REVIEW: Reactive Oxygen and Nitrogen Species: Friends or Foes?
D. B. Zorov1*, S. Y. Bannikova1, V. V. Belousov2, M. Y. Vyssokikh1, L. D. Zorova1, N. K. Isaev1, B. F. Krasnikov1,3, and E. Y. Plotnikov1
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; fax: (7-095) 939-3181; E-mail: zorov@genebee.msu.ru2Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, 117997 Moscow, Russia
3Burke Medical Research Institute, White Plains, New York, USA
* To whom correspondence should be addressed.
Received October 4, 2004
Chemical and physiological functions of molecular oxygen and reactive oxygen species (ROS) and existing equilibrium between pools of pro-oxidants and anti-oxidants providing steady state ROS level vital for normal mitochondrial and cell functioning are reviewed. The presence of intracellular oxygen and ROS sensors is postulated and few candidates for this role are suggested. Possible involvement of ROS in the process of fragmentation of mitochondrial reticulum made of long mitochondrial filaments serving in the cell as “electric cables”, as well as the role of ROS in apoptosis and programmed mitochondrial destruction (mitoptosis) are reviewed. The critical role of ROS in destructive processes under ischemia/reoxygenation and ischemic preconditioning is discussed. Mitochondrial permeability transition gets special consideration as a possible component of the apoptotic cascade, resulting in excessive “ROS-induced ROS release”.
KEY WORDS: reactive oxygen species, nitric oxide, mitochondria, apoptosis, ischemia/reoxygenation, ischemic preconditioning, fragmentation, electric cable
The very first published paper of V. P. Skulachev's scientific career [1] and a large set of papers he has published within recent years all demonstrate his great devotion to the elucidation of the mechanism of oxidative processes in organisms, namely of those for which the participation of oxygen is obligatory. The role of this chemical element that gave the birth to the entire aerobic World looks quite simple--oxygen itself is a rather efficient oxidant, the cell has systems of its utilization, and one useful output from this cooperation is that an energy buffer is built up in the cell as a result of oxidative phosphorylation. But the general significance of oxygen-consuming cellular systems is not limited by these bioenergetic properties--oxygen is also successfully used in processes of detoxication and generation of signaling molecules. Considering a protean manifestation of signaling mechanisms, i.e., from constructive to destructive, one can say that oxygen is a double-edge sword; however, the destructive function is not widely accepted as a vital expediency, thus creating the image of a friend and a foe at the same time.
REACTIVE OXYGEN SPECIES
The image of oxygen as an enemy to a great extent was created because a quite large set of compounds called “reactive oxygen species” (ROS) originate from molecular oxygen. The most abundant ROS representative in the living cell, superoxide, as well as hydrogen peroxide, singlet oxygen, and hydroxyl anion radical, all fell into one group [2]. The special feature of these agents is their high oxidative reactivity, creating a significant threat for redox-sensitive cellular components, primarily for proteins, lipids, and nucleic acids. The natural systems protecting against an excess of ROS (redox-buffering systems such as reduced glutathione, vitamins, albumins and globulins, free fatty acids (especially unsaturated), and many others) are considered as an essential pool of antioxidants. Apparently, pro-oxidative and anti-oxidative systems coexist in a state of dynamic equilibrium, thus providing some steady state levels of pro-oxidants, which are critically important for cell functioning. It is known that an excess of antioxidant results in fatal consequences [3] as well as pro-oxidant excess determining the phenomenon of oxidative stress with all unwanted effects (aging and death) [4-6]. It is worth emphasizing here that the mutual transformations of ROS have been demonstrated (through dismutation, from superoxide to hydrogen peroxide, by the Fenton reaction, from superoxide to hydrogen anion radical, etc.), which results in a significant change in the oxidative potential of a given ROS member. Also, these transitions can result in a conversion of ROS action from anti-oxidative to pro-oxidative. It is obvious that one Golden Rule, multet nocem (excess is harmful), is valid for ROS [7].
SENSORS FOR OXYGEN AND ROS
For realization of the mentioned rule, it is necessary to assume a priori that a cell keeps systems measuring ROS level. The same is valid also for molecular oxygen as a ROS forefather--we must believe that there are systems for oxygen detection.
There are many candidates for the role of intracellular oxygen sensor. One of the earliest dogmas is that oxygen sensors are heme-containing proteins (e.g., different cytochromes) [8]. Due to the limited scope of the current review, we will list only some of them. Although there have been attempts to divide all oxygen sensors into real (measuring oxygen concentration) and pseudo-sensors (measuring the absence of oxygen) [9], sometimes it is quite difficult to delimit the definition of ROS or O2·- sensors. And it is surely very difficult to discriminate sensors for ROS and so-called redox sensors [10], which potentially may be all the same. Increasingly spreading opinion that the basics for prevention of aging and cellular pathologies is in a strict maintenance of cellular redox homeostasis at minimum explains the physiological importance of knowledge how these regulators act in cells [11]. One indisputable fact is that the gene expression mechanism is redox dependent. Delayed effect of protein synthesis under hypoxic conditions, and among these proteins the hematopoietic growth factor and erythropoietin have particular importance, helped to discover the transcription factor HIF (hypoxia-induced factor). This factor serves a few tens of different genes involved in hypoxia [12]. The activity of this protein is regulated by posttranslational enzymatic oxygen-dependent hydroxylation of prolines and asparagines in its oxygen-sensitive domain. Proteins from the family of 2-oxoglutarate-dependent oxygenases (non-heme Fe2+-dependent enzymes) are responsible for such hydroxylation activity. Direct effect of molecular oxygen on the hydroxylase activity explains why HIF is placed in the group of oxygen sensors.
Another although less explored example of possible involvement of non-heme proteins in oxygen reception is the mitochondrial benzodiazepine receptor (MBR). This protein has quite high homology with tryptophan-rich sensor protein TspO (CrtK, according to earlier nomenclature) from Rhodobacter sphaeroides. TspO is a negative regulator of expression of a number of genes encoding components of the photosynthetic complex (structural proteins, bacteriochlorophyll, carotenoids) in these bacteria. It has been demonstrated that MBR substitutes for TspO function in bacteria and in the presence of oxygen inhibits the expression of photosynthetic genes. Moreover, the effect was abolished by MBR ligands [13], which was interpreted as MBR being an oxygen sensor. At the same time, the fact that mitochondrial megachannel can be equally blocked by either MBR ligands [14] or inhibitors of the mitochondrial permeability transition (MPT) allowed the speculation that MPT is a functional state of MBR. Because MBR is a multi-protein complex [15, 16] and assuming, that it can carry O2- or ROS-sensing function, such activity may be determined by the flexible interactions between proteins, components of the mitochondrial contact site [17].
FRAGMENTATION OF THE MITOCHONDRIAL RETICULUM
We predicted the existence of the MBR more than 20 years ago based on the discovery of the phenomenon of generalized fragmentation of the mitochondrial reticulum after cells were treated with MBR ligand [18]. Although it was known much before that mitochondria can constantly and spontaneously generate small round fragments that can subsequently fuse with other mitochondrial bodies to form specific elongated structures, the total fragmentation resulting in a complete loss of filamentous mitochondria had never been described. Later we found that a whole set of inhibitors (rotenone (Fig. 1), antimycin A, myxothiazol, cyanide, azide, uncouplers, oligomycin, tetraphenylphosphonium, and other agents [19]) after just 30-60 min of exposure begin to induce a global conversion of mitochondrial filaments into small fragments. A similar result but on the time scale of seconds was observed in mitochondria of fibroblasts under photodynamic effect (irradiation of the cell stained by mitochondrial dyes with light at the absorption maximum of the dye). It became clear that without any doubt, ROS are essential players in the mitochondrial fragmentation. However, we failed to prevent the fragmentation by supplementing with antioxidants such as alpha-tocopherol, which pointed to either low efficiency of such antioxidant or its unavailability at the sites responsible for the fragmentation.
This shows that the mitochondrion itself, or better to say its structure, is the sensor for ROS. It seems that we are facing a paradox--mitochondria themselves are ROS generators, and if so, how they can be their sensors. First, let us correct one common statement defining the mitochondrion as the major source of ROS. It is true that mitochondria do significantly contribute to the basal levels of intracellular ROS. However, besides their own production of ROS, mitochondria regulate their generation by other systems through the regulation of the oxygen content in the cellular compartments by its active consumption, and mitochondria are known to be the main oxygen consumers. But still in the early studies of B. Chance and collaborators the partial contribution of each cellular component into net hydrogen peroxide production was evaluated, and it was concluded that in liver (of course, in specialized tissues the share of each compartment to total ROS production can be different) the main ROS generator are peroxisomes, followed by microsomes, but mitochondria take only third place [20].Fig. 1. Fragmentation of mitochondrial reticulum in pig embryo kidney epithelial cells. a) Control culture stained with permeant fluorescent cation, rhodamine 123 (10 µM), which is preferentially accumulated in mitochondria; b) same culture stained with rhodamine 123 after rotenone (2 µM) treatment for 6 h; c, d) three-dimensional reconstruction made from electron microscopic serial section images for (a) and (b), correspondingly.
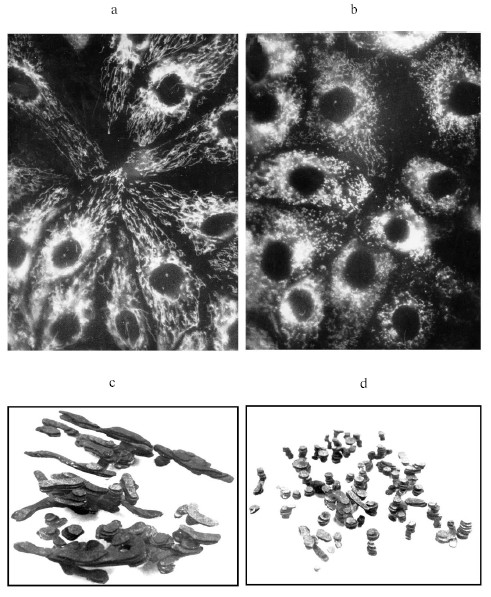
One more time, we repeat that the reciprocal reaction of the mitochondrial structure on the elevated level of ROS in its environment confirms the principle multet nocem. Apparently, induced fragmentation is the maximally possible and critical response to the elevation of ROS levels exceeding the physiological range (it will not exclude the existence of more delicate oxygen sensing mechanisms in mitochondria providing the self-regulation of ROS production in mitochondria, which suggests more than one oxygen or ROS sensing system in mitochondria with different affinity and different reciprocal reactions).
In recent years, the phenomenon of fragmentation of the mitochondrial reticulum that nobody could explain for many years has become more or less clear as a transient step preceding cell death. It became obvious that the process is active with many specific proteins responsible for the mitochondrial fusion and fission involved [21]. It is also clear that the process cannot run without ROS.
SOLUTION OF THE PROBLEM OF ENERGY TRANSDUCTION
It is quite obvious that adequate energy supply to all cellular compartments may become a problem particularly under conditions of activated metabolism or limited access to oxidative substrates or oxygen. As an example, the first condition occurs under high muscle load, which results in cellular hypoxia and transition from oxidative to anaerobic metabolism. The second condition in practice is a consequence of vascular pathologies that may limit or block the tissue supply with oxygen and substrates (tissue ischemia as a result of vasoconstriction, thrombosis, or surgical procedure, for instance under organ transplantation). The second condition (possibly, the first as well) is fraught with irreversible tissue degradation due to that in a majority of cases ischemia or hypoxia is usually following by the restoration of adequate blood supply and proper delivery of nutrients to the cell. The solution of two problems is sought: how to provide an adequate energy supply for all cellular compartments and how to protect the cell from damage caused by the hypoxia/normoxia transitions.
V. P. Skulachev suggested that the extended coupling membranes might serve as intracellular energetic cables [22-24]. The first suggestion successfully proven under his guidance [25-27] defines that coupling membranes (i.e., inner mitochondrial membranes) are energized in the loci favorable for generation of proton motive force having good access to sources for oxidizable substrates and oxygen-transporting roots. Because of the equipotentiality, coupling membranes can use the energy of the proton potential for ATP synthesis along the entire membrane length. This will be beneficial in terms of providing energy supply of remote cellular compartments, thus eliminating the dependence of these compartments on oxygen. The second suggestion (which still needs to be proven) is that due to a high affinity of phospholipid membranes to oxygen, extended mitochondria can serve as oxygen transmitters, at the same time being its users, and may be carrying on itself sensors for oxygen or for its active forms. So, membranes are absorbing oxygen thus to some extent lowering oxygen danger for water-soluble cellular components but making this danger higher for lipid-soluble components, first of all membrane proteins and lipids themselves.
ISCHEMIA, REOXYGENATION, AND ISCHEMIC PRECONDITIONING
High metabolic activity of mitochondria decreases the oxygen content in the cell, thus balancing on the edge of developing hypoxia.
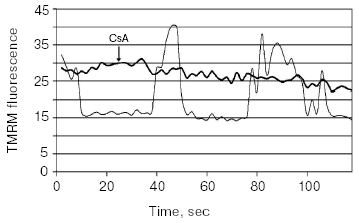
We note that while under normoxic conditions mitochondrial energization is driven by respiration, nevertheless, under hypoxic conditions for unknown reasons, mitochondria still keep their energization, which is supported by the reversal of the ATP-synthase reaction [28]. That will result in a waste of ATP accumulated under aerobic period and also formed in glycolysis in the absence of oxygen. It is obvious that the mitochondrial membrane potential becomes the very last line of defense that after its loss the cell is anticipated to die. Apparent resistance of the cell to the irreversibly loss of the membrane potential apparently can be observed under photodynamic ROS generation in mitochondria when cells (cardiac myocytes, astrocytes), stained by permeant mitochondrial fluorescent dyes (i.e., methyl ester of tetramethylrhodamine (TMRM)), after illumination with the excitation light of a moderate intensity showed long-term oscillations of the membrane potential in their mitochondria (Fig. 2) [29, 30], which still finally results in cell death. In astrocytes, these oscillations are prevented by cyclosporine A (Fig. 2), which suggested that oscillations occur on the level of a megachannel. Similar, but apparently different in its nature, oscillations of the mitochondrial membrane potential were observed by others [31-33]. Without any doubt, such oscillations of the membrane potential, which (as we emphasized above) is significantly buffered by cellular ATP, may be referred to as pathological events with possibly fatal result for the cell.
Hypoxia itself as well as hypoxia/normoxia transitions is extremely dangerous for the cell. In the nerve tissue, hypoxia rather quickly results in a release of neurotoxic amounts of glutamate (for review, see [34]) that irreversibly initiates neuronal death with an important role of the MPT [35]. In other tissues like heart and kidney, hypoxia is not itself as dangerous as these cytotoxic consequences, which brings up tissue reoxygenation resulting in a burst of ROS generation [36-38]. In principle, cells can be prepared to meet the change of hypoxia to normoxia through adaptation by a short pulses of hypoxia following by short normoxic periods, i.e., through the process of so-called ischemic (hypoxic) preconditioning [39, 40]. The mechanism of ischemic preconditioning is among the most studied problems because it seems to be one of the most perfect mechanisms designed by nature. Separate deciphered fragments of the entire mechanism are presently used for protection by pharmacological preconditioning, i.e., when pharmacological drugs activating one or another partial reaction of a physiological cascade are used. To put in order known fragments is highly difficult, but what became clear is that protecting drugs we can refer to as activators of plasmalemmal or mitochondrial K/ATP channel, of protein kinase C, ligands of opioid receptor, bradykinin, activators of tyrosine kinase receptors (insulin and IGF-1), A1 receptor ligands, and others. We succeeded in a proper organization of the entire system of complex protective reactions into two separate mechanisms converging at the specific pivotal kinase (glycogen synthase kinase-3beta), located in mitochondria [41]. One of these protecting mechanisms proceeds with obligatory participation of ROS and it is clear that the use of antioxidants can be fatal for patients especially in the pre-infarct period. The most essential detail of both mechanisms is that the pivotal kinase regulates the induction of the MPT that is blocked after inhibition of the kinase activity. Thus, the strategy for defense consists in abolishing all possible mechanisms of MPT induction.Fig. 2. Time-dependent fluctuations in fluorescence recorded from a single cell in cultured astrocytes stained with methyl ester of tetramethylrhodamine (TMRM) (125 nM) and exposed with the excitation light for 30 sec. Thin line, without inhibitors; thick line, with cyclosporin A (4 µM).
ROLE OF MITOCHONDRIAL PERMEABILITY TRANSITION AND ROS IN
APOPTOSIS AND MITOPTOSIS
The data on the role of ROS in MPT induction are contradictory. On isolated mitochondria, we succeeded in demonstration that MPT induction by calcium ions takes place even under very low partial oxygen pressure [42, 43]. From another side, using rat cardiac myocyte we observed that ROS can induce MPT in situ [30]. It is quite possible that a ROS-regulating motif can be bypassed being located upstream of other inducers or, there is a duplication of induction pathways when the absence of one pathway cannot exclude the induction by another pathway.
Currently, it is believed that the MPT in many cases is apparently the necessary element of an apoptotic cascade [44]. One of the most possible damaging consequences of the MPT induction is excessive mitochondrial ROS generation, which we called ROS-induced ROS-release [43] representing a cascade of accelerated generation of ROS by mitochondria with still unclear mechanism.
The identity of the mitochondrial megachannel and MPT induction [44, 45] gave us impetus to suggest that the megachannel opening (possibly through excessive ROS generation) is a signal for programmed mitochondrial destruction [46], thus creating a basis for the solution the problem of degradation firstly of mitochondria and later, of the entire cell. This concept further developed by Skulachev was given the term “mitoptosis” [47] as an initial phase of a structural organization of the death cascade in the living system, from mitoptosis through apoptosis to phenoptosis, i.e., the death of the whole individual [48, 49]. Recently we received the first confirmations supporting the correctness of the concept of mitoptosis [50]. In the entity of the concept and model on the level of mitochondria, the pivotal role of ROS is obvious or suggested. The proofs of phenoptosis and the critical role of ROS in it are waiting for their turn.
REACTIVE NITROGEN SPECIES (RNS)
The expedient destructive function of ROS is tightly linked to the function of another radical, nitric oxide. Besides NO, the RNS group contains diverse nitrogen derivatives (N2O3, NO2-, peroxynitrite, and others), that all possess a strong oxidative capacity. Protein modification (i.e., nitrosylation of cysteines and tyrosines) is one of the most powerful elements of signaling transduction. Currently, NO takes as much attention as ROS do. Both destructive and signaling functions of NO are suggested, but a clear-cut border between these two definitions is not obvious. The discovery in mitochondria of its own machinery for NO production, mitochondrial NO-synthase [51], places this enzyme among other pivotal mitochondrial enzymes but still with obscure function. Cross talk between ROS and RNS (interaction and common targets) allows both forms to be referred to as a general group of small signaling molecules [52, 53]. In particular, mitochondrial cytochrome oxidase is the common target for NO and O2 [54], which suggests the regulatory role of NO in mitochondrial respiration, and when taking into account the proximity of mitochondrial NO-synthase to cytochrome oxidase, it becomes clear that such regulation may be of urgent character.
We have demonstrated that oxidative stress caused by ischemia/reperfusion of the kidney is accompanied by a nitrosative stress, which is characterized by enhanced NO production [55]. Such production is provided by NO-synthase since it was inhibited by a specific inhibitor, nitrorarginine. On the microscopic level, NO generation was co-localized with mitochondria, which gave strong evidence in support of the idea that nitrosative stress under these conditions is provided by the activation of the mitochondrial isoform of NO-synthase.
Different interpretation of the role of ROS and RNS in cell functioning sometimes results in a polar scattering of existing opinions on this role. In this review, we attempted to discuss only a few aspects of cell functioning with apparent emphasis focusing on mitochondria, organelles with multiple functions [7]. It is obvious that mitochondria are generators, sensors, and targets for ROS and RNS. The delicate balance between their production levels and their utilization rate is provided by a complex set of measures localized inside as well as outside of mitochondria. The coordination of this balance is the result of the proper tuning of the sensors with possible involvement of other cellular components, for example of cytoskeleton.
This work was supported by the Russian Foundation for Basic Research (grants 02-04-48845, 05-04-49697, and 05-04-48412).
REFERENCES
1.Skulachev, V. P. (1958) Uspekhi Sovr. Biol.,
46, 241-245.
2.Nohl, H., and Jordan, W. (1980) Eur. J.
Biochem., 111, 203-210.
3.Heart Protection Study Collaborative Group MRC/BHF
(2002) Lancet, 360, 23-33.
4.Harman, D. (1956) J. Gerontol., 11,
298-300.
5.Zorov, D. B. (1996) Biochim. Biophys. Acta,
1275, 10-15.
6.Papa, S., and Skulachev, V. P. (1997) Mol. Cell.
Biochem., 174, 305-319.
7.Zorov, D. B., Krasnikov, B. F., Kuzminova, A. E.,
Vysokikh, M. Yu., and Zorova, L. D. (1997) Biosci. Rep.,
17, 507-520.
8.Schumacker, P. T. (2003) Adv. Exp. Med.
Biol., 543, 57-71.
9.Richalet, J. P. (1997) Comp. Biochem. Physiol.
A. Physiol., 118, 9-14.
10.Forsberg, J., Rosenquist, M., Fraysse, L., and
Allen, J. F. (2001) Biochem. Soc. Trans., 29,
403-407.
11.Droge, W. (2002) Physiol. Rev., 82,
47-95.
12.Schofield, C. J., and Ratcliffe, P. J. (2004)
Nat. Rev. Mol. Cell. Biol., 5, 343-354.
13.Yeliseev, A. A., Krueger, K. E., and Kaplan, S.
(1997) Proc. Natl. Acad. Sci. USA, 94, 5101-5106.
14.Kinnally, K. W., Zorov, D. B., Antonenko, Y. N.,
Snyder, S. H., McEnery, M. W., and Tedeschi, H. (1993) Proc. Natl.
Acad. Sci. USA, 90, 1374-1378.
15.McEnery, M. W., Snowman, A. M., Trifiletti, R.
R., and Snyder, S. H. (1992) Proc. Natl. Acad. Sci. USA,
89, 3170-3174.
16.Vyssokikh, M. Y., Goncharova, N. Y., Zhuravlyova,
A. V., Zorova, L. D., Kirichenko, V. V., Krasnikov, B. F., Kuzminova,
A. E., Melikov, K. C., Melik-Nubarov, N. S., Samsonov, A. V., Belousov,
V. V., Prischepova, A. E., and Zorov, D. B. (1999) Biochemistry
(Moscow), 64, 390-398.
17.Vyssokikh, M. Y., Katz, A., Rueck, A., Wuensch,
C., Dorner, A., Zorov, D. B., and Brdiczka, D. (2001) Biochem.
J., 358, 349-358.
18.Vorobjev, I. A., and Zorov, D. B. (1983) FEBS
Lett., 163, 311-314.
19.Avad, S. A., Vorobjev, I. A., and Zorov, D. B.
(1984) 16th Congr. of FEBS, Moscow, Abst. XI-080.
20.Boveris, A., and Chance, B. (1973) Biochem.
J., 134, 707-716.
21.Bossy-Wetzel, E., Barsoum, M. J., Godzik, A.,
Schwarzenbacher, R., and Lipton, S. A. (2003) Curr. Opin. Cell.
Biol., 15, 706-716.
22.Skulachev, V. P. (1969) Energy Accumulation in
Cell [in Russian], Nauka, Moscow.
23.Skulachev, V. P. (1971) Curr. Top.
Bioenerg., 4, 127-190.
24.Skulachev, V. P. (1980) Biochim. Biophys.
Acta, 604, 297-310.
25.Drachev, V. P., and Zorov, D. B. (1986) Dokl.
Akad. Nauk SSSR, 287, 1237-1238.
26.Amchenkova, A. A., Bakeeva, L. E., Chentsov, Y.
S., Skulachev, V. P., and Zorov, D. B. (1988) J. Cell. Biol.,
107, 481-495.
27.Severina, I. I., Skulachev, V. P., and Zorov, D.
B. (1988) J. Cell. Biol., 107, 497-501.
28.Di Lisa, F., Blank, P. S., Colonna, R., Gambassi,
G., Silverman, H. S., Stern, M. D., and Hansford, R. G. (1995) J.
Physiol., 486, 1-13.
29.Belousov, V. V., Bambrick, L. L., Starkov, A. A.,
Zorov, D. B., Skulachev, V. P., and Fiskum, G. (2001) Abst. Soc.
Neurosci., 27, 205-218.
30.Zorov, D. B., Filburn, C. R., Klotz, L. O.,
Zweier, J. L., and Sollott, S. J. (2000) J. Exp. Med.,
192, 1001-1014.
31.Romashko, D. N., Marban, E., and O'Rourke, B.
(1998) Proc. Natl. Acad. Sci. USA, 95, 618-623.
32.Huser, J., Rechenmacher, C. E., and Blatter, L.
A. (1998) Biophys. J., 74, 2129-2137.
33.Aon, M. A., Cortassa, S., Marban, E., and
O'Rourke, B. (2003) J. Biol. Chem., 278, 44735-44744.
34.Isaev, N. K., Andreeva, N. A., Stelmashook, E.
V., and Zorov, D. B. (2005) Biochemistry (Moscow), in press.
35.Isaev, N. K., Zorov, D. B., Stelmashook, E. V.,
Uzbekov, R. E., Kozhemyakin, M. B., and Victorov, I. V. (1996) FEBS
Lett., 392, 143-147.
36.Kloner, R. A., Ellis, S. G., Carlson, N. V., and
Braunwald, E. (1983) Cardiology, 70, 233-246.
37.Zweier, J. L., Flaherty, J. T., and Weisfeldt, M.
L. (1987) Proc. Natl. Acad. Sci. USA, 84, 1404-1407.
38.Darley-Usmar, V. M., Stone, D., Smith, D., and
Martin, J. F. (1991) Ann. Med., 23, 583-588.
39.Murry, C. E., Jennings, R. B., and Reimer, K. A.
(1986) Circulation, 74, 1124-1136.
40.Kloner, R. A., Bolli, R., Marban, E., Reinlib,
L., and Braunwald, E. (1998) Circulation, 97,
1848-1867.
41.Juhaszova, M., Zorov, D. B., Kim, S. H., Pepe,
S., Fu, Q., Fishbein, K. W., Ziman, B. D., Wang, S., Ytrehus, K.,
Antos, C. L., Olson, E. N., and Sollott, S. J. (2004) J. Clin.
Invest., 113, 1535-1549.
42.Krasnikov, B. F., Kuzminova, A. E., and Zorov, D.
B. (1997) FEBS Lett., 419, 137-140.
43.Kuzminova, A. E., Zhuravlyova, A. V., Vyssokikh,
M. Yu., Zorova, L. D., Krasnikov, B. F., and Zorov, D. B. (1998)
FEBS Lett., 434, 313-316.
44.Zamzami, N., Susin, S. A., Marchetti, P., Hirsch,
T., Gomez-Monterrey, I., Castedo, M., and Kroemer, G. (1996) J. Exp.
Med., 183, 1533-1544.
45.Szabo, I., and Zoratti, M. (1991) J. Biol.
Chem., 266, 3376-3379.
46.Kinnally, K. W., Zorov, D., Antonenko, Y., and
Perini, S. (1991) Biochem. Biophys. Res. Commun., 176,
1183-1188.
47.Skulachev, V. P. (1999) Mol. Aspects Med.,
20, 139-184.
48.Skulachev, V. P. (1999) Biochemistry
(Moscow), 64, 1418-1426.
49.Skulachev, V. P. (2002) Ann. N. Y. Acad.
Sci., 959, 214-237.
50.Zorov, D. B., Kobrinsky, E., Juhaszova, M., and
Sollott, S. J. (2004) Circ. Res., 95, 239-252.
51.Ghafourifar, P., and Richter, C. (1997) FEBS
Lett., 418, 291-296.
52.Inoue, M., Sato, E. F., Nishikawa, M., Park, A.
M., Kira, Y., Imada, I., and Utsumi, K. (2003) Antioxid. Redox.
Signal., 5, 475-484.
53.Forman, H. J., Fukuto, J. M., and Torres, M.
(2004) Am. J. Physiol. Cell Physiol., 287, 246-256.
54.Cooper, C. E., Davies, N. A., Psychoulis, M.,
Canevari, L., Bates, T. E., Dobbie, M. S., Casley, C. S., and Sharpe,
M. A. (2003) Biochim. Biophys. Acta, 1607, 27-34.
55.Plotnikov, E. Y., Vyssokikh, M. Y., Tcvirkun, D.
V., Kazachenko, A. V., and Kirpatovsky, V. I. (2005) Dokl. Ros.
Akad. Nauk, in press.