev,
A. V., and Smirnov, V. V. (1993) Tsitologiya, 35,
58-63.
138.Zoccarato, F., Cavallini, L., and Alexandre, A.
(2004) J. Biol. Chem., 279, 4166-4174.
139.Panfili, E., Sandri, G., and Ernster, L. (1991)
FEBS Lett., 290, 35-37.
140.Kelner, M. J., and Montoya, M. A. (2000)
Biochem. Biophys. Res. Commun., 269, 366-368.
141.Hoek, J. B., and Rydstrom, J. (1988)
Biochem. J., 254, 1-10.
142.Kirsch, M., and De Groot, H. (2001) FASEB
J., 15, 1569-1574.
143.Tischler, M. E., Hecht, P., and Williamson, J.
R. (1977) FEBS Lett., 76, 99-104.
144.Skulachev, V. P. (2001) FEBS Lett.,
492, 1-3.
145.Lenzen, S., Drinkgern, J., and Tiedge, M.
(1996) Free Rad. Biol. Med., 20, 463-466.
146.Asayama, K., Yokota, S., Dobashi, K.,
Hayashibe, H., Kawaoi, A., and Nakazawa, S. (1994)
Histochemistry, 102, 213-219.
147.Utsunomiya, H., Komatsu, N., Yoshimura, S.,
Tsutsumi, Y., and Watanabe, K. (1991) J. Histochem. Cytochem.,
39, 1167-1174.
148.Esworthy, R. S., Ho, Y. S., and Chu, F. F.
(1997) Arch. Biochem. Biophys., 340, 59-63.
149.Ho, Y. S., Magnenat, J. L., Bronson, R. T.,
Cao, J., Gargano, M., Sugawara, M., and Funk, C. D. (1997) J. Biol.
Chem., 272, 16644-16651.
150.Spector, A., Yang, Y., Ho, Y. S., Magnenat, J.
L., Wang, R. R., Ma, W., and Li, W. C. (1996) Exp. Eye Res.,
62, 521-540.
151.Cheng, W. H., Ho, Y. S., Valentine, B. A.,
Ross, D. A., Combs, G. F., Jr., and Lei, X. G. (1998) J. Nutr.,
128, 1070-1076.
152.Cheng, W. H., Ho, Y. S., Ross, D. A.,
Valentine, B. A., Combs, G. F., and Lei, X. G. (1997) J. Nutr.,
127, 1445-1450.
153.De Haan, J. B., Bladier, C., Griffiths, P.,
Kelner, M., O'Shea, R. D., Cheung, N. S., Bronson, R. T., Silvestro, M.
J., Wild, S., Zheng, S. S., Beart, P. M., Hertzog, P. J., and Kola, I.
(1998) J. Biol. Chem., 273, 22528-22536.
154.Zhang, J., Graham, D. G., Montine, T. J., and
Ho, Y. S. (2000) J. Neuropathol. Exp. Neurol., 59,
53-61.
155.Klivenyi, P., Andreassen, O. A., Ferrante, R.
J., Dedeoglu, A., Mueller, G., Lancelot, E., Bogdanov, M., Andersen, J.
K., Jiang, D., and Beal, M. F. (2000) J. Neurosci., 20,
1-7.
156.Fujii, J., and Ikeda, Y. (2002) Redox
Rep., 7, 123-130.
157.Wood, Z. A., Schroder, E., Robin Harris, J.,
and Poole, L. B. (2003) Trends Biochem. Sci., 28,
32-40.
158.Chae, H. Z., Kim, H. J., Kang, S. W., and Rhee,
S. G. (1999) Diabetes Res. Clin. Pract., 45, 101-112.
159.Leyens, G., Donnay, I., and Knoops, B. (2003)
Comp. Biochem. Physiol. B. Biochem. Mol. Biol., 136,
943-955.
160.Araki, M., Nanri, H., Ejima, K., Murasato, Y.,
Fujiwara, T., Nakashima, Y., and Ikeda, M. (1999) J. Biol.
Chem., 274, 2271-2278.
161.Hattori, F., Murayama, N., Noshita, T., and
Oikawa, S. (2003) J. Neurochem., 86, 860-868.
162.Banmeyer, I., Marchand, C., Verhaeghe, C.,
Vucic, B., Rees, J. F., and Knoops, B. (2004) Free Rad. Biol.
Med., 36, 65-77.
163.Johansson, C., Lillig, C. H., and Holmgren, A.
(2004) J. Biol. Chem., 279, 7537-7543.
164.Gromer, S., Urig, S., and Becker, K. (2004)
Med. Res. Rev., 24, 40-89.
165.Fernandes, A. P., and Holmgren, A. (2004)
Antioxid. Redox Signal, 6, 63-74.
166.Nonn, L., Williams, R. R., Erickson, R. P., and
Powis, G. (2003) Mol. Cell Biol., 23, 916-922.
167.Patenaude, A., Murthy, M. R., and Mirault, M.
E. (2004) J. Biol. Chem., 279, 27302-27314.
168.Staniek, K., and Nohl, H. (1999) Biochim.
Biophys. Acta, 1413, 70-80.
169.Savage, M. K., Jones, D. P., and Reed, D. J.
(1991) Arch. Biochem. Biophys., 290, 51-56.
170.Nicholls, D. G., and Budd, S. L. (2000)
Physiol. Rev., 80, 315-360.
171.Fiskum, G. (2000) J. Neurotrauma,
17, 843-855.
172.Maciel, E. N., Vercesi, A. E., and Castilho, R.
F. (2001) J. Neurochem., 79, 1237-1245.
173.Perez Velazquez, J. L., Frantseva, M. V., and
Carlen, P. L. (1997) J. Neurosci., 17, 9085-9094.
174.Anderson, M. F., and Sims, N. R. (2002) J.
Neurochem., 81, 541-549.
175.Starkov, A. A. (1997) Biosci. Rep.,
17, 273-279.
176.Zhang, Z., Huang, L., Shulmeister, V. M., Chi,
Y. I., Kim, K. K., Hung, L. W., Crofts, A. R., Berry, E. A., and Kim,
S. H. (1998) Nature, 392, 677-684.
177.Novgorodov, S. A., Gudz, T. I., Kushnareva, Y.
E., Roginsky, V. A., and Kudrjashov, Y. B. (1991) Biochim. Biophys.
Acta, 1058, 242-248.