
|
DNA Methylation and Demethylation as Targets for Anticancer TherapyM. SzyfDepartment of Pharmacology and Therapeutics, McGill University, 3655 Sir Wiliam Osler Promenade, Montreal PQ H3G 1Y6, Canada; fax: (1-514) 398-6690; E-mail: moshe.szyf@mcgill.ca
|
Received November 17, 2004
Cancer growth and metastasis require the coordinate change in gene expression of different sets of genes. While genetic alterations can account for some of these changes, it is becoming evident that many of the changes in gene expression observed are caused by epigenetic modifications. The epigenome consists of the chromatin and its modifications, the “histone code” as well as the pattern of distribution of covalent modifications of cytosines residing in the dinucleotide sequence CG by methylation. Although hypermethylation of tumor suppressor genes has attracted a significant amount of attention and inhibitors of DNA methylation were shown to activate methylated tumor suppressor genes and inhibit tumor growth, demethylation of critical genes plays a critical role in cancer as well. This review discusses the emerging role of demethylation in activation of pro-metastatic genes and the potential therapeutic implications of the demethylation machinery in metastasis.
KEY WORDS: DNA methylation, DNA demethylation, DNA methyltransferase (DNMT), DNA demethylase, MeCP2, MBD2, histone acetylation, epigenetics, epigenome, histone deacetylase (HDAC), TSA, HDAC inhibitors, metastasis
The development of cancer involves the concurrent disruption of regulation of expression of multiple genes. This switch in gene expression first allows the cancer cell to override normal breaks on uncontrolled growth and at a later stage enables cell invasion and motility, which allows the cancer to metastasize to distal organs. Such a concerted change in regulation of expression of numerous genes must involve some basic processes responsible for the normal programming of gene expression. Since the genome contains all the potential information required to manage a living organism but only a fraction of this information is active at different time points and in different cell types and individual cells, this information must be regulated.
The epigenome is responsible for regulation of the genome. In contrast to the genome, which is identical in different cell types and through life, the epigenome is dynamic and varies from cell type to cell type and from time point to time point in life. It is responsive to developmental, physiological, environmental, and pathological signals and confers cell type and temporal identities of gene expression programs. The epigenome consists of the chromatin and its modifications [1, 2] as well as a covalent modification of cytosine rings found at the dinucleotide sequence CG [3].
The basic building block of chromatin is the nucleosome, which is formed of an octamer of histone proteins containing a H3-H4 tetramer flanked on either side with a H2A-H2B dimer [4]. The N-terminal tails of these histones are extensively modified by methylation [5, 6], phosphorylation, and acetylation [7, 8]. Different histone variants also play a regulatory role [9]. The pattern of histone modification creates a “histone code”, which dictates, which parts of the genome are expressed at a given point in time [2].
Specific transcription factors and transcription repressors recruit histone-modifying enzymes to specific genes and thus define the profile of histone modification around genes [10]. The best-studied examples are histone acetyltransferases (HAT), which acetylate histone H3 and H4 tails at the K9 residue, and histone deacetylases (HDAC), which deacetylate histone tails [11].
Histone acetylation is believed to be a predominant signal for an active chromatin configuration. Deacetylated histones signal inactive chromatin. Many repressors and repressor complexes recruit HDACs to genes, thus causing their inactivation. Histone tail acetylation is believed to enhance the accessibility of a gene to the transcription machinery, whereas deacetylated tails are tightly interacting with DNA and limit its accessibility to transcription factors [11]. Histone methylation at K9 of their N-terminal tail signals inactivity and is also determined by the recruitment of histone methyltransferases such as SUV39 to genes [12]. The heterochromatin protein HP-1, which binds methylated histones and precipitates an inactive chromatin structure [12], recognizes the methylated histones. Chromatin remodeling complexes, which are ATP dependent, alter the position of nucleosomes around the transcription initiation site and define its accessibility to the transcription machinery [13]. The different combinations of these changes determine the diverse programs of gene expression. Chromatin is dynamic and plastic and could respond by altered recruitment of the different histone modification enzymes in response to different signals. Nevertheless, the majority of epigenomic programs is laid down during development and remains relatively stable for the entire life span of an organism. Some parts of the epigenome such as heterochromatin are particularly stably repressed [6].
In addition to chromatin, which is associated with DNA, the DNA molecule itself is chemically modified by methyl residues at the 5´ position of the cytosine rings in the sequence CG in vertebrates [14]. What distinguishes DNA methylation in vertebrate genomes is the fact that not all CGs are methylated in any given cell type [3]. Different CGs are methylated in different cell types, generating cell type specific patterns of methylation.
Thus, the DNA methylation pattern confers upon a genome itself its cell type identity. Since DNA methylation is part of the chemical structure of the DNA itself, it remains long after all other proteins and epigenomic marks are degraded, and thus it has extremely important diagnostic potential [15, 16].
It was originally believed that the DNA methylation pattern is established during development and is then maintained faithfully through life by the maintenance DNA methyltransferase [3]. The DNA methylation reaction was believed to be irreversible, thus the only way methyl residues could be lost was believed to be through replication in the absence of DNA methyltransferase (DNMT) by passive demethylation [14]. This mechanism is not applicable to postmitotic tissue such as neurons in the brain. However, I will propose here based on our own data and data of others that the DNA methylation pattern is dynamic and is an equilibrium of methylation and demethylation reactions [17].
We have proposed that DNA methylation is a reversible signal like any other biological signal and could therefore potentially change in response to environmental and physiological signals [18]. A hallmark of DNA methylation patterns is the correlation between chromatin and the DNA methylation pattern. Active chromatin is usually associated with unmethylated DNA while inactive chromatin is associated with methylated DNA [19]. This linkage between DNA methylation and chromatin structure has important implications for our understanding of the function of DNA methylation as well as the processes responsible for generating, maintaining, and altering DNA methylation patterns under physiological and pathological conditions. It was originally believed that DNA methylation precedes and is dominant over chromatin structure changes. Methylation was believed to be generated independently of chromatin structure. Methylated DNA attracts methylated DNA binding proteins, which recruit repressor complexes containing histone deacetylases, which results in inactive chromatin [20, 21]. This model, positioning DNA methylation as driving chromatin inactivation is widespread and has profoundly influenced our understanding of how altered DNA methylation is involved in cancer. Nevertheless, there is currently data suggesting that the state of chromatin structure could also determine DNA methylation and that chromatin can affect DNA methylation in both directions triggering either de novo DNA methylation or demethylation as will be discussed extensively below. This data forces us to revisit the classic model of a DNA methylation pattern which is predetermined during development and is then maintained through life and adopt a more dynamic view of the DNA methylation pattern. This has obviously implications for our understanding of how DNA methylation patterns are altered in cancer and on how to approach the DNA methylation pattern therapeutically. This review will focus on the implication of this new understanding of the dynamic epigenome on our therapeutic approach to cancer.
Since the epigenome is responsible for coordinating gene expression programs and DNA methylation is so tightly linked to chromatin structure and since cancer progression involves concurrent change in expression of numerous genes, it is not surprising that DNA methylation patterns are altered in cancers and that changes in the DNA methylation enzymes are noted as well [22]. Hypermethylation of tumor suppressor genes received considerable attention and there are a number of clinical trials with inhibitors of DNMT aiming at demethylating these genes resulting in their activation and arrest of cell growth [23]. Nevertheless, a hallmark of cancer cells is also global hypomethylation [24], which has received very limited attention. However, recent studies point to a role for hypomethylation in activating genes required for metastasis [25].
This review will discuss the notion that demethylase(s), which catalyze replication-independent demethylation, are responsible for demethylation of pro-metastatic genes and that they can be targeted in anti-metastasis therapy [25]. An additional implication of the emerging role of hypomethylation is that although DNA methylation inhibitors might be excellent agents to inhibit growth through demethylation of tumor suppressor genes, they might nevertheless also induce pro-metastatic genes.
An additional issue, which is emerging from recent data, is the potential role of DNMT in transformation [17]. It was proposed a decade ago that DNMT are candidate anticancer targets [26]. It is widely accepted that DNMT play a causal role in cancer through their DNA methylation activity, which brings about methylation of tumor suppressor genes. Therefore, the idea that dominates is that inhibitors of the catalytic activity of DNMT DNA methylation should be used in anticancer therapy. However, recent data points to DNA methylation independent activities of DNMT, which are important for transformation [17]. This obviously has implications on designing inhibitors of DNMT1 as anticancer agents.
Another interesting question is which DNMT should be targeted. It was originally believed that DNMT1 is responsible for maintaining the entire DNA methylation pattern because of its ability to faithfully copy a methylation pattern from the mother strand to the daughter strand. It is therefore still widely believed that inhibition of DNMT1 should be sufficient to demethylate and activate all methylated genes. However, recent data suggests that other DNMTs are involved in maintaining the DNA methylation pattern [27]. The implications of this new data on DNA methylation directed therapeutics will be discussed.
It is clear that recent changes in our understanding of the interaction between chromatin and DNA methylation and a more dynamic concept of the DNA methylation pattern and the merging role of hypomethylation in metastasis obliges us to reconsider some of our strategies for DNA methylation therapeutics [28].
MECHANISMS RESPONSIBLE FOR GENERATION AND MAINTENANCE OF THE DNA
METHYLATION PATTERN
DNA methyltransferases. DNA methylation is laid down by enzymes, which catalyze the transfer of a methyl group from the methyl donor S-adenosyl methionine (SAM) onto the 5´ position on the cytosine ring, called DNA methyltransferases (DNMT) [29, 30]. In vertebrates, the majority of DNA methylation occurs when the cytosine is found at the dinucleotide sequence CG [31]. Not all CGs are methylated and they are distributed in a cell specific pattern [3]. This raises three basic questions. First, how is this pattern generated during development and cellular differentiation? Second, is the generated pattern static or dynamic through life? Third, how is this pattern maintained? Answering these questions is required before we could understand the mechanisms driving the DNA methylation patterns observed during differentiation.
De novo and maintenance DNMTs. The dominant theory in the field is based on differentiation between maintenance and de novo methylation. It has been proposed that there are two kinds of DNA methyltransferases. First, a maintenance DNMT, which can copy a methylation pattern from a methylated parental strand template to an unmethylated daughter strand [14] (Fig. 1). This DNMT could do so since it discriminates between a hemimethylated site, which is generated during replication of a methylated CG, and a site that is nonmethylated on both strands of DNA. Second, de novo DNMTs do not discriminate between unmethylated and hemimethylated sites and could therefore introduce new methylation sites. Originally, this model limited de novo DNMTs temporally to early development, the period when extensive de novo methylation takes place and when DNA methylation patterns are laid down [14]. After development is completed, the pattern of methylation is faithfully conserved through life since the maintenance DNMT faithfully replicates the DNA methylation pattern during cell division in replicating cells and since the DNA methylation is an irreversible reaction, no loss of methylation could occur in nondividing postmitotic cells such as CNS neurons.
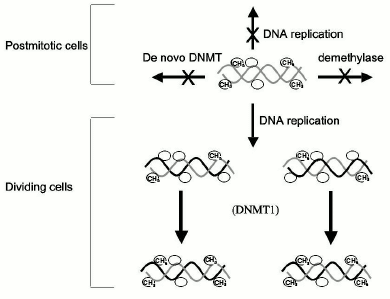
This model therefore establishes a qualitative distinction between DNA methylation during development, which is dynamic, and DNA methylation later in life, which is static and permanent (Fig. 2). Several lines of evidence support this model of maintenance of methylation post-development. DNMT1, the candidate maintenance DNMT, has shown preference to hemimethylated DNA in vitro [32], and transfection experiments revealed that ectopically methylated DNA maintained its pattern of methylation when introduced into somatic cells, supporting the idea that maintenance DNMT is the main DNA methylation reaction taking place in somatic cells [33].Fig. 1. DNA methylation reactions. DNA methyltransferases (DNMT) catalyze the transfer of methyl groups onto DNA. De novo methyltransferases introduce methyl groups (CH3) onto CG sites (indicated by a circle), which were not previously methylated on the parental template strands of DNA. All DNMTs were shown to possess de novo methylation activity, but DNMT1 is very inefficient in de novo methylation. Demethylases remove methyl groups to create unmethylated CG sites. Once a DNA methylation pattern is carved by de novo methyltransferases and demethylases it is maintained during DNA replication by maintenance DNA methyltransferase. The enzyme copies the DNA methylation pattern from the template strand to the unmethylated daughter nascent strand. It was believed that DNMT1 preferentially methylates hemimethylated strands generated during replication, thus copying the DNA methylation pattern.
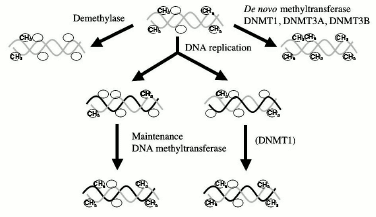
The preference that DNMT1 exhibits towards hemimethylated DNA has been recently reconfirmed [34]. DNMT1 preference to hemimethylated substrate has been localized to its N terminal region [35]. DNMT1 is localized to the DNA replication fork [36], is associated with PCNA the replication processivity protein that is present in the replication fork [37] and is processive in vitro as expected from its predicted function in the replication fork [34]. Methylation of replicating DNA occurs concurrently with DNA replication, which is consistent with the semi-conservative model of copying of the methylation pattern during cell division [38]. According to this model, the only manner by which a change in DNA methylation might occur in a somatic cell is by blocking the progressing DNMT1 during DNA replication by high affinity binding of a trans-acting protein to specific sequences. Recent studies have confirmed that high-affinity binding of proteins to CG containing sequences can cause passive loss of methylation at the time of DNA replication [39]. Fifty percent of methyl groups will be lost at each round of replication as long as the protein is bound to the site. Knock out of DNMT1 in mice results in extensive but not complete loss of methylation, which is consistent with its primary role in maintaining the DNA methylation pattern [40]. Similarly, knock down of DNMT1 in somatic cell lines was shown to cause demethylation [41-43] and activation of methylated genes and knock down of DNMT1 in human cancer cell lines causes demethylation and activation of tumor suppressor genes [44].Fig. 2. The classic model of maintenance of DNA methylation patterns in somatic cells. DNA methylation patterns are maintained in somatic cells post development. In nondividing cells such as neurons, the DNA methylation pattern was believed to be fixed since it was thought that demethylase and de novo methyltransferases did not exist in somatic cells. In dividing cells, the DNA methylation pattern was believed to be maintained by a maintenance DNA methyltransferase, which copied faithfully the DNA methylation pattern from the paternal strand in the absence of de novo methyltransferase and demethylase.
De novo methyltransferases have also been discovered. DNMT3A and -3B were cloned and did not show preference to hemimethylated DNA substrate [45]. Knock out of these genes in mice results in loss of methylation of specific sequences during early development [27, 46, 47]. Interestingly, mutations of human DNMT3B are found in ICF syndrome, a developmental defect characterized by hypomethylation of pericentromeric repeats [27].
It was anticipated that if DNA methylation patterns are just maintained in somatic cells that these de novo DNMTs would not be required in cells post development and that their inhibition in somatic cells would have no consequence. However, recent data suggests that the idea that DNA methylation is determined exclusively by the methylation pattern of the template must be revisited. In addition, the clear differentiation between de novo and maintenance DNMT is not fully supported by recent data. Recent experiments suggests that the putative de novo DNMT, DNMT3B and the proposed “maintenance” DNMT1, interact in maintaining the DNA methylation pattern [48], that they are found together in multiprotein complexes [49], and that knock down of both activities is required to cause global loss of DNA methylation [50-53].
There is some controversy regarding the extent of demethylation that is caused by knock down of DNMT1 in human cancer cell lines. Initial reports suggested that antisense knock down of DNMT1 resulted in demethylation of tumor suppressor genes such as P16 [54] while other reports showed that a complete knock out of DNMT1 in a colorectal cancer cell line did not cause either global demethylation or demethylation of P16 [51], whereas combined knock out of DNMT1 and DNMT3B resulted in demethylation and activation of P16 [52]. These results were then challenged by a later report, which showed that either siRNA or antisense RNA knock down resulted in demethylation of P16 [44]. However, these results were not reproduced in a more recent publication [53]. Perhaps the best way to resolve the contradiction between these studies is that the differences in the extent of demethylation caused by knock out of DNMT1 resulted from different sensitivities of detection of P16 demethylation by MS-PCR. Nevertheless, it is clear that total inhibition of DNMT1 does not result in erasure of the entire DNA methylation pattern, suggesting that “maintenance” DNA methylation must be catalyzed by additional enzymes. It is also quite clear that the presence of a so-called de novo methyltransferase is required for maintaining the DNA methylation pattern. These DNMTs, DNMT3A and DNMT3B, are maintaining DNA methylation patterns without being able to discriminate a hemimethylated site from an unmethylated site. These data strongly support the conclusion that the DNA methylation of nascent replicating strand in somatic cells is not determined by the methylation pattern of the template exclusively as described by the “semi-conservative” model of inheritance of DNA methylation patterns. Other mechanisms must be involved in defining the inheritance of the methylation pattern even in somatic cells.
Another important concept which is now emerging from the analyses of DNA methylation patterns of specific DNMT knock outs and knock downs is that the different DNMTs play different relative roles in the methylation of different sequences. In addition, different splice variants of DNMT were discovered, these might add specificity to the DNMT repertoire [55, 56]. For example, although a DNMT1-/- human colorectal cancer line maintains P16 gene methylation, other repetitive sequences such as pericentromeric sequences and ribosomal RNA genes become unmethylated [57]. It is clear however that there is redundancy amongst the DNMTs since knock out of both DNMT1 and DNMT3B is required to abolish P16 methylation, suggesting that the presence of either DNMT is sufficient to maintain the DNA methylation pattern.
Similar conclusions could be derived from mouse DNMT knock out experiments. Comparisons of DNA methylation patterns of DNMT1, DNMT3A, and DNMT3B deficient embryonal cells revealed that although there are CpG islands targets which are shared by Dnmt1 and Dnmt3a/3b, each Dnmt has target preferences depending on the genomic context of the CpG islands as illustrated by the differences in the extent of methylation loss in the specific DNMT knock outs [58]. The knock out data also supports the conclusion that all three DNMTs participate in maintenance of the DNA methylation pattern since deficiency of DNMT1, the putative maintenance DNMT, results only in loss of 16-48% of methylation of CG sites [58]. This recent data has important implications on the future use of DNMT inhibitors in therapy and the possibility of preferred inhibition of certain classes of genes using DNMT specific inhibitors such as DNMT1 antisense oligonucleotides [59-61].
Our understanding of the specificity of the different DNMTs and their splice variants in cancer is thus far very limited. A critical challenge is to develop DNMT isoform specific inhibitors, which would demethylate tumor suppressor genes without demethylating pro-metastatic genes. The most common inhibitor currently in research and therapy is 5-aza-cytidine (5-aza-CdR). The trinucleotide metabolite of this nucleoside analog is incorporated into DNA and covalently traps DNMTs during the progression of replication fork resulting in DNA synthesis in the absence of DNMT [62]. However, since this drug does not discriminate between the different DNMTs, most of our information on the consequences of DNMT inhibition does not take into account the differential specificities of DNMTs. In addition, the concept that replication of DNA methylation patterns is copied by a semi-conservative maintenance DNMT presumes that the entire methylation pattern is copied during the progression of the replication fork by the same DNMT. The catalytic activity of DNMT, which is common to all DNMTs, is targeted by most current screens rather than the unique functions of particular DNMTs. This approach needs to be modified if we desire to limit the adverse effects of DNA demethylation. In summary, recent data suggests that DNA methylation patterns are maintained by a combination of DNMT activities which target and recognize different sequence context rather than by copying of the methylation pattern of the template as predicted by the semi-conservative model of methylation (see Fig. 3 for a model).
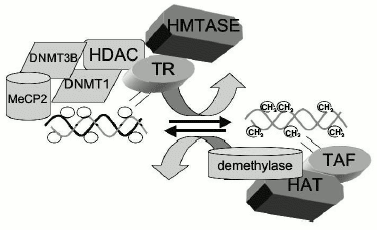
DNA demethylases. Our understanding of the mechanisms responsible for maintenance of DNA methylation patterns in somatic tissues had been based on the supposition that DNA methyltransferases(s) are exclusively responsible for DNA methylation. However, most biological processes such as phosphorylation and acetylation are reversible. Enzymes exist which catalyze biological modification and demodifications in both directions. One of the longstanding controversies in DNA methylation is the question of whether DNA methylation is reversible in somatic tissues and whether enzymes exists which could convert a methylated cytosine to an unmethylated cytosine [18]. Perhaps the strongest dogma in the field has been the idea that DNA methylation is an irreversible reaction. This idea of irreversibility of DNA methylation and the concept of semi-conservative maintenance DNMT has entrenched the notion that DNA methylation is fixed post development and that it plays no significant role in adaptive responses in developed tissue (see Fig. 2 for model). A methylation pattern that is exclusively dictated by the methylation pattern of the template does not allow for change. However, the correlation between chromatin and DNA methylation and the inarguable fact that chromatin is dynamic and responsive to different signals is somewhat inconsistent with a fixed post-development methylation pattern. If DNA methylation patterns act as biological signals in post development cells, these patterns must be reversible [18]. Though it is possible to reverse DNA methylation in replicating cells by passive demethylation, by blocking DNMT during DNA synthesis, demethylation in post mitotic tissues could potentially only be achieved by an active process which converts methylated cytosine to cytosine. Similarly, de novo methylation must also be possible in post-development tissues if DNA methylation is dynamic through life, and it could not be restricted to development as previously proposed.Fig. 3. The DNA methylation pattern is a steady-state balance of DNA methylation and demethylation, a model. In contrast to the classic model, it is proposed here that DNA methylation pattern is in a dynamic steady state of methylation and demethylation catalyzed by DNA methyltransferases and demethylases. The state of DNA methylation is determined by a combination of factors. Interaction of trans-acting repressors (TR) recruits histone modification enzymes such as histone deacetylases (HDAC) and histone methyltransferases (HMTASE) to specific genes. HDACs and HMTASE in turn recruit DNA methyltransferases (DNMT) and methylated DNA binding proteins such as MeCP2. These will tilt the balance of the reaction toward DNA methylation. The occupancy of a gene with some or all of these factors determines the state of methylation of the gene. Trans-acting activating factors (TAF) on the other hand recruit histone acetyl transferases (HAT) to specific genes. The presence of HATs results in acetylation of histones, opening up of chromatin configuration, and increased accessibility to demethylases tilting the steady state balance of the DNA methylation equilibrium toward demethylation.
There is now convincing evidence that active demethylation which is independent of DNA replication occurs during development. The paternal genome is almost completely demethylated hours after fertilization well before replication of DNA is initiated [63]. Ectopic DNA introduced into mouse zygotes underwent active demethylation [64]. Other studies have previously shown that the process of differentiation of erythroleukemia cells [65] and Epstein-Barr virus (EBV) producer cell lines was associated with genome wide replication-independent demethylation [66]. Specific genes were shown to undergo active demethylation upon differentiation. Examples include the vitellogenin genes in estrogen responsive chicken tissues [67], Ig gene locus during B cell maturation [68, 69], and muscle specific alpha-actin gene, which are actively demethylated upon differentiation [70]. There is however new evidence suggesting that active demethylation takes place in somatic cells post differentiation [25]. Perhaps the most significant illustration that demethylation is an adaptive response in postmitotic tissues is our recent demonstration that the exon 17 promoter of the glucocorticoid receptor in the hippocampus undergoes active demethylation as a response to maternal care six days after birth. Moreover, demethylation of this promoter could be induced in the adult brain by the HDAC inhibitor TSA [71]. Similarly, TSA [72] and the mood stabilizing drug valproate [73] can induce active replication-independent demethylation of ectopically methylated nonreplicating DNA transiently transfected into somatic cells such as HEK 293 cells, thus demonstrating that the DNA methylation is reversible. These studies demonstrated that the steady state of methylation was not defined exclusively by the methylation pattern of the template DNA and that DNA methylation states could be modulated outside of the elongating replication fork [72, 74].
The identity of demethylase(s) and the nature of their biochemical activity is a matter of controversy. Three different activities, which could potentially bring about demethylation of DNA in the absence of DNA replication, were previously proposed. The first two did not stray from the dogma that it was impossible to revert a methyl cytosine to a cytosine and proposed a repair based mechanism which is a variation on the passive-demethylation theme; the methylated bases are first removed and then replaced with unmethylated cytosines in the absence of DNMT. Since repair processes could take place in the absence of cell division, such mechanisms could explain loss of methylation in nondividing cells such as neurons.
Two repair processes were proposed. One mechanism implicates a glycosylase activity, which cleaves the bond between the 5-methylcytosine base and the deoxyribose moiety in DNA. The abasic site is then repaired by resident repair activity in the absence of DNA methyltransferase resulting in replacement of a 5-methylcytosine with an unmethylated cytosine [65, 75-77]. A second mechanism proposed that the methylated nucleotide was removed by nucleotide excision and was then replaced by an unmethylated cytosine [78, 79]. There are data to support the model that specific glycosylase(s) participates in part at least in demethylation. Two mismatch-repair glycosylases were shown to possess methyl-CG-DNA-glycosylase (5-MCDG) activity that results in demethylation in vitro, the cloned G/T mismatch repair enzyme [80], and the methylated binding protein MBD4 [81]. Ectopic expression of the 5-MCDG glycosylase in human embryonal kidney cells results in the specific demethylation of a stably integrated ecodysone-retinoic acid responsive enhancer-promoter linked to a beta-galactosidase reporter gene [82], suggesting that this glycosylase could cause demethylation in living cells. It was also shown that this glycosylase participates in global demethylation during differentiation of C2C12 cells since transfection of cells with an antisense oligonucleotide to 5-methylcytosine-DNA-glycosylase (G/T-mismatch-DNA-glycosylase) decreases both the activity of the enzyme and genome-wide demethylation [82]. Genetic evidence from plants suggests that specific glycosylases such as DEMETER are involved in locus-specific demethylation [83, 84].
One interesting property of the 5-MCDG-glycosylase is that it demethylates specifically hemimethylated mCGs. The mCG/CG hemimethylated site resembles the TG/CG mismatch. Both T and mC are pyrimidines, which are methylated at their 5´ position. The critical problem is to define the principal activity of this enzyme in living cells. One possibility is that the bona fide substrate of the enzyme is a G/T mismatch and its in vivo function is mismatch repair. In this case, the mCG/CG repair activity is an artifact that is a consequence of high expression in transfection experiments or the high concentrations present in vitro. Alternatively, the main function of this enzyme is demethylation of hemimethylated DNA. It is also possible that the enzyme performs both tasks in the cell. In any case, global demethylation and demethylation during differentiation and tumorigenesis involve demethylation of both strands of DNA, which must be therefore catalyzed by a different enzyme. A possible role for the 5-MCDG-glycosylase might be correction of aberrant methylated sites introduced during replication and added to one strand of DNA. Methyl groups found only on one strand of the DNA might signal that they are not authentically inherited and should be removed.
One main problem with repair mediated passive demethylation mechanisms is explaining global hypomethylation, especially the global hypomethylation that takes place at the very early stages of embryogenesis as discussed above [63]. If demethylation occurs by a repair mechanism, global hypomethylation would involve global DNA damage. This might significantly harm the integrity of the genome and it stands to reason that such a mechanism would not be utilized early in development.
We have extracted from human lung cancer cells an activity that demethylates DNA by removing the methyl residue, which is released as a volatile product [18]. We also cloned a cDNA that we showed to possess demethylase activity which was concurrently cloned and characterized as a methylated DNA binding protein MBD2 by Hendrich et al. [85]. It was proposed by Hendrich et al. that MBD2 functions as a typical methylated DNA binding protein suppressing methylated promoters by recruitment of the chromatin remodeling complexes NuRd containing HDACs to methylated DNA [86]. The assignment of a transcriptional suppression function to a protein which we characterized as a demethylase triggered a controversy which has not yet been resolved.
A number of publications have shown that MBD2 is part of the methylated gene-repression MeCP1 complex [87-90]. Our laboratory has since provided additional evidence that MBD2 is a demethylase; exogenous expression of MBD2 increased replication-independent active demethylation of ectopically methylated DNA in HEK cells [72]; antisense inhibition of MBD2 reduced replication-independent active demethylation induced by valproate [73]; and recombinant MBD2 expressed in insect cells was purified and shown to possess demethylase activity in vitro [91]. Perhaps one resolution of this discrepancy is that MBD2 has two functions in the cell a demethylase and a suppressor of certain methylated genes and that it selects either of these functions by the context of proteins interacting with each promoter. This hypothesis has been supported by our observation showing that MBD2 activity is promoter dependent [92]. It is clear nevertheless that there must be demethylases in addition to MBD2 since active demethylation of the parental genome, which normally occurs immediately after fertilization takes place in an mbd2-/- mouse [63]. The search for additional demethylases should become a priority in coming years. Without a full understanding of the repertoire of demethylases, we would be unable to grasp the basic mechanisms defining the DNA methylation pattern and what causes their alteration in cancer.
Bilateral relation between DNA methylation and chromatin. Since as discussed above the DNA methylation pattern cannot be explained just by semi-conservative inheritance by a maintenance DNA methyltransferase, there must be additional factors. We have proposed that the DNA methylation pattern at any point in life could be considered as a steady state, balance of DNA methylation and demethylation. The direction of this balance is directed by the chromatin structure. That is, DNA methylation is enhanced by factors, which are responsible for an inactive chromatin structure while demethylation is triggered by factors, which cause activation of chromatin [25] (Fig. 3).
The tight relation between chromatin and DNA methylation was first understood in the context of the mechanisms through which DNA methylation suppresses gene expression. DNA methylation was considered the primary event in silencing of gene expression through chromatin inactivation and a unidirectional relation between DNA methylation and repressed chromatin was proposed that DNA methylation determined chromatin structure [93]. A molecular causal link between DNA methylation and inactive chromatin structure was described [93]. Methylated DNA attracts methylated DNA binding proteins such as MeCP2, which in turn recruit histone modification complexes containing histone deacetylases and other chromatin remodeling activities [93].
However, it is becoming clear from both genetic and biochemical evidence that the relation between chromatin and DNA methylation is bidirectional and that chromatin plays a vital role in defining DNA methylation patterns. There are genetic data supporting the hypothesis that DNA methylation is dependent on chromatin structure from Neurospora to humans. In the plant Arabidopsis, a gene required for maintenance of DNA methylation DDM1 has similarity to the SWI/SNF family of adenosine triphosphate-dependent chromatin remodeling proteins [94, 95]. A mouse gene that is required for genomic DNA methylation, LSH, is a member of the SNF2 ATPase chromatin modeling protein family [96], and in humans deficiency of ATRX encoding a SW1/SNF chromatin remodeling protein results in distinct hypo- and hypermethylation of specific sites while there is no change in global methylation levels [97]. A histone methyltransferase is required for DNA methylation in Neurospora crassa [12] and Kryptonite, a histone methyltransferase gene specific to H3 Lys9, is required for CpNpG methylation by the Arabidopsis DNA methyltransferase chromomethylase 3 (CMT3) [98]. Although these genetic data indicate that chromatin modification proteins are required for generating a DNA methylation pattern, it is clear however that the relation between DNA methylation machinery and chromatin status is bilateral. DNA methylation enzymes are required as well for forming chromatin structure. Knock out of DNMT1 in a human colorectal cancer cell line results in a global change in chromatin structure; an increase in K9 acetylation and a decrease in K9 methylation [57].
While this genetic evidence derived from different species indicates that proper chromatin structure is critical for establishing DNA methylation patterns during development, it does not necessarily imply that chromatin is involved in DNA methylation changes post development. Evidence that changes in chromatin result in bidirectional alterations in DNA methylation states in somatic cells was provided by pharmacological, biochemical, and cellular studies. A good paradigm for how repressor complexes might cause de novo methylation in somatic cells is the PML-RAR fusion protein which recruits histone deacetylases and DNMTs to its target binding sequences resulting in de novo DNA methylation of target genes [99]. Although it is not yet known whether histone deacetylation indeed precedes DNA methylation, nevertheless this example illustrates that a cis-acting repressor, which targets genes for chromatin inactivation also causes de novo methylation and that the DNA methylation pattern is actively defined in somatic cells by cis-acting factors. Moreover, this study also illustrates the dynamic nature of DNA methylation in somatic cells since treatment with retinoic acid, which reverses the fusion protein from a repressor to an activator results in chromatin activation and demethylation [99]. Thus, interaction of trans-acting factors can bidirectionally determine the state of methylation in somatic cells (see model in Fig. 3). Although it is not known whether demethylation in this example was active or not, this case illustrated how the DNA methylation pattern might be dynamically determined by the repertoire of trans-acting repressor and physiological signaling molecules such as retinoids, rather than exclusively defined by the inherited methylation pattern.
Thus, a simple mechanism through which inactivation of chromatin structure could possibly bring about de novo methylation is that the different chromatin modifying enzymes recruit DNMTs to genes. There is experimental support for the idea that DNMTs form protein-protein interactions with histone modifying enzymes such as histone methyltransferases and histone deacetylases [100-102]. These enzymes are recruited to specific genes by trans-acting repressors recognizing cis-acting sequences juxtaposed to the genes. The repertoire of trans-acting repressor interacting with a gene could be altered in response to different physiological and pathological signals causing changes in the methylation pattern. On the other hand, the continuous presence of such factors on a gene ensures that it is maintained in its methylated state. Whereas this model can convincingly explain how de novo methylation might occur in somatic cells in response to a chromatin inactivation event it does not instruct us as to whether DNA methylation pattern could be reversed if chromatin is activated. However, if DNA methylation is as dynamic as chromatin in somatic cells, demethylation must take place in somatic cells as well. Since methyltransferases were known to be present in somatic cells, the idea that chromatin inactivation might bring about recruitment of DNMTs to a gene and cause de novo methylation in somatic cells did not encounter significant resistance. In contrast, the idea that demethylation of DNA could be triggered in response to chromatin activation was more problematic as discussed above.
The first indication that active chromatin structure could trigger active demethylation of DNA in somatic cells came from studies showing that the histone deacetylase inhibitor sodium butyrate can trigger replication independent active DNA demethylation in the EBV producer line P3HR-1 [66]. We have more recently shown in a series of studies that the state of chromatin modification associated with an ectopically methylated gene determines whether it maintains its methylation pattern or whether it is demethylated. Methylated genes, which are controlled by highly active promoters, are demethylated when introduced into HEK-293 cells whereas weak promoters remain methylated [72]. Acetylation of histones induced by the HDAC inhibitor TSA would cause demethylation of ectopically promoterless methylated genes suggesting that the critical event directing demethylation is histone acetylation [72]. This hypothesis was confirmed by showing that proteins, which inhibit histone acetylation by blocking access of HATs to histone tails, also inhibit demethylation induced by TSA [74].
It is unclear yet what is the mechanism through which histone acetylation brings about demethylation. A possible model that we proposed is that the tight interaction of nonacetylated histone tails blocked the access of demethylases to a gene [74]. These experiments illustrate that demethylase is constitutively present in cells and that maintaining a gene in its methylated state requires protection from demethylase by maintaining an inaccessible chromatin structure. Once this block is removed, a gene is actively demethylated. The fact that activation of chromatin by TSA could cause demethylation stands in contrast to the commonly accepted assumption that TSA acts only on chromatin and not on the DNA methylation pattern and that histone deacetylase inhibitors and DNA methylation inhibitors target different processes and that therefore TSA does not contribute to demethylation of genes [103]. This is important since both classes of drugs are currently in clinical trials and current interpretation of the data is based on the incorrect assumption that histone deacetylase inhibitors do not affect DNA methylation. For example, as discussed above the mood stabilizer drug valproate, which was recently shown to inhibit histone deacetylases, also triggers active replication independent demethylation [73]. Thus, the long-term effects of DNA methylation changes must be taken into account when considering the possible outcome of valproate treatment.
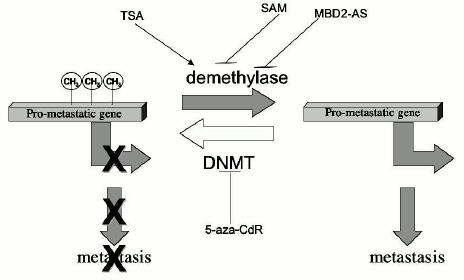
We have very recently shown that TSA triggers DNA demethylation in the hippocampus in adult rats demonstrating that genes in nondividing tissues are protected from demethylation by inactive chromatin structure and that any point in life this steady state could be potentially altered by altering chromatin structure [71]. The steady state methylation pattern is maintained by the steady state chromatin structure. Very recent comparative analyses of the effects of 5-aza-CdR, a DNA methylation inhibitor, and TSA, a histone deacetylase inhibitor, on the transcriptome of a colorectal cancer cell line HCT116 revealed a strong correlation between these effects suggesting that both drugs act by a similar mechanism, which is most probably DNA demethylation [104]. Furthermore, the authors suggested that since the effects of 5-aza-CdR were similar at one and five days exposure, the changes in gene expression were caused by active demethylation [104]. If the DNA methylation pattern was exclusively maintained by DNMT as was widely believed, then 5-aza-CdR should have caused only passive demethylation, a consequence of synthesis of new DNA in the absence of DNMT. The fact that 5-aza-CdR causes active demethylation rather than passive demethylation as originally thought is consistent with the working hypothesis presented in this review that the DNA methylation pattern in mature tissues is a steady state balance of constitutive methylation and demethylation reactions. According to this model, inhibition of DNMT by 5-aza-CdR leaves the demethylation reaction unopposed by DNA methylation resulting in a new unmethylated state (Fig. 4).
This view that the DNA methylation pattern is a steady state of methylation and demethylation implies that the DNA methylation pattern is dynamic and could be potentially responsive to any signaling pathway which might alter the chromatin structure in both dividing and nondividing tissues. Our model also proposes that the DNA methylation pattern must be understood in the context of all epigenomic factors and therefore inheritance of DNA methylation patterns involves the inheritance of the entire chromatin structure (Fig. 3). The inheritance of DNA methylation patterns and their maintenance in nondividing tissues cannot be explained exclusively by the maintenance DNMT1; it involves a balance of multiple DNMTs chromatin remodeling and modification complexes and demethylases (Fig. 3). The inheritance of a DNA methylation pattern is not a passive copying of a template but involves active methylation and demethylation decisions, which are determined by the repertoire of factors defining the chromatin structure in a given cell at a given time point. Demethylation or de novo methylation would occur when this repertoire is changed. Interaction of a factor that recruits HATs to a gene would cause acetylation and increase the accessibility of the gene to the constitutive demethylase. An elegant demonstration of how an interaction of a transcription factor with an enhancer sequence precipitates active demethylation is provided by the study of demethylation of the immunoglobulin kappa-light-chain gene during B cell differentiation [105]. The demethylation and activation of this gene requires both the presence of the intronic enhancer element and the binding of the NF-kappaB transcription factor [69]. On the other hand, an example of how de novo methylation could be brought about by interaction of a repressor, which recruits HDACs, is provided by the leukemia-promoting PML-RAR fusion protein that induces hypermethylation of genes containing its target sequences which was discussed above [99]. Our model offers a unifying hypothesis for maintenance and de novo methylation and demethylation during development and at any time point later in life.Fig. 4. Regulation of pro-metastatic gene by DNA hypomethylation, effects of epigenomic drugs. A balance of DNA methylation and demethylation determines the state of methylation of pro-metastatic genes. Pro-metastatic genes such as the uPA gene are silenced and methylated (CH3) in normal cells and non-metastatic breast cancer cells. The balance of the DNA methylation reactions in non-metastatic cancer cells points to extensive DNA methylation. Inhibition of DNMTs by 5-aza-CdR would inhibit the DNA methylation reaction resulting in unopposed demethylation and activation of pro-metastatic genes. Induction of histone acetylation by HDAC inhibitor TSA would increase accessibility to demethylases and increase demethylation. In metastatic cancer cells, the demethylation reaction is enhanced resulting in demethylation and activation of pro-metastatic genes. Inhibition of demethylase by either SAM or MBD2-antisense knock down oligonucleotides would enable unopposed DNA methylation to proceed, tilting the balance to methylation and silencing of pro-metastatic genes such as uPA.
The role of DNA methylation patterns in epigenomic regulation. Epigenetic regulation is possible in the absence of DNA methylation as illustrated by other species such as Saccharomyces cerevisiae, which possess complex and sophisticated epigenetic regulation in the absence of significant DNA methylation. Moreover, since, as proposed here, the DNA methylation pattern is determined by chromatin structure, what is the advantage that DNA methylation confers upon a cell over chromatin structure per se? What additional information does it convey that could not have been encoded by the “histone code” per se?
A partial answer to this question is that methylation of vertebrate genes not only reflects but can also precipitate an inactive chromatin structure. DNA methylation attracts methylated DNA proteins which in turn recruit histone modifying enzymes causing inactivation of chromatin [86, 93] and it has also been shown that DNMT1 knock down leads to inhibition of histone acetylation and increase in histone K9 methylation [57]. It is therefore clear that in some instances DNA methylation plays a primary role in setting up silencing of gene expression. It is critical to determine under which circumstances either DNA methylation or chromatin modification play a primary role? Even so the basic problem remains; why do vertebrates require this additional layer of epigenomic regulation if other organisms can maintain chromatin inactivation in the absence of DNA methylation? There is no definitive resolution as yet to this difficulty. A plausible answer is that since DNA methylation is a significantly more stable than protein modification it serves as a long-term memory of epigenomic states. It is proposed that whereas chromatin changes in euchromatic regions might be dynamic and transient, de novo methylation and demethylation would occur only in response to a persistent and significant change in the “histone code”. This change in DNA methylation once accomplished would then serve as a long-term memory of the initial event. DNA methylation would serve as a stable guardian of the silenced state. For example, a methylated gene which is also found in an inactive chromatin structure would be protected from loss of the silenced chromatin state since methylated binding proteins interacting with the methylated CGs recruit histone deacetylases and histone methyltransferases. Thus, in a case where a chromatin modification is aberrantly lost, DNA methylation would direct chromatin structure and restore the correct state of chromatin modification.
An illustration of this relationship between chromatin and DNA methylation could be found in the epigenetic modulation of stress response by maternal care, which was recently described [71]. In this model DNA methylation of the glucocorticoid receptor gene promoter in the hippocampus serves as a long-term memory of chromatin states triggered by maternal care early after birth. Although this DNA methylation pattern remains stable through life, it could be reversed in the adult by treatment with the HDAC inhibitor TSA [71]. This study illustrates on one hand the stability of DNA methylation marks and how they bear the memory of early changes in chromatin states induced by signaling pathways triggered by maternal licking and grooming and, on the other hand, it illustrates how a methylation mark is altered by persistent alteration of chromatin state with a pharmacological HDAC inhibitor.
ABERRATIONS OF DNA METHYLATION IN CANCER
Hypermethylation of specific genes in cancer. Both hyper- and hypomethylation and deregulation of DNMTs have been documented in cancer. As cancer progression and metastasis involve multiple changes in gene expression, it is not surprising that all tumors are distinguished by DNA methylation aberrations [22]. The main attention has been directed at hypermethylation of critical genes such as tumor suppressor genes, adhesion molecules, DNA repair, inhibitors of angiogenesis, and inhibitors of metastasis. DNA methylation of these genes serves a similar role to genetic alterations since it silences gene expression similar to deletions and mutations [106]. In difference, however, from genetic mutation or deletion, it is possible to reverse a methylation silencing event by using DNMT inhibitors [26]. Therefore, the understanding that critical cancer genes are silenced by methylation has led to a number of attempts to develop and test DNMT inhibitors. A number of clinical trials [23, 107, 108] are underway with either catalytic inhibitors of DNMT or an antisense oligonucleotide, which knocks down DNMT1 [43, 54]. Profiles of methylated CG islands containing genes correlate with specific classes and stages of cancer [15, 109, 110]. Different whole genome and high throughput approaches are being utilized to profile the methylation state of CG islands in different cancers. The CGs islands methylation profiles are now being tested as diagnostic marker for different cancers, staging of cancer, and as predictors of response to chemotherapy [15, 111, 112].
Since it is clear that cancer also involves activation of genes such as oncogenes as well as pro-metastatic genes, it is not surprising that hypomethylation is also a hallmark of cancer. Oncogenes and genes involved in metastasis such as S100P [113], S100A4 [114], uPA [115], and HEPARANASE [116] were shown to be hypomethylated in metastatic cancer in comparison with normal tissue and non-metastatic cancer. In addition to gene specific hypomethylation, global hypomethylation of repetitive sequences such as LINE1 [117] sequences is consistently observed. Hypomethylation of specific genes might be secondary to local chromatin changes perhaps induced by activation of a cellular signaling pathway leading to activation of transcription activators. Global hypomethylation on the other hand suggests a global defect in the DNA methylation machinery or a global change in chromatin structure.
Possible mechanisms responsible for coexistence of global hypomethylation and regional hypermethylation in cancer. The classic model of maintenance methylation, which is carried out by a single maintenance DNMT, cannot explain the coexistence of hypomethylation and hypermethylation in cancer. If DNMT1 was the only enzyme responsible for DNA methylation, then global hypomethylation should have resulted from a reduction in DNMT1 levels. However, the opposite is true since increased DNMT levels [118] and cell cycle deregulated activity of DNMT1 in tumors [119-121] are widely documented in many but not all reports [122]. However, if DNA methylation is determined and maintained by a more complex mechanism, which involves different combinations of DNMT and demethylase enzymes as well as histone modifying enzymes, then diversity in alterations of DNA methylation patterns in response to cancer could be anticipated.
Although there is lack of evidence as to the details of the mechanisms responsible for hypermethylation and demethylation in cancer, some of the data available point to the model presented here. It is proposed here that hypermethylation in cancer is a consequence of regional inactivation of the chromatin associated with certain tumor suppressor genes. Such inactivation might be triggered by interaction of a transcriptional repressor with specific genes, which in turn recruits HDAC and other histone modifying enzymes to the genes. The idea that DNA methylation follows rather than precedes chromatin inactivation is supported by data showing that in a human colorectal cancer cell line which is deficient of DNMT1 and DNMT3B chromatin inactivation of tumor suppressor genes occurs prior to DNA methylation [123]. An analysis of the state of methylation, chromatin and expression of E CADHERIN in different cancer lines is consistent with the hypothesis that chromatin modification occurs prior to DNA methylation since the gene is associated with deacetylated histones in all cancer cell lines where it is silenced but DNA hypermethylation only occurs in a subset of these lines [124]. Certain lines bear a silenced but unmethylated E CADHERIN. It is therefore proposed that the first event involved in silencing of a tumor suppressor is triggered by recruitment of HDAC to a promoter by a trans-acting repressor(s) [124]. The residence of a HDAC or a histone methyltransferase on a gene would result in recruitment of DNMTs to the gene since HDACs [101, 102, 125, 126] and histone methyltransferases [100] were shown to form protein-protein interactions with DNMTs as discussed above [101, 102, 125, 126]. In support of this hypothesis, it was shown that the leukemia-promoting PML-RAR fusion protein recruits HDAC and DNMT to target genes [99]. Similarly, it was shown that the repression of ESTROGEN RECEPTOR alpha repression in ER-breast cancer cells involved recruitment of a multiprotein complex containing the trans-acting repressor complex pRb2/p130-E2F4/5, HDACs, histone methyltransferases, and DNMT1 [126].
Global hypomethylation could be a consequence of either activation of demethylase or a global change in a chromatin modifying protein. A general activation of demethylase should not affect regionally hypermethylated genes since these genes are inaccessible to demethylase by the suppressed chromatin associated with these genes [25]. This suppressed chromatin is actively maintained by resident HDACs and other chromatin modifying enzymes recruited by specific repressors interacting with these genes. It has been shown that interaction of an inhibitor of histone acetylation InHAT with chromatin associated with a methylated gene could protect it from demethylation [74]. It is unclear yet, which demethylases are responsible for demethylation of critical genes in cancer. We have recently shown that MBD2 is abundantly expressed in the highly metastatic breast cancer cell line MDA-MB-231 but its expression is reduced in the low invasive breast cancer cell line MCF-7 [127]. Antisense inhibition of MBD2 results in hypermethylation and silencing of the pro-metastatic gene uPA. Similarly, we have shown that antisense knock down of MBD2 inhibits active demethylation induced by valproate in HEK 293 cells [73]. High MBD2 levels correlated with hypomethylation and cancer in ovarian cancers [128] and lung cancers [129]. However, this was not seen in colorectal cancer, where an inverse correlation was observed [130]. There is a raging controversy on whether MBD2 is a demethylase, a silencer of methylated genes, or bears both activities as discussed above. It is clear that future studies are required to identify the proteins and the multiprotein machineries required for demethylation in cancer. Another possibility, which must be considered, is that local targeted activation of chromatin rather than a global rise in demethylase activity might be involved in demethylation of specific genes in cancer, especially genes which are critical for metastasis.
Roles of hyper- and hypomethylation in cancer. Hypermethylation of CG islands is widely recognized as a mechanism for silencing gene expression in cancer cells [131]. DNA methylation silences genes involved in cell adhesion [132], mismatch repair [133], and glutathione transferases, which confer protection against cytotoxic agents [134] and inhibitors of invasion and angiogenesis [135]. All these changes in gene expression confer selective advantage, which is probably involved in promoting growth and evading normal growth control mechanisms. Silencing of tumor suppressor genes such as P16 could result in uncontrolled cell growth. The evidence for this role of methylation of tumor suppressor genes comes from experiments with the DNA methylation inhibitors 5-aza-CdR [136] and zebularine [137]. Nevertheless, it is still unclear whether the cell cycle arrest by DNA methylation inhibitors is caused by activation by demethylation of genes like P16 or by a methylation independent mechanism [59-61]. It was shown for example that growth arrest following DNMT1 antisense knock down preceded activation of P16 by demethylation [54]. Based on this data we proposed that the DNA methylation independent activities of DNMT1 rather than the catalytic activity might be targeted in anticancer therapy [17].
Global hypomethylation was previously proposed to be responsible for the chromosomal instability and defective chromosomal integrity of cancer cells [138]. Gene specific hypomethylation was originally proposed to be responsible for activation of oncogenes and hypomethylation of C-MYC and C-JUN protooncogenes observed in livers of mice exposed to hypomethylating diets [139-141]. Hypomethylation is now emerging as an important mechanism for promoting metastasis [25, 28]. We have previously proposed that hypermethylation and hypomethylation might be important for different stages of cancer. Hypermethylation might play a critical role in promoting cell growth while later in progression of a cancer to a metastatic state hypomethylation of pro-metastatic genes takes over as the primary epigenetic aberration [28, 127]. This model has obvious therapeutic implications, which will be discussed below.
THERAPEUTIC IMPLICATIONS OF DNA HYPOMETHYLATION AND
HYPERMETHYLATION IN CANCER
The use of DNA methylation inhibitors. The original drive for use of DNMT inhibitors in anticancer therapy [26] came from the discovery that oncogenic signaling pathways induced DNMT1 expression through AP-1 sites in DNMT1 regulatory regions [142, 143] and that knock down of DNMT1 by antisense inhibitors blocked Ras-mediated transformation [42, 43]. The involvement of DNMT1 in transformation mediated by the downstream effector of Ras, c-Fos was later confirmed [144]. Similarly, it was shown that oncogenic proteins such as SV40, T antigen, which suppress tumor suppressor activity, also induce DNMT1 and that knock down of DNMT1 blocked transformation induced by T antigen [145]. Thus, DNMT1 activity is controlled both by oncogenic and tumor suppressor pathways. Antisense inhibitors of DNMT1 were shown to block tumor growth in vivo and in vitro [43]. An attractively simple explanation for the anticancer activity of DNMT1 antisense inhibitors was that inhibition of DNMT1 resulted in activation of tumor suppressor genes by passive demethylation.
Similarly to the results obtained with mRNA knock-down agents, inhibitors of the catalytic activity of DNMT such as 5-aza-cytidine [146] and zebularine demethylated and activated tumor suppressor genes and inhibited tumorigenesis [137]. Different clinical trials are now being conducted with DNA methylation inhibitors. Two lines of data raise some doubt however regarding the suitability of catalytic DNA methylation inhibitors in anticancer therapy. First, DNMT1 suppresses gene expression by a mechanism that does not involve DNA methylation. Inhibition of DNMT1 in human lung cancer cells A549 induced expression of the tumor suppressor P21 by a methylation independent mechanism [60]. Antisense knock down of DNMT1 results in inhibition of firing of origins of replication [147], an intra-S phase arrest of DNA replication [61], and activation of promoters bearing the Sp1 response element by methylation independent mechanism [59].
These data lead to the conclusion that inhibiting the DNA methylation independent activities of DNMT1 might be more critical for suppression of tumor growth than inhibiting its catalytic DNA methylating activity. DNMT1 bears a large N-terminal regulatory domain, which forms protein-protein interactions with HDAC1 [102], HDAC2 [148], Rb [149], and replication fork protein PCNA [37]. It is not clear which of these interactions are critical for cellular transformation but it is imperative to clarify this issue for further progress with designing DNMT inhibitors as therapeutic anticancer agents. The second issue is the risk that catalytic inhibitors of DNMT such as 5-aza-CdR and zebularine which cause demethylation would also trigger the expression of pro-metastatic genes as discussed above. 5-aza-CdR induces the expression of uPA in non-metastatic breast cancer cells and transforms them into highly invasive metastatic cancer cells [25, 28, 115]. Similarly, it was shown that 5-aza-CdR induces the heparanase gene [116], S100P [113], and S1004 [150], all of which play a critical role in metastasis. Taking into account the issues raised here, it appears that it is desirable to target the regulatory protein-protein interactions of DNMT1 rather than the catalytic activity and thus avoid the adverse effects of DNA demethylation and the activation of genes leading to metastasis [17]. Accomplishment of this goal would require a better understanding of the structure function relation between DNMT1 and regulation of tumor suppressor gene expression.
Protection against hypomethylation; the use of methyl-rich diets. There is evidence from rodents that hypomethylating diets result in hypomethylation of DNA and increased incidence of liver cancer [151, 152]. S-Adenosyl-methionine (SAM) (methyl donor, cofactor of the DNA methylation reaction) or methionine (methyl donor) could protect from liver carcinogenesis [153, 154]. This protection is lost by co-administration of the DNA demethylating agent 5-aza-CdR suggesting that SAM acts through stimulation of DNA methylation [153]. Low folate intake, which is required for cellular methylation reactions and the generation of SAM, could cause global hypomethylation in humans. Alcohol consumption could further reduce SAM levels. A number of studies have shown an increased risk for colorectal adenocarcinoma with low folate intake, which is increased even further with alcohol [154-156]. An association between low folate intake and breast cancer was found in alcohol consuming women although the data are inconclusive in establishing a clear causal correlation between cervical cancer and low folate intake because of multiple confounding factors [157]. Similarly polymorphisms in the gene encoding methylenetetrahydrofolate reductase enzyme (MTHFR) involved in folate metabolism and as a consequence in SAM production were also shown to cause genomic hypomethylation [158-160] and in some studies were shown to correlate with increased incidence of cancer [161-163] but not in cervical cancer [164].
The epidemiological studies though not yet conclusive point to a potential role of low methyl content diets in cancer risk. This question has to be critically addressed and the possibility of dietary supplementation of folates as a protection against different cancers and specifically cancer metastasis should be seriously considered. The adverse effects of highly folate-enriched diets must be considered as well. If indeed hypomethylation plays a causal role in expression of pro-metastatic genes as previously suggested then it might be interesting to test whether high concentrations of methyl donors such as methionine or folates could be used pharmacologically to inhibit or slow down metastasis.
Demethylase inhibitors as anti-metastatic therapy. Although the focus of attention in DNA methylation therapy is on DNA methylation inhibitors, the discussion presented here suggests that we should shift our attention to a diametrically opposed process, inhibition of demethylation. The basic assumption is that the state of methylation of any given gene is not fixed but is a dynamic balance of methylation and demethylation. The hypomethylated state of metastatic genes such as uPA is maintained in metastatic cancer because demethylase activity persistently demethylates it at a higher rate than it is methylated by DNMT. If we inhibit the demethylase process, we will shift the reaction toward DNA methylation and silencing of the gene (Fig. 4). Although as discussed above the identity of the demethylase responsible for preserving metastatic genes in an unmethylated state is unclear, we have some data suggesting that MBD2 might play a causal role in cancer and a critical role in metastasis. MBD2 anti-sense mRNA suppressed tumorigenesis of human cancer cell lines in vitro and in vivo [165, 166] and knock out of the MBD2 gene suppressed intestinal tumorigenesis in mice [167].
We have developed sequence specific antisense inhibitors of MBD2 mRNA and we have shown that they inhibit tumorigenesis of human cancer cell lines in vivo [168]. We also showed that MBD2 antisense inhibitors bring about hypermethylation and inhibit the expression of the uPA gene in metastatic breast cancer cell lines [127]. MBD2 anti-sense treated breast cancer cells had reduced invisibility in vitro and inhibited metastasis in vivo [127]. These data point to MBD2 as a candidate regulator of methylation state of some pro-metastatic genes perhaps through its demethylase activity [25, 28]. A correlation was found between expression of MBD2/demethylase and expression of lung resistance protein LRP [129] and demethylation and expression of C-ERB2 and SURVIVIN in ovarian cancer [128] and MYC and hMSH2 in gastric cancerous specimens [169]. These clinical studies are consistent with MBD2 playing a role in demethylation of these genes in cancerous tissue.
Future studies should establish whether MBD2 plays a critical role in metastasis of other cancers and test the feasibility of using MBD2 antisense oligonucleotides in clinical trials. In addition, it will be crucial to develop small molecule inhibitors of MBD2 demethylase and test their potential as anti-metastatic agents. MBD2 might unfold to become an extremely important target for anti-metastatic therapeutics.
Another approach to inhibit demethylase activity and suppress pro-metastatic genes such as uPA, which has been recently demonstrated in our laboratory, is the use of the methyl donor SAM [127]. We have shown that SAM inhibits MBD2-demethylase activity in vitro and active replication-independent demethylation in cells [91]. SAM is a highly effective inhibitor of metastasis of breast cancer cell lines in vitro and in vivo [127]. Thus, SAM could be considered the first small molecule inhibitor of MBD2 demethylase. SAM is an extremely attractive agent since it is a natural metabolite and could be modulated as discussed above by diet. However, SAM is unlikely to be used as a pharmacological agent per se since it is highly unstable under physiological conditions. Future structure function analyses should identify the active moiety on SAM involved in inhibition of demethylase and establish active and stable analogs, which could serve as MBD2 demethylase inhibitors.
One important consideration in further utilization of inhibitors of the DNA methylation machinery in anticancer therapy is to define the relative adverse effects of either DNA methylation inhibition or DNA demethylation inhibition. Our current understanding is that DNA methylation inhibition is effective in blocking tumor growth by activation of tumor suppressor genes but it carries the risk of induction of metastasis. Inhibition of demethylation on the other hand, would inhibit metastasis by hypermethylation of pro-metastatic genes. It is not clear yet whether inhibition of demethylation would also result in hypermethylation and suppression of tumor suppressor genes and increased growth. This has to be carefully addressed by analyzing the effects of MBD2 antisense knock down and SAM on the state of methylation and expression of tumor suppressor genes. Nevertheless, since metastasis is most probably the most devastating aspect of cancer, it is imperative upon us to shift our attention to the potential consequences of DNA demethylating agents as inhibitors of metastasis. Even if one takes into account the possibility that demethylase inhibitors might also stimulate tumor growth, inhibition of metastasis is nevertheless a paramount goal.
The work described here from the laboratory of M. S. was supported by the National Cancer Institute of Canada and the Canadian Institutes of Health Research.
REFERENCES
1.Strahl, B. D., and Allis, C. D. (2000)
Nature, 403, 41-45.
2.Jenuwein, T., and Allis, C. D. (2001)
Science, 293, 1074-1180.
3.Razin, A., and Szyf, M. (1984) Biochim. Biophys.
Acta, 782, 331-342.
4.Finch, J. T., Lutter, L. C., Rhodes, D., Brown, R.
S., Rushton, B., Levitt, M., and Klug, A. (1977) Nature,
269, 29-36.
5.Rea, S., Eisenhaber, F., O'Carroll, D., Strahl, B.
D., Sun, Z. W., Schmid, M., Opravil, S., Mechtler, K., Ponting, C. P.,
Allis, C. D., and Jenuwein, T. (2000) Nature, 406,
593-559.
6.Jenuwein, T. (2001) Trends Cell Biol.,
11, 266-273.
7.Wade, P. A., Pruss, D., and Wolffe, A. P. (1997)
Trends Biochem. Sci., 22, 128-132.
8.Wade, P. A., and Wolffe, A. P. (1997) Curr.
Biol., 7, 82-48.
9.Henikoff, S., Furuyama, T., and Ahmad, K. (2004)
Trends Genet., 20, 320-326.
10.Roth, S. Y., Denu, J. M., and Allis, C. D. (2001)
Annu. Rev. Biochem., 70, 81-120.
11.Kuo, M. H., and Allis, C. D. (1998)
BioEssays, 20, 615-626.
12.Lachner, M., O'Carroll, D., Rea, S., Mechtler,
K., and Jenuwein, T. (2001) Nature, 410, 116-120.
13.Tsukiyama, T., and Wu, C. (1997) Curr. Opin.
Genet. Dev., 7, 182-191.
14.Razin, A., and Riggs, A. D. (1980)
Science, 210, 604-610.
15.Shi, H., Maier, S., Nimmrich, I., Yan, P. S.,
Caldwell, C. W., Olek, A., and Huang, T. H. (2003) J. Cell.
Biochem., 88, 138-143.
16.Beck, S., Olek, A., and Walter, J. (1999) Nat.
Biotechnol., 17, 1144.
17.Szyf, M. (2001) Trends Pharmacol. Sci.,
22, 350-354.
18.Ramchandani, S., Bhattacharya, S. K., Cervoni,
N., and Szyf, M. (1999) Proc. Natl. Acad. Sci. USA, 96,
6107-6112.
19.Razin, A., and Cedar, H. (1977) Proc. Natl.
Acad. Sci. USA, 74, 2725-2728.
20.Nan, X., Ng, H. H., Johnson, C. A., Laherty, C.
D., Turner, B. M., Eisenman, R. N., and Bird, A. (1998) Nature,
393, 386-389.
21.Jones, P. L., Veenstra, G. J., Wade, P. A.,
Vermaak, D., Kass, S. U., Landsberger, N., Strouboulis, J., and Wolffe,
A. P. (1998) Nat. Genet., 19, 187-191.
22.Baylin, S. B., Esteller, M., Rountree, M. R.,
Bachman, K. E., Schuebel, K., and Herman, J. G. (2001) Hum. Mol.
Genet., 10, 687-692.
23.Goffin, J., and Eisenhauer, E. (2002) Ann.
Oncol., 13, 1699-1716.
24.Ehrlich, M. (2002) Oncogene, 21,
5400-5413.
25.Szyf, M., Pakneshan, P., and Rabbani, S. A.
(2004) Cancer Lett., 211, 133-143.
26.Szyf, M. (1994) Trends Pharmacol. Sci.,
15, 233-238.
27.Okano, M., Bell, D. W., Haber, D. A., and Li, E.
(1999) Cell, 99, 247-257.
28.Szyf, M., Pakneshan, P., and Rabbani, S. A.
(2004) Biochem. Pharmacol., 68, 1187-1197.
29.Turnbull, J. F., and Adams, R. L. (1976)
Nucleic Acids Res., 3, 677-695.
30.Pradhan, S., and Esteve, P. O. (2003) Clin.
Immunol., 109, 6-16.
31.Gruenbaum, Y., Stein, R., Cedar, H., and Razin,
A. (1981) FEBS Lett., 124, 67-71.
32.Gruenbaum, Y., Cedar, H., and Razin, A. (1982)
Nature, 295, 620-622.
33.Stein, R., Gruenbaum, Y., Pollack, Y., Razin, A.,
and Cedar, H. (1982) Proc. Natl. Acad. Sci. USA, 79,
61-65.
34.Hermann, A., Goyal, R., and Jeltsch, A. (2004)
J. Biol. Chem., 279, 48350-48359.
35.Araujo, F. D., Croteau, S., Slack, A. D.,
Milutinovic, S., Bigey, P., Price, G. B., Zannis-Hadjopoulos, M., and
Szyf, M. (2000) J. Biol. Chem., 276, 6930-6936.
36.Leonhardt, H., Page, A. W., Weier, H. U., and
Bestor, T. H. (1992) Cell, 71, 865-873.
37.Chuang, L. S., Ian, H. I., Koh, T. W., Ng, H. H.,
Xu, G., and Li, B. F. (1997) Science, 277, 1996-2000.
38.Araujo, F. D., Knox, J. D., Szyf, M., Price, G.
B., and Zannis-Hadjopoulos, M. (1998) Mol. Cell. Biol.,
18, 3475-3482.
39.Hsieh, C. L. (1999) Mol. Cell. Biol.,
19, 46-56.
40.Li, E., Bestor, T. H., and Jaenisch, R. (1992)
Cell, 69, 915-926.
41.Szyf, M., Rouleau, J., Theberge, J., and Bozovic,
V. (1992) J. Biol. Chem., 267, 12831-12836.
42.MacLeod, A. R., and Szyf, M. (1995) J. Biol.
Chem., 270, 8037-8043.
43.Ramchandani, S., MacLeod, A. R., Pinard, M., von
Hofe, E., and Szyf, M. (1997) Proc. Natl. Acad. Sci. USA,
94, 684-689.
44.Robert, M. F., Morin, S., Beaulieu, N., Gauthier,
F., Chute, I. C., Barsalou, A., and MacLeod, A. R. (2003) Nat.
Genet., 33, 61-65.
45.Xie, S., Wang, Z., Okano, M., Nogami, M., Li, Y.,
He, W. W., Okumura, K., and Li, E. (1999) Gene, 236,
87-95.
46.Hansen, R. S., Wijmenga, C., Luo, P., Stanek, A.
M., Canfield, T. K., Weemaes, C. M., and Gartler, S. M. (1999) Proc.
Natl. Acad. Sci. USA, 96, 14412-14417.
47.Okano, M., Xie, S., and Li, E. (1998) Nat.
Genet., 19, 219-220.
48.Liang, G., Chan, M. F., Tomigahara, Y., Tsai, Y.
C., Gonzales, F. A., Li, E., Laird, P. W., and Jones, P. A. (2002)
Mol. Cell. Biol., 22, 480-491.
49.Kim, G. D., Ni, J., Kelesoglu, N., Roberts, R.
J., and Pradhan, S. (2002) EMBO J., 21, 4183-4195.
50.Leu, Y. W., Rahmatpanah, F., Shi, H., Wei, S. H.,
Liu, J. C., Yan, P. S., and Huang, T. H. (2003) Cancer Res.,
63, 6110-6115.
51.Rhee, I., Jair, K. W., Yen, R. W., Lengauer, C.,
Herman, J. G., Kinzler, K. W., Vogelstein, B., Baylin, S. B., and
Schuebel, K. E. (2000) Nature, 404, 1003-1007.
52.Rhee, I., Bachman, K. E., Park, B. H., Jair, K.
W., Yen, R. W., Schuebel, K. E., Cui, H., Feinberg, A. P., Lengauer,
C., Kinzler, K. W., Baylin, S. B., and Vogelstein, B. (2002)
Nature, 416, 552-556.
53.Ting, A. H., Jair, K. W., Suzuki, H., Yen, R. W.,
Baylin, S. B., and Schuebel, K. E. (2004) Nat. Genet.,
36, 582-584.
54.Fournel, M., Sapieha, P., Beaulieu, N.,
Besterman, J. M., and MacLeod, A. R. (1999) J. Biol. Chem.,
274, 24250-24256.
55.Deng, J., and Szyf, M. (1998) J. Biol.
Chem., 273, 22869-22872.
56.Weisenberger, D. J., Velicescu, M., Cheng, J. C.,
Gonzales, F. A., Liang, G., and Jones, P. A. (2004) Mol. Cancer
Res., 2, 62-72.
57.Espada, J., Ballestar, E., Fraga, M. F.,
Villar-Garea, A., Juarranz, A., Stockert, J. C., Robertson, K. D.,
Fuks, F., and Esteller, M. (2004) J. Biol. Chem., 279,
37175-37184.
58.Hattori, N., Abe, T., Hattori, N., Suzuki, M.,
Matsuyama, T., Yoshida, S., Li, E., and Shiota, K. (2004) Genome
Res., 14, 1733-1740.
59.Milutinovic, S., Brown, S. E., Zhuang, Q., and
Szyf, M. (2004) J. Biol. Chem., 279, 27915-27927.
60.Milutinovic, S., Knox, J. D., and Szyf, M. (2000)
J. Biol. Chem., 275, 6353-6359.
61.Milutinovic, S., Zhuang, Q., Niveleau, A., and
Szyf, M. (2003) J. Biol. Chem., 278, 14985-14995.
62.Wu, J. C., and Santi, D. V. (1985) Progr.
Clin. Biol. Res., 198, 119-129.
63.Oswald, J., Engemann, S., Lane, N., Mayer, W.,
Olek, A., Fundele, R., Dean, W., Reik, W., and Walter, J. (2000)
Curr. Biol., 10, 475-478.
64.Kafri, T., Gao, X., and Razin, A. (1993) Proc.
Natl. Acad. Sci. USA, 90, 10558-10562.
65.Razin, A., Feldmesser, E., Kafri, T., and Szyf,
M. (1985) Progr. Clin. Biol. Res., 198, 239-253.
66.Szyf, M., Eliasson, L., Mann, V., Klein, G., and
Razin, A. (1985) Proc. Natl. Acad. Sci. USA, 82,
8090-8094.
67.Wilks, A. F., Cozens, P. J., Mattaj, I. W., and
Jost, J. P. (1982) Proc. Natl. Acad. Sci. USA, 79,
4252-4255.
68.Frank, D., Lichtenstein, M., Paroush, Z.,
Bergman, Y., Shani, M., Razin, A., and Cedar, H. (1990) Philos.
Trans. R. Soc. Lond. B. Biol. Sci., 326, 241-251.
69.Kirillov, A., Kistler, B., Mostoslavsky, R.,
Cedar, H., Wirth, T., and Bergman, Y. (1996) Nat. Genet.,
13, 435-441.
70.Paroush, Z., Keshet, I., Yisraeli, J., and Cedar,
H. (1990) Cell, 63, 1229-1237.
71.Weaver, I. C., Cervoni, N., Champagne, F. A.,
D'Alessio, A. C., Sharma, S., Seckl, J. R., Dymov, S., Szyf, M., and
Meaney, M. J. (2004) Nat. Neurosci., 7, 847-854.
72.Cervoni, N., and Szyf, M. (2001) J. Biol.
Chem., 276, 40778-4087.
73.Detich, N., Bovenzi, V., and Szyf, M. (2003)
J. Biol. Chem., 278, 27586-27592.
74.Cervoni, N., Detich, N., Seo, S. B., Chakravarti,
D., and Szyf, M. (2002) J. Biol. Chem., 277,
25026-25031.
75.Razin, A., Szyf, M., Kafri, T., Roll, M., Giloh,
H., Scarpa, S., Carotti, D., and Cantoni, G. L. (1986) Proc. Natl.
Acad. Sci. USA, 83, 2827-2831.
76.Jost, J. P., and Jost, Y. C. (1995) Gene,
157, 265-266.
77.Jost, J. P., Siegmann, M., Sun, L., and Leung, R.
(1995) J. Biol. Chem., 270, 9734-9739.
78.Weiss, A., Keshet, I., Razin, A., and Cedar, H.
(1996) Cell, 86, 709-718.
79.Weiss, A., and Cedar, H. (1997) Genes
Cells, 2, 481-486.
80.Zhu, B., Zheng, Y., Hess, D., Angliker, H.,
Schwarz, S., Siegmann, M., Thiry, S., and Jost, J. P. (2000) Proc.
Natl. Acad. Sci. USA, 97, 5135-5139.
81.Zhu, B., Benjamin, D., Zheng, Y., Angliker, H.,
Thiry, S., Siegmann, M., and Jost, J. P. (2001) Proc. Natl. Acad.
Sci. USA, 98, 5031-5036.
82.Jost, J. P., Oakeley, E. J., Zhu, B., Benjamin,
D., Thiry, S., Siegmann, M., and Jost, Y. C. (2001) Nucleic Acids
Res., 29, 4452-4461.
83.Dickinson, H., and Scott, R. (2002) Mol.
Cell, 10, 5-7.
84.Choi, Y., Gehring, M., Johnson, L., Hannon, M.,
Harada, J. J., Goldberg, R. B., Jacobsen, S. E., and Fischer, R. L.
(2002) Cell, 110, 33-42.
85.Hendrich, B., and Bird, A. (1998) Mol. Cell.
Biol., 18, 6538-6547.
86.Ng, H. H., Zhang, Y., Hendrich, B., Johnson, C.
A., Turner, B. M., Erdjument-Bromage, H., Tempst, P., Reinberg, D., and
Bird, A. (1999) Nat. Genet., 23, 58-61.
87.Stirzaker, C., Song, J. Z., Davidson, B., and
Clark, S. J. (2004) Cancer Res., 64, 3871-3877.
88.David, G. L., Yegnasubramanian, S., Kumar, A.,
Marchi, V. L., De Marzo, A. M., Lin, X., and Nelson, W. G. (2004)
Cancer Biol. Ther., 3, 540-548.
89.Lin, X., and Nelson, W. G. (2003) Cancer
Res., 63, 498-504.
90.Bakker, J., Lin, X., and Nelson, W. G. (2002)
J. Biol. Chem., 277, 22573-22580.
91.Detich, N., Hamm, S., Just, G., Knox, J. D., and
Szyf, M. (2003) J. Biol. Chem., 278, 20812-20820.
92.Detich, N., Theberge, J., and Szyf, M. (2002)
J. Biol. Chem., 277, 35791-35794.
93.Nan, X., Campoy, F. J., and Bird, A. (1997)
Cell, 88, 471-481.
94.Jeddeloh, J. A., Bender, J., and Richards, E. J.
(1998) Genes Dev., 12, 1714-1725.
95.Jeddeloh, J. A., Stokes, T. L., and Richards, E.
J. (1999) Nat. Genet., 22, 94-97.
96.Dennis, K., Fan, T., Geiman, T., Yan, Q., and
Muegge, K. (2001) Genes Dev., 15, 2940-2944.
97.Gibbons, R. J., McDowell, T. L., Raman, S.,
O'Rourke, D. M., Garrick, D., Ayyub, H., and Higgs, D. R. (2000)
Nat. Genet., 24, 368-371.
98.Jackson, J. P., Lindroth, A. M., Cao, X., and
Jacobsen, S. E. (2002) Nature, 416, 556-560.
99.Di Croce, L., Raker, V. A., Corsaro, M., Fazi,
F., Fanelli, M., Faretta, M., Fuks, F., Lo Coco, F., Kouzarides, T.,
Nervi, C., Minucci, S., and Pelicci, P. G. (2002) Science,
295, 1079-1082.
100.Fuks, F., Hurd, P. J., Deplus, R., and
Kouzarides, T. (2003) Nucleic Acids Res., 31,
2305-2312.
101.Fuks, F., Burgers, W. A., Godin, N., Kasai, M.,
and Kouzarides, T. (2001) EMBO J., 20, 2536-2544.
102.Fuks, F., Burgers, W. A., Brehm, A.,
Hughes-Davies, L., and Kouzarides, T. (2000) Nat. Genet.,
24, 88-91.
103.Cameron, E. E., Bachman, K. E., Myohanen, S.,
Herman, J. G., and Baylin, S. B. (1999) Nat. Genet., 21,
103-107.
104.Gius, D., Cui, H., Bradbury, C. M., Cook, J.,
Smart, D. K., Zhao, S., Young, L., Brandenburg, S. A., Hu, Y., Bisht,
K. S., Ho, A. S., Mattson, D., Sun, L., Munson, P. J., Chuang, E. Y.,
Mitchell, J. B., and Feinberg, A. P. (2004) Cancer Cell,
6, 361-371.
105.Kistler, B., Baumann, B., Bergman, Y., and
Wirth, T. (1997) Immunobiology, 198, 24-34.
106.Esteller, M., Fraga, M. F., Guo, M.,
Garcia-Foncillas, J., Hedenfalk, I., Godwin, A. K., Trojan, J.,
Vaurs-Barriere, C., Bignon, Y. J., Ramus, S., Benitez, J., Caldes, T.,
Akiyama, Y., Yuasa, Y., Launonen, V., Canal, M. J., Rodriguez, R.,
Capella, G., Peinado, M. A., Borg, A., Aaltonen, L. A., Ponder, B. A.,
Baylin, S. B., and Herman, J. G. (2001) Hum. Mol. Genet.,
10, 3001-3007.
107.News (2002) Expert Rev. Anticancer
Ther., 2, 620-621.
108.Aparicio, A., and Weber, J. S. (2002) Curr.
Opin. Investig. Drugs, 3, 627-633.
109.Paz, M. F., Fraga, M. F., Avila, S., Guo, M.,
Pollan, M., Herman, J. G., and Esteller, M. (2003) Cancer Res.,
63, 1114-1121.
110.Uhlmann, K., Rohde, K., Zeller, C., Szymas, J.,
Vogel, S., Marczinek, K., Thiel, G., Nurnberg, P., and Laird, P. W.
(2003) Int. J. Cancer, 106, 52-59.
111.Adorjan, P., Distler, J., Lipscher, E., Model,
F., Muller, J., Pelet, C., Braun, A., Florl, A. R., Gutig, D., Grabs,
G., Howe, A., Kursar, M., Lesche, R., Leu, E., Lewin, A., Maier, S.,
Muller, V., Otto, T., Scholz, C., Schulz, W. A., Seifert, H. H.,
Schwope, I., Ziebarth, H., Berlin, K., Piepenbrock, C., and Olek, A.
(2002) Nucleic Acids Res., 30, 21.
112.Chen, C. M., Chen, H. L., Hsiau, T. H., Hsiau,
A. H., Shi, H., Brock, G. J., Wei, S. H., Caldwell, C. W., Yan, P. S.,
and Huang, T. H. (2003) Am. J. Pathol., 163, 37-45.
113.Sato, N., Fukushima, N., Matsubayashi, H., and
Goggins, M. (2003) Oncogene, 22, 5021-5030.
114.Rosty, C., Ueki, T., Argani, P., Jansen, M.,
Yeo, C. J., Cameron, J. L., Hruban, R. H., and Goggins, M. (2002)
Am. J. Pathol., 160, 45-50.
115.Guo, Y., Pakneshan, P., Gladu, J., Slack, A.,
Szyf, M., and Rabbani, S. A. (2002) J. Biol. Chem., 277,
41571-41579.
116.Shteper, P. J., Zcharia, E., Ashhab, Y.,
Peretz, T., Vlodavsky, I., and Ben-Yehuda, D. (2003) Oncogene,
22, 7737-7749.
117.Chalitchagorn, K., Shuangshoti, S., Hourpai,
N., Kongruttanachok, N., Tangkijvanich, P., Thong-Ngam, D., Voravud,
N., Sriuranpong, V., and Mutirangura, A. (2004) Oncogene,
23.
118.El-Deiry, W. S., Nelkin, B. D., Celano, P.,
Yen, R. W., Falco, J. P., Hamilton, S. R., and Baylin, S. B. (1991)
Proc. Natl. Acad. Sci. USA, 88, 3470-3474.
119.Nass, S. J., Ferguson, A. T., El-Ashry, D.,
Nelson, W. G., and Davidson, N. E. (1999) Oncogene, 18,
7453-7461.
120.De Marzo, A. M., Marchi, V. L., Yang, E. S.,
Veeraswamy, R., Lin, X., and Nelson, W. G. (1999) Cancer Res.,
59, 3855-3860.
121.Szyf, M. (2001) Front. Biosci.,
6, 599-609.
122.Lee, P. J., Washer, L. L., Law, D. J., Boland,
C. R., Horon, I. L., and Feinberg, A. P. (1996) Proc. Natl. Acad.
Sci. USA, 93, 10366-10370.
123.Bachman, K. E., Park, B. H., Rhee, I.,
Rajagopalan, H., Herman, J. G., Baylin, S. B., Kinzler, K. W., and
Vogelstein, B. (2003) Cancer Cell, 3, 89-95.
124.Koizume, S., Tachibana, K., Sekiya, T.,
Hirohashi, S., and Shiraishi, M. (2002) Nucleic Acids Res.,
30, 4770-4780.
125.Datta, J., Ghoshal, K., Sharma, S. M., Tajima,
S., and Jacob, S. T. (2003) J. Cell. Biochem., 88,
855-864.
126.Macaluso, M., Cinti, C., Russo, G., Russo, A.,
and Giordano, A. (2003) Oncogene, 22, 3511-3517.
127.Pakneshan, P., Szyf, M., Farias-Eisner, R., and
Rabbani, S. A. (2004) J. Biol. Chem., 279,
31735-31744.
128.Hattori, M., Sakamoto, H., Satoh, K., and
Yamamoto, T. (2001) Cancer Lett., 169, 155-164.
129.Hattori, M., Sakamoto, H., and Yamamoto, T.
(2001) J. Int. Med. Res., 29, 204-213.
130.Kanai, Y., Ushijima, S., Nakanishi, Y., and
Hirohashi, S. (1999) Biochem. Biophys. Res. Commun., 264,
962-966.
131.Baylin, S. B., Herman, J. G., Graff, J. R.,
Vertino, P. M., and Issa, J. P. (1998) Adv. Cancer Res.,
72, 141-196.
132.Graff, J. R., Herman, J. G., Lapidus, R. G.,
Chopra, H., Xu, R., Jarrard, D. F., Isaacs, W. B., Pitha, P. M.,
Davidson, N. E., and Baylin, S. B. (1995) Cancer Res.,
55, 5195-5199.
133.Esteller, M., Levine, R., Baylin, S. B.,
Ellenson, L. H., and Herman, J. G. (1998) Oncogene, 17,
2413-2417.
134.Esteller, M., Corn, P. G., Urena, J. M.,
Gabrielson, E., Baylin, S. B., and Herman, J. G. (1998) Cancer
Res., 58, 4515-4518.
135.Bachman, K. E., Herman, J. G., Corn, P. G.,
Merlo, A., Costello, J. F., Cavenee, W. K., Baylin, S. B., and Graff,
J. R. (1999) Cancer Res., 59, 798-802.
136.Bender, C. M., Pao, M. M., and Jones, P. A.
(1998) Cancer Res., 58, 95-101.
137.Cheng, J. C., Matsen, C. B., Gonzales, F. A.,
Ye, W., Greer, S., Marquez, V. E., Jones, P. A., and Selker, E. U.
(2003) J. Natl. Cancer Inst., 95, 399-409.
138.Eden, A., Gaudet, F., Waghmare, A., and
Jaenisch, R. (2003) Science, 300, 455.
139.Pascale, R. M., Simile, M. M., De Miglio, M.
R., and Feo, F. (2002) Alcohol, 27, 193-198.
140.Ge, R., Wang, W., Kramer, P. M., Yang, S., Tao,
L., and Pereira, M. A. (2001) Toxicol. Sci., 62,
28-35.
141.Tao, L., Yang, S., Xie, M., Kramer, P. M., and
Pereira, M. A. (2000) Toxicol. Sci., 54, 399-407.
142.MacLeod, A. R., Rouleau, J., and Szyf, M.
(1995) J. Biol. Chem., 270, 11327-11337.
143.Rouleau, J., MacLeod, A. R., and Szyf, M.
(1995) J. Biol. Chem., 270, 1595-1601.
144.Bakin, A. V., and Curran, T. (1999)
Science, 283, 387-390.
145.Slack, A., Cervoni, N., Pinard, M., and Szyf,
M. (1999) J. Biol. Chem., 274, 10105-10112.
146.Momparler, R. L., and Bovenzi, V. (2000) J.
Cell. Physiol., 183, 145-154.
147.Knox, J. D., Araujo, F. D., Bigey, P., Slack,
A. D., Price, G. B., Zannis-Hadjopoulos, M., and Szyf, M. (2000) J.
Biol. Chem., 275, 17986-17990.
148.Rountree, M. R., Bachman, K. E., and Baylin, S.
B. (2000) Nat. Genet., 25, 269-277.
149.Robertson, K. D., Ait-Si-Ali, S., Yokochi, T.,
Wade, P. A., Jones, P. L., and Wolffe, A. P. (2000) Nat. Genet.,
25, 338-342.
150.Nakamura, N., and Takenaga, K. (1998) Clin.
Exp. Metastasis, 16, 471-479.
151.Wainfan, E., Dizik, M., Stender, M., and
Christman, J. K. (1989) Cancer Res., 49, 4094-4097.
152.Kanduc, D., Ghoshal, A., Quagliariello, E., and
Farber, E. (1988) Biochem. Biophys. Res. Commun., 150,
739-744.
153.Pascale, R., Simile, M. M., Ruggiu, M. E.,
Seddaiu, M. A., Satta, G., Sequenza, M. J., Daino, L., Vannini, M. G.,
Lai, P., and Feo, F. (1991) Cancer Lett., 56,
259-265.
154.Pereira, M. A., Wang, W., Kramer, P. M., and
Tao, L. (2004) Toxicol. Sci., 77, 243-248.
155.Giovannucci, E., Stampfer, M. J., Colditz, G.
A., Rimm, E. B., Trichopoulos, D., Rosner, B. A., Speizer, F. E., and
Willett, W. C. (1993) J. Natl. Cancer Inst., 85,
875-884.
156.Potter, J. D. (1999) J. Natl. Cancer
Inst., 91, 916-932.
157.Eichholzer, M., Luthy, J., Moser, U., and
Fowler, B. (2001) Swiss Med. Wkly., 131, 539-549.
158.Kamiya, H., Kawakami, K., Miyanaga, T., Omura,
K., Oda, M., Murakami, S., and Watanabe, Y. (1998) Oncol. Rep.,
5, 911-914.
159.Stern, L. L., Mason, J. B., Selhub, J., and
Choi, S. W. (2000) Cancer Epidemiol. Biomarkers Prev., 9,
849-853.
160.Friso, S., Choi, S. W., Girelli, D., Mason, J.
B., Dolnikowski, G. G., Bagley, P. J., Olivieri, O., Jacques, P. F.,
Rosenberg, I. H., Corrocher, R., and Selhub, J. (2002) Proc. Natl.
Acad. Sci. USA, 99, 5606-5611.
161.Shannon, B., Gnanasampanthan, S., Beilby, J.,
and Iacopetta, B. (2002) Gut, 50, 520-524.
162.Fang, J. Y., and Xiao, S. D. (2003) J.
Gastroenterol., 38, 821-829.
163.Heijmans, B. T., Boer, J. M., Suchiman, H. E.,
Cornelisse, C. J., Westendorp, R. G., Kromhout, D., Feskens, E. J., and
Slagboom, P. E. (2003) Cancer Res., 63, 1249-1253.
164.Lambropoulos, A. F., Agorastos, T., Foka, Z.
J., Chrisafi, S., Constantinidis, T. C., Bontis, J., and Kotsis, A.
(2003) Cancer Lett., 191, 187-191.
165.Slack, A., Bovenzi, V., Bigey, P., Ivanov, M.
A., Ramchandani, S., Bhattacharya, S., tenOever, B., Lamrihi, B.,
Scherman, D., and Szyf, M. (2002) J. Gene Med., 4,
381-389.
166.Ivanov, M. A., Lamrihi, B., Szyf, M., Scherman,
D., and Bigey, P. (2003) J. Gene Med., 5, 893-899.
167.Sansom, O. J., Berger, J., Bishop, S. M.,
Hendrich, B., Bird, A., and Clarke, A. R. (2003) Nat. Genet.,
34, 145-147.
168.Campbell, P. M., Bovenzi, V., and Szyf, M.
(2004) Carcinogenesis, 25, 499-507.
169.Fang, J. Y., Cheng, Z. H., Chen, Y. X., Lu, R.,
Yang, L., Zhu, H. Y., and Lu, L. G. (2004) World J.
Gastroenterol., 10, 3394-3398.