Cancer as a Programmed Death of an Organism
A. V. Lichtenstein
Institute of Carcinogenesis, Blokhin Cancer Research Center, Russian Academy of Medical Sciences, 115478 Moscow, Russia; fax: (7-095) 324-1205; E-mail: alicht@online.ru
Received March 16, 2005
The hypothesis introduces the idea that there is a critical level of mutagenesis that triggers a program of organism death by means of proliferation of killer cells. Similarly to apoptosis, which is an altruistic suicidal act of a faulty cell threatening the stability of a multicellular organism, a malignant tumor is an altruistic suicide of an individual carrier of harmful alleles threatening genetic stability of the population.
KEY WORDS: carcinogenesis, apoptosis, programmed organism death, evolution, mutagenesis
“The Samurai law”: It is better to die than to be wrong!
V. P. Skulachev [1]
During recent decades, enormous progress has been achieved in elucidation of molecular mechanisms of cancer (all malignant tumors are kept in mind). In particular, genetic defects have been identified that determine necessary and sufficient properties of a cancer cell. It is strange, but the sufficient properties do not include an apparently crucial property, the ability to kill the “host”. This property is believed to be a self-evident consequence of other features of the tumor phenotype, although there are data indicating the existence of a special “killer” function.
DNA is the most protected cell component. Its integrity is one of the essential priorities of living beings. Depending on the level of DNA damage, there are de facto three putative outcomes: (i) a weak damage initiates repair mechanisms which restore the initial state; (ii) a severe damage triggers a lethal program which prevents appearance of a faulty cell clone; (iii) if apoptosis is impossible, accumulated unrepaired damages induce cancer which kills the individual. Similarly to apoptosis, cancer is a program, i.e., a certain sequence of active events with a regular termination [2].
Carcinogenesis is a multistage process of accumulation in the cell of genetic defects resulting in its “asocial” behavior: constant mitogenic stimulation, insensitivity to antigrowth and proapoptotic signals, unlimited proliferative potential, angiogenesis, invasion, and metastasizing [3]. The totality of these properties determines the tumor phenotype of the cell but does not explain the clinical picture of cancer: there is a conceptual gap in our understanding of carcinogenesis, on one hand, and of oncology illness pathogenesis, on the other. In particular, it is unclear why arising of a small handful of cells (0.01-0.1% of the total weight), very slightly different from the other cells, is incompatible with the life of the organism. Relatively seldom, the cause of the patient's death is obvious (brain squeezing, hemorrhage, intestinal perforation, etc.), but in most cases, it is unknown.
Although every variety of cancer has its specific features, the course of the illness and its final outcome are generally the same. Therefore, the statement that the cancer cell, in addition to canonical features [3], has special killer properties directed against the total organism seems to be only a declaration of the doubtless situation. But although the manner of its actuation is unknown [4], its existence causes no doubt. Paradoxically, the current paradigm of carcinogenesis ignores the apparently obvious “killer” function of the tumor cell [3]. Nevertheless, assuming such a function significantly changes the concept of cancer phenomenon as it is. A short version of the proposed hypothesis was published earlier [5].
THE EVOLUTIONARY NECESSITY OF CANCER
Cancer occurs in all animal species, from mollusks to mammals [6]. The evolutionary conservatism confirms the necessity of the phenomenon because it performs some important tasks. Moreover, this also indicates a principle insufficiency of the cellular anticancer defense (see below).
The Peto paradox. Fifty years ago a fundamental concept of oncology was formulated that carcinogenesis is a multistage process [7]. On analyzing statistics of age dependence of cancer incidence in humans, seven stages were revealed (today it is clear that they are associated with accumulation in the cellular DNA of mutations which induce discrete and irreversible changes in the cell genotype and, respectively, phenotype). Obviously, the more stages are required for the cell transformation, the higher is its anticancer defense [8]; thus, this value (the number of stages and, respectively, mutations which underlie them) may be used to quantitatively characterize “transformational resistance” of the cell.
The multistage pattern of carcinogenesis shows a very low probability of transformation of a normal cell to a cancer cell: mutations (which are extremely rare events, ~10-7 gene/division [9-11]) have to hit several times a very small target (~300 genes, or ~1% of the genome [12]) and be accumulated in the genome of the same cell. Nevertheless, cancer is rather common in humans (the probability of the illness during the lifetime is ~20% and 40-50% at the age of 60-80 years [13]). The discrepancy between the low probability of cancer transformation of the cell and the high probability of cancer morbidity in humans is explained by a high number of proliferating cells in the human body (~10-12-10-13) and a prolonged lifetime: even a highly improbable event can occur under such conditions. In accordance with this logic, cancer should not occur in small and short-living animals (such as mice), whereas big and long-living animals (elephants, whales) should be ill even in uterus and die out as species. However, these theoretical predictions do not correspond to the reality: cancer occurs in all animals, independently of size and longevity, but, on the other hand, cancer does not threaten the existence of species (the known Peto paradox [14]).
The Peto paradox is explained by lack of the definitely fixed transformational resistance of the cell. On the contrary, it is very movable, changeable, and susceptible to evolutionary adaptation: and cells of different species significantly vary in this respect [6, 15]. These variations can be caused, first, by different efficiency of the DNA repair systems and, as a result, different frequency of mutations [16]; second, species-specific differences in the degree of selective advantages gained by the cell as a result of the same mutations (for example, a different degree of haplo-insufficiency on inactivation of one allele of the suppressor gene [6]); and, finally, different number of transformation stages (thus, human fibroblasts are transformed on damaging five signal pathways instead of three pathways in mice [17]). The last finding has been empirically known for a long time: cultured primary cells of rodents can be transformed much easier than human cells [18]. The transformational resistance seems to be the highest in whale cells and the lowest in mouse cells.
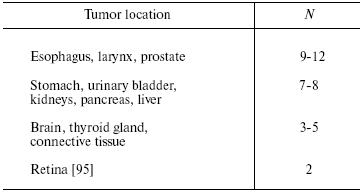
The Peto paradox is also true for different tissues of the same organism [15]. Thus, man has ~100 types of tissues, which considerably vary in both number of cells and their proliferative activity, but they all are susceptible to cancer transformation. The transformational resistance depends on both the species and tissue (table) [19]. Thus, the anticancer defense of the cell adapts to conditions of its existence in the body not only on the genome level (as it seems to be for species-associated differences), but also on the transcriptional level, which is markedly more changeable. Therefore, if the cancer phenomenon had even a slight negative effect on the species survival and reproduction, it would be easily eliminated during evolution. By contrast, the stubborn maintaining of this phenomenon in Nature (proved by its universality) suggests its evolutionary reasonability. It seems that the anticancer defense on the organism's level is balanced by the opposite tendency (i.e., directed for its preservation), which acts on the level of the population (see below).
Transformational resistance of human cells [19]
(N is the number of mutations required for cell transformation)
Cancer as an “organ”. Mechanisms of transformation are considered in thousands of works, and nearly all of them describe cancer as an entity, without attempts to clarify the nature of this phenomenon. The acknowledgment that cancer in its essence is a programmed death of an organism (phenoptosis [20]) would be the first step in this direction: the existence of such a program (i.e., the sequence of events connected by causal-consequence relations and leading to the definite result) and its lethal character for the organism in this case are obvious.
Although cancer is thought to be an age-related illness, it is fundamentally different from all other diseases. Cardiovascular diseases, diabetes, Parkinson's and Alzheimer's diseases, etc. are passive consequences of the loss of function of the appropriate organ caused by its degeneration and/or death of its cells. Cancer is cardinally different because of active character of the process, namely, the transformed cells are gaining new, previously not inherent functions. Playing cat-and-mouse (the term of R. Weinberg [21]) with the organism, the cancer tumor uses a sophisticated strategy, which combines aggression, adaptation to environmental conditions, escaping the organism's defense, and a striking ability to recruit the surrounding tissues. The cancer tumor converts its natural opponents (normal cells) to unnatural allies because it can grow only under conditions of such a paradoxical support [22-27]. The structure of tumor and its functional connections with the normal environment are so complicated and multiform that it seems reasonable to liken it to a special “organ” [23]. And this “organ” obviously arose during phylogenesis to perform an important function (that is confirmed by its evolutionary conservatism), namely, to kill the “host's” organism.
KILLER FUNCTION OF CANCER CELL
The above-presented data allow us to state that the tumor cell has a special “killer” function directed not to individual cells but the total organism. It is rather paradoxical that such an important property is neglected in the current paradigm of carcinogenesis [3].
Paraneoplastic syndromes. The doubtless ability of cancer tumor for distant and generalized effects on the body is evidenced by paraneoplastic syndromes extremely varying in manifestations and affecting virtually all organs and tissues [4, 28-32]. Patients with cancer are found to have anemia, hypoalbuminemia, hypercalcemia, hyponatremia, hypoglycemia, and elevated ESR. Cachexia, anorexia, neuropathies, retinopathies, general indisposition, and changes in vascular, endocrine, neuromuscular systems, blood, and bones are the most common clinical manifestations. Long before the diagnosis (16-20 months earlier), some patients are recorded to increasingly lose weight, which suggests the generalized and progressing effect of the tumor on the body even at the early stage of its growth [33, 34].
Some of these effects (in particular, neuropathies and retinopathies) are caused by a perverted immune reaction of the organism to antigens expressed by the tumor, and this finally results in autoimmune disease [35]. The appearance of cancer-associated cachexia is supposed to involve both the primary products generated by the tumor itself (tumor necrosis factor TNF-alpha [36, 37], lipid mobilization factor LMF, proteolysis-inducing factor PIF) and the secondary products, which are synthesized by normal cells under influence of the tumor (uncoupling protein UCP3, interleukins IL-1 and IL-6) [4]. However, pronounced cachexia (more than 5% weight loss) occurs in about one third of the patients, but causes death only in 20% of the cases [4]; obviously, the tumor also has other, still unidentified, approaches for killing.
It is unclear to what extent paraneoplastic syndromes are direct manifestations of the killer function of the tumor or its side consequences, exemplified, in particular, by autoimmune diseases. The second idea seems to be more likely, because cancer is far from always accompanied by paraneoplastic syndromes [38], whereas the killer function never betrays it. On the other hand, paraneoplastic syndromes completely disappear on successful antitumor therapy, whereas the alternative approach (symptomatic treatment aimed to eliminate particular manifestations of the tumor growth) only seldom gives positive results and never cures completely. Thus, most of the syndromes justify their name of paraneoplastic because seem to be beyond the main programmed pathway of death, and its mechanism still remains unknown. Therefore, afterwards we implicate the killer function (not penetrating its mechanism) of the tumor cell to be its obvious ability of killing the organism.
Killer function as a crucial property of the cancer cell. It is generally accepted that the cancer cell phenotype is determined by a set of necessary and sufficient properties: constant mitogenic stimulation, insensitivity to antigrowth and proapoptotic signals, unlimited proliferative potential, ability for inducing angiogenesis, invasion, and metastasizing [3]. The necessity of each property is evident, but their sufficiency induces a doubt: among them there is no the killer function which seems to play a crucial role in the picture of oncological illness (just this function is responsible for the “mission” for which the programmed death of an organism seemed to arise during evolution). From this viewpoint, all other properties gained by the cancer cell play only an auxiliary role increasing the number of cell killers and promoting their expansion over the body.
The killer function of cancer cell is its specific feature, which is not settled with its other properties, such as unregulated proliferation or metastasizing. Indeed, the active proliferation of cancer cells seems unlikely to be as disastrous as it is, because every day in the organism of an adult human dozens of billions of cells are dividing compensating the loss of dead cells, and this is manifold higher than the proliferative pool of the largest tumor. And even metastases, which are foci of ectopic growth of relatively small numbers of cells, do not explain the death of the organism (except the above-mentioned rare situations when just the tumor location is fatal). In most cases, not local but general manifestations of tumor growth play the leading role in the clinical picture of cancer.
The killer function of a cancer cell is a universal property: in the absence of treatment the lethal outcome is inevitable, independently of the tumor type and location, its ability to relapse and metastasize, induce cachexia, and affect biochemical parameters. And finally, this function is specific for cancer cell, because normal cell physiology presents no examples of similar activity (nearly all other inherent features of the cancer cell are found in normal cells during some developmental stages, in particular, in stem cells [39]). Green and Evan in their “paradoxical hypothesis” [40] supposed that the platform necessary and sufficient for carcinogenesis could be provided only by two main processes, deregulation of proliferation and suppression of apoptosis. Other properties of tumor (invasion, stimulation of angiogenesis, escaping the immune response, etc.) are secondary and inherent to any tissue during its expansion. Assuming similarity of many features of tumor and normal growing tissue, it should be noted that neither the growing nor resting normal tissue displays the killer ability.
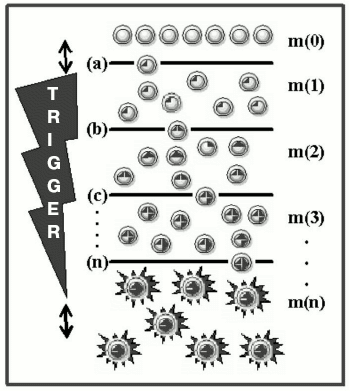
Mutagenesis as a trigger of the death program. Carcinogenesis is a Darwin's evolutionary process of selection and stepwise accumulation in a somatic cell of mutations and epimutations [13], which promote expansion of the appropriate clone [41]. However, the killer function is fundamentally different from all other features, because it does not give selective advantages to the cancer cell. On the contrary, realization of this function equalizes all cells of the body because it results in their common catastrophe. And just in this feature (the lack of selective advantages and self-destruction) the killer function and apoptosis are alike. These two phenomena developed during evolution represent built-in and ready-for-use programs of self-destruction in response to a specific trigger. These programs are similar in their altruistic character, but different in direction of the effect (against the cell itself in the case of apoptotic cell and against “the host” in the case of cancer cell) and the trigger switching on: after a singular event (the ligand-receptor interaction or damage to DNA) on initiation of apoptosis; after multiple influences (accumulation of several mutations) on initiation of the programmed death of the organism. Figure 1 presents a scheme of cell transformation, with mutagenesis acting as a trigger with a countdown mechanism.
Fig. 1. The scheme of malignant transformation. Mutations in the cellular DNA (dark areas inside the circles) trigger the inherent program of the organism destruction (asterisks); m(0) are normal cells; m(1), m(2), m(3), and m(n) are mutant cells with 1, 2, 3, and n defects, respectively, in the “cancerous” genes; (a), (b), (c), ..., (n) are selection “sieves” which determine stages of the transformation. To the left the trigger is shown with an inverse time count (the two-directional arrow shows that trigger steps are amenable to species- and tissue-specific variations).
CANCER AS AN ALTRUISTIC PROGRAM
Genetic defects determining the tumor phenotype are characterized by their inability to decrease the cell viability (contrasting to the cell specific functions, its basic functions are activated). So to say, these are defects not of the cell itself but of the cell/organism “interface” which mediates the subordination of cell functions to interests of the total organism. In fact, all manifestations of the cell activity (proliferation, differentiation, death, mobility, synthesis and secretion of specific products) are performed “on order from the outside” via activation of specific receptors on the cell surface and, as a consequence, of the inner signaling pathways. This results in a corresponding cellular reaction.
In the described system of cell/organism, the receptor signaling pathways are fetters, which fasten the cell to the system of tissue homeostasis robbing it of “free will” and subordinating to the organism's needs. From this viewpoint, the cancer-inducing mutations, which break somewhere signaling pathways present a gradual, step-by-step liberation of the cell from its fetters. Their number (and, respectively, the number of mutations liberating the cell) determines its transformational resistance (see above). Thus, the cell transformation is its road “from slavery to freedom”, or, in other words, transition of cancer cells to “egoistic” behavior dictated solely by intracellular impulses. However, the concept about cancer cells as cells-egoists [15] misrepresents the essence of the events because it underestimates their killer function. Indeed, the acquisition of this function means a suicide for the cancer cell as a component of the organism, and this contradicts the egoistic behavior, which is mainly characterized by satisfactory and imperceptible existence. On the contrary, cancer cells may be considered as cells-altruists, which execute a mission and sacrifice themselves and the total organism to some higher (populational?) interests. Similarly to apoptosis, which is an act of the cell self-sacrifice for genetic stability of the cell community (the multicellular organism) [2], cancer is an altruistic act of self-destruction of the individual carrier of harmful alleles in favor of genetic stability of the population (see below).
The general concept about the programmed death of an organism (phenoptosis) and carcinogenesis as a particular mechanism of its realization has been formulated by V. P. Skulachev [42]. According to the so-called Samurai law (“it is better to die than to be wrong”), complicated biological systems are provided with self-destruction programs. Suicidal mechanisms are activated in the cases when the system became threatening for the above-standing system, i.e., the system which is higher in biological hierarchy [43]. Cancer seems to be a particular case of realization of phenoptosis.
Hypotheses of evolution, which attempt to explain appearance of cooperation and altruistic behavior, are based on ideas of kin or group selection [44, 45]. According to the known Hamilton's rule, altruistic behavior can strike root in the population at the inequality RB > C, if the cost of losses (C) of an acting individual is lower than the product of benefits (B) on degree of his genetic relatedness (R) with social partners [46]. Apoptosis which appeared during the evolution as a consequence of joining of unicellular organisms into a multicellular association is the most striking example of an altruistic program. Evolution of apoptosis during four billion years was followed in the recent work [2], and a slow transformation was shown of separate and egoistic (parasitic) genetic elements into an altruistically aimed genetic “ensemble” of the programmed cell death. Possibly, similar genetic “mining” concerning the killer function of cancer cell will be available when its mechanism is completely understood.
CANCER IS A LOCAL MANIFESTATION OF GENERALIZED MUTAGENESIS
According to a fundamental concept of oncology, tumor has a clonal origin, i.e., consists of descendants of a single transformed cell. Moreover, the presence of a single tumor in most patients results in the idea that carcinogenesis is a purely local process which arises in the site of the cell contact with carcinogens. Experiments with chemical carcinogens applied to skin in animals and cases of occupational cancer clearly show the association between the exposed area and the tumor location, and this supports the concept. No doubt, these observations are important for theory and practice; nevertheless, they are exclusive and limited (see below). Not the environment, but the internal medium of the organism, seems to be the main source of mutagenic influences [47].
Mutagenesis is closely linked with vital activities and, therefore, is omnipresent: in every cell of the organism thousands of acts of the genome injury occur because of errors in replication and repair, spontaneous depurinization, deamination of methylcytosine, influence of reactive oxygen species, shortening of telomeres [9, 11, 48-54]. And mutagenic effect of apoptosis also should be added during which phagocytizing cells uptake the genetic material of dying cells. Considering, first, the involvement of all cells of the organism in this process, second, scale of this natural transfection (every day ~1011 cells dies in the body of an adult human and ~0.6 g DNA is liberated), and, third, continuity of this transfection during the whole life, its essential contribution, although yet unestimated quantitatively, to general mutagenesis is very likely. In any case, a possibility was shown of such a “horizontal” transmission of genetic information and tumor transformation as a consequence [55, 56]. As DNA repair mechanisms are imperfect, the mutation rate varies in the limits of 10-4-10-8 mutation/gene per cell division [9, 11, 57], increases with age, and affects all tissues [11, 50, 58-61]. According to calculations, in a normal stem cell of the intestinal crypt more than 105 mutations are accumulated by the age of 65 years [62]. In intact mice, the mutation frequency rapidly increases with age, and this parameter is similar in all tissues of adult animals [63].
Based on the above-presented data mutagenesis, which is a driving force
of carcinogenesis, is considered to be a natural process affecting all
tissues of the organism and increasing with age. The dynamics of this
process may be estimated quantitatively. Based on general ideas about
the multistage pattern of carcinogenesis [15], in
the exponentially growing cell population the number of mutant cells
increases according to the formula:
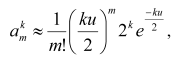
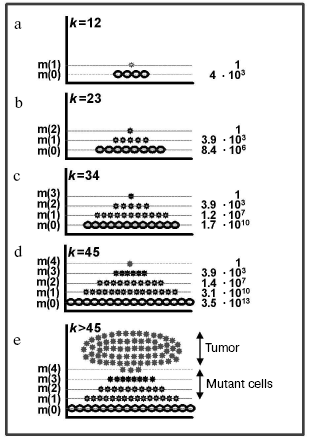
Figure 2 shows that, depending on the number of cell generations k and, respectively, size of the population (the total number of cells in it is presented as the m(0) layer), the number of cells increases with one, two, three, and four mutations (presented as the m(1), m(2), m(3), and m(4) layers, respectively; the last layer is conventionally taken as a threshold of the complete cellular transformation). Thus, on every stage of carcinogenesis, the law of quantity (of cells) transmission to quality is realized (the appearance of a cell with an additional mutation and, thus, having gone one more stage of the transformation). Each subsequent cell layer grows faster than the previous one, and the growth of the “pyramid” is accelerated due to reasons as follows: a) every new mutation provides the cells with a selective advantage which induces them to divide faster [10]; b) each subsequent layer is constantly fed up with the cell of the previous layer which is also constantly growing; c) on arising chromosomal instability [65] or a mutator phenotype [9, 66], the process gets an additional stimulus to the process. Thus, a preclinical stage of carcinogenesis may be likened to a pyramid growing out and up until appearance on its summit of the completely transformed cell which gives rise to tumor; and the whole process, including its clinical stage, is mushroom-like [67]: its “leg” and “cap” are hidden and manifested stages of the illness, respectively (Fig. 2e).
The completely transformed cell (a founder of the tumor clone) appears in this model system after 45 cell divisions (k = 45, the total number of cells in the population is 245 = 3.5*1013, transformational resistance of the cells is 4). At this moment, the population contains 3.9*103 cells with three mutations, 1.4*107 cells with two mutations, and 3.1*1010 cells with one mutation (Fig. 2d). In other words, the population is flooded with mutant cells (every thousandth cell has at least one mutation in one of the “cancerous” genes). But in a real situation the number of mutant cells has to be even higher, because this model neglects the cell death and the compensating cell divisions which are significantly increasing k. These calculations, although rather approximate, give an idea about the scale of the phenomenon (see also [62]) and are in agreement with experimental data [11].Fig. 2. Accumulation of mutant cells in the exponentially growing cell population: a-e) stages of carcinogenesis; k is the number of cell cycles; m(0) are normal cells; m(1), m(2), m(3), and m(4) are mutant cells with one, two, three, and four mutations, respectively, in the “cancerous” genes. The number of cells in each layer is indicated to the right (see text).
Thus, the tumor focus at the moment of its appearance is only the summit of an iceberg which has been maturing in the organism for decades and contains abundance of mutant cells at different stages of transformation [67]. This is also confirmed by clinical experience: clear signs of the illness are preceded by precancerous changes, such as hyperplasia, metaplasia, displasia [68, 69], which occupy a large tissue areas [70].
CANCER AS A MECHANISM OF NEGATIVE SELECTION OF MUTANT
ALLELES
Obviously, mutagenesis is concurrently an extensive and intensive process (characterized by the total number of mutant cells in the organism and the number of mutations in individual cells, respectively), and these parameters correlate: the larger is the damaged zone, the deeper is the injury of individual cells. And the opposite correlation is also true: the deeper is the mutational injury of individual, the most “advanced” cells (just from them tumor develops), the larger is, under similar other conditions, the zone of damage. Thus, in the general case, just the tumor emergence is a manifestation of a considerable damage of the organism by mutagenesis. Moreover, because the height and width of the mutational “pyramid” (Fig. 2) correlate quantitatively, the cell on the summit (i.e., the tumor cell) objectively functions as a “sensor” of mutagenesis. This means that the degree of mutational damage of the cell (the number of mutations accumulated in its genome) reflects the degree of mutational damage of the organism (the number of mutant cells accumulated in it).
It seems that transformational resistance of the cell during evolution adapts to specific features of a species (body weight, longevity) in such a way that tumor emerges just at the moment when the degree of mutational damage of the organism reaches a certain threshold. The death program is realized, and the genetically compromised individual threatening the population is eliminated. This process may be considered as altruistic (because the self-destruction of an individual occurs) and protective (because faulty alleles of crucially important genes undergo negative selection).
Obviously, the threat presented by the mutant individual for the population is not “horizontal” because cancer is not infectious, but “vertical”, i.e. associated with a possible appearance of genetically defective posterity and, thus, destabilization of the genofund. And indeed, if the tumor is a manifestation of mutagenesis which has occupied the total organism, germ cells should be also affected (available data support the concept that “the mutant seed is in the mutant soma”). Thus, spontaneous mutations are similarly frequent in cultures of somatic and germinal human cells [58, 71]. Moreover, female germ cells are relatively stable (i.e., mutate similarly to somatic cells because of the usual cell structure and absence of postnatal proliferation), whereas sperm cells display an extreme mutability because of a relative lack of protective cytoplasmic structures, DNA repair, and apoptosis at certain stages of development [11, 72-74]. There is also an unclear phenomenon: sometimes mutant sperm cells are selectively preferable as compared to the intact cells for fertilization of an oocyte, and this increases the probability of transmission of faulty alleles to the posterity [75, 76].
Thus, the essence of the proposed hypothesis is as follows: on accumulation in the somatic “sensor” cell of a critical set of genetic errors indicating a menacingly high level of mutagenesis of the organism's cells (including most importantly the germinal cells), a mechanism is triggered of self-elimination of the faulty individual by means of proliferation and spreading over the total organism of cells possessing killer function. Thus, the transformed cell plays a triple role: a) sensor of mutagenesis; b) executioner of the killer function; c) founder of the clone of killer cells that accelerates the realization of the program. If on the cellular level cancer cells appear as aggressive egoists (“cheats” [15]), on the organism's level the process appears as an altruistic act of elimination of mutant alleles of crucially important genes from the population genofund.
Inheritable syndromes. If the concept about cancer as of a local manifestation of generalized mutagenesis is a more or less reasonable hypothesis in the case of sporadic tumors, it is a statement of the generally known situation in the case of inheritable tumors [77, 78]. In a carrier of such tumors, all cells including the germinal ones are initially mutant, and carcinogenesis gets great “odds” as compared to wild type individuals [13] (in other words, the trigger of killer function is initially shifted one position ahead, and this decreases the chance of the carrier escaping the illness). It is also clear that inevitable in this case mutations of germ cells are the most threatening for the population genofund. A stable association of these two phenomena (the high menace for the population genofund, on one hand, and the high risk of cancer development, on the other hand) seems to be not casual, but, on the contrary, is based on causal-consequence relationships. Apparently, persistent mutagenesis of key genes of germ cells gave birth to the cancer phenomenon as a mechanism of their elimination from the population genofund. Hereditary tumors with their hard association of “mutant seed in mutant soma” seem to be the most convincing argument in favor of the hypothesis about the altruistic function of cancer (the altruism is presented by self-elimination of a carrier of the mutant allele during reproduction age). Germinal mutations of some crucial genes, which inevitably appear in the population with a certain background frequency, seem to be a motive force of the evolution of cancer.
There are many germinal mutations with phenotypic manifestations, but only a few of them (perhaps ~50 [79]) are associated with hereditary tumors. This prohibition seems to concern not every mutation but only those, which affect crucial genes (gatekeepers and caretakers, in terms of Kinzler and Vogelstein [80]) and are the most menacing for genetic stability of the population. Many germinal mutations are simply unable to surpass the “sieve” of embryogenesis and result in an early interruption of pregnancy [11]. It seems that there are a number of barriers, which prevent the spreading of mutant alleles in the population, and cancer is only one of them.
Influence on genetic structure of the population. This hypothesis predicts that cancer has to prevent the expansion in the population of mutant alleles of some vitally important genes. The paradox is that such an effect, namely, the strong effect of cancer morbidity on genetic structure of the population has been well known for a long time. Thus, risk of cancer during the life of persons with germinal mutations of suppressor genes is 50-80%, and tumors, sometimes multiple, appear during the reproduction period [13]. Due to extreme selective pressure, the alleles predisposing to the illness are very rare (~1 : 1000 or lower), and the fraction of these forms in the total morbidity is no more than 1-2%, which seems to indicate the efficiency of cancer as a mechanism of negative selection.
Germinal mutations of functionally important genes should strongly stimulate the appearance of a mechanism to prevent their spreading over the population. A less strong but also important stimulus is presented by gene polymorphism and existence of many normal but slightly attenuated forms [81-84]. In addition to rare alleles with a strong inherited predisposition to cancer, which is the reason for their being literally extirpated from the population, there are many alleles with a weak predisposition, but their cumulative effect can be rather considerable [13, 81, 84]. The frequency of such alleles in the population inversely depends on degree of the cancer risk associated with them.
What would be if cancer did not exist? Most likely, it would be unfavorable for the species because it would promote destabilization of the genofund (see the above-presented data on population genetics). A recent theoretical study also confirms this conclusion: mathematical modeling and computerized simulation were used to assess consequences of increased transformational resistance of human cells during 104 generations [85]. Along with a slight initial decrease in the cancer-caused mortality, it would lead to spreading of mutant alleles in the population and a significant increase in general morbidity.
Cancer in youth and old age. A favorable trait is fixed in the population if it manifests itself during the reproduction period. But cancer is mainly an illness of elderly humans and relatively seldom occurs in young persons, and this seems to obviously contradict the hypothesis that postulates its evolutionary reasonability. However, this contradiction is only apparent: cancer incidence in young persons is not high just due to effective negative selection of harmful alleles that constantly arise in the population but are present in it only on the background level. As to high cancer incidence in elderly humans, which is clearly unreasonable for evolution, it may be explained from the viewpoint of the theory of antagonistic pleiotropy [86]. The postponed negative consequences of genetic gains, which were advantageous in youth, are manifested in old age. In other words, the genetic program which was positive in youth continues its operating in old age, even if it seems not reasonable, simply because of lack of correction (mechanisms of evolution do not act during the post-reproduction period [87]). The age-related accumulation of mutations should be also added, as well as weakening of repair systems, and these result in exponential growth of cancer morbidity at the age of 40-80 years [58, 60].
Local effects in carcinogenesis. The concept that tumor is a local manifestation of generalized mutagenesis apparently contradicts numerous cases of tumors that obviously developed because of a local exposure: inflammation, bacterial infection [54, 88], ultraviolet radiation [47]. Occupational cancer and induction of tumors in animals confirm that this association is doubtless.
We think that the association of local exposure with appearance of the local tumor is a side consequence of the double role played by the cancer cell: it is a sensor of the general mutagenesis and executor of the death program. In this mechanism, local fluctuations can trigger a fatal cascade even at the low mutational background (the situation is similar to a thermoregulator which has to react to temperature in the total system but is operating in response to a local heating). In fact, a hypothetical sensor/executor seems to operate under conditions of constant “noise”: hindrances and false switchings on caused by the presence of multiple local mutagens in both external and internal media of the organism. On assuming the hypothesis that cancer has appeared during evolution to protect the population genofund, data on human cancer morbidity make us agree that this mechanism is operating too actively and exceeds the limits of its initial purpose. Both the high incidence of cancer in old age and numerous cases of cancer in response to local exposure are manifestations of this “hyper-reactivity” which shades the true nature of this phenomenon. Cancer seems to operate preventively, according to the principle: “better to kill millions of innocents than to omit a guilty one”. Apoptosis acts also preventively, and weakening of such an outstripping defense can lead to very bad consequences [89, 90]. From the evolutionary viewpoint, to provide the genetic stability of the population seems to be the most important, notwithstanding considerable individual losses associated with the rather rude mechanism chosen for realization of the purpose.
During recent years, views on carcinogenesis have changed in turn. Earlier all “beginnings and ends” of carcinogenesis were thought to be hidden inside the cancer cell itself, but today studies are extended far beyond the cancer cell. It has been a discovery to comprehend that a tumor can survive, grow, and spread over the organism only due to unnatural support by normal tissues [26, 91-93]. However, to elucidate tumor/organism interrelationship, it is still necessary to find the mechanism of the killer function of the cancer cell. Just this unique specific feature imparts malignancies their extreme significance. Being deprived of this function (i.e. becoming benign), the tumor cell attracts no special attention either of society or basic science and clinical medicine. The current paradigm of malignant growth does not consider the killer function as a specific feature because it is believed to be an intrinsic property of tumor growth as it is and metastasizing; therefore, this function is not given the interest that it deserves.
The modern DNA array technology allows us to detect genetic changes and variations in gene expression [94] responsible for the acquisition of killer function by tumor cells. This can be realized by comparison, first, of benign and malignant tumors, and, second, malignant tumors with differently expressed killer function. The ability for invasion and metastasizing is the main difference of malignant tumors from benign ones. And although these features themselves do not explain the mechanism of the death, there is no doubt that their appearance is associated with a new quality of the process. Possibly, the ability for invasion and metastasizing, on one hand, and the killer function, on the other, are co-regulated and constitute the same “genetic module”. We think that just the coupling of these properties hides the killer function as a specific property of the cancer cell. This hypothesis can be tested by identifying the corresponding metabolic pathways followed by attempts to artificially uncouple them.
For applied purpose, discovery of the killer function mechanism is promising for detection of a new target for therapy, and this target would be specific just for the tumor (that is, foreign for normal tissues), and, as a result, unassociated with side effects. The current antitumor therapy is based, without especial success, on the imperative of “destroying the vile creature” (the tumor cell). To neutralize this cell, that is, inhibit its pathways of affecting normal tissues, is the essence of a supposed alternative strategy, which might be more efficient.
The author is grateful to G. A. Sardanashvily for help in mathematical calculations and to V. E. Gurtsevitch and B. P. Kopnin for critical discussion.
This work was supported by the Russian Foundation for Basic Research (project No. 04-04-48094).
REFERENCES
1.Skulachev, V. P. (2001) Exp. Gerontol.,
36, 995-1024.
2.Ameisen, J. C. (2002) Cell Death Differ.,
9, 367-393.
3.Hanahan, D., and Weinberg, R. A. (2000)
Cell, 100, 57-70.
4.Tisdale, M. J. (2002) Nat. Rev. Cancer,
2, 862-871.
5.Lichtenstein, A. V. (2005) Cancer Cell Int.,
5, 5 (Open access http://www.cancerci.com/content/5/1/5).
6.Leroi, A. M., Koufopanou, V., and Burt, A. (2003)
Nat. Rev. Cancer, 3, 226-231.
7.Armitage, P., and Doll, B. (1954) Br. J.
Cancer, 8, 1-12.
8.Frank, S. A. (2004) Proc. Natl. Acad. Sci.
USA, 101, 8061-8065.
9.Loeb, L. A. (2001) Cancer Res., 61,
3230-3239.
10.Tomlinson, I. P., Novelli, M. R., and Bodmer, W.
F. (1996) Proc. Natl. Acad. Sci. USA, 93,
14800-14803.
11.Vijg, J. (2000) Mutat. Res., 447,
117-135.
12.Futreal, P. A., Coin, L., Marshall, M., Down, T.,
Hubbard, T., Wooster, R., Rahman, N., and Stratton, M. R. (2004)
Nat. Rev. Cancer, 4, 177-183.
13.Ponder, B. A. (2001) Nature, 411,
336-341.
14.Peto, R., Roe, F. J., Lee, P. N., Levy, L., and
Clack, J. (1975) Br. J. Cancer, 32, 411-426.
15.Nunney, L. (1999) Proc. Roy. Soc. (L.) Br.
Biol. Sci., 266, 493-498.
16.Hoeijmakers, J. H. (2001) Nature,
411, 366-374.
17.Hahn, W. C., and Weinberg, R. A. (2002) Nat.
Rev. Cancer, 2, 331-341.
18.Hahn, W. C., Counter, C. M., Lundberg, A. S.,
Beijersbergen, R. L., Brooks, M. W., and Weinberg, R. A. (1999)
Nature, 400, 464-468.
19.Renan, M. J. (1993) Mol. Carcinog.,
7, 139-146.
20.Skulachev, V. P. (2002) Ann. N. Y. Acad.
Sci., 959, 214-237.
21.Weinberg, R. A. (1997) Cell, 88,
573-575.
22.Bergers, G., and Benjamin, L. E. (2003) Nat.
Rev. Cancer, 3, 401-410.
23.Bissell, M. J., and Radisky, D. (2001) Nat.
Rev. Cancer, 1, 46-54.
24.Elenbaas, B., and Weinberg, R. A. (2001) Exp.
Cell Res., 264, 169-184.
25.Friedl, P., and Wolf, K. (2003) Nat. Rev.
Cancer, 3, 362-374.
26.Jacks, T., and Weinberg, R. A. (2002)
Cell, 111, 923-925.
27.Liotta, L. A., and Kohn, E. C. (2001)
Nature, 411, 375-379.
28.Finora, K. (2003) Clin. Tech. Small Anim.
Pract., 18, 123-126.
29.Posner, J. B. (2003) Ann. N. Y. Acad.
Sci., 998, 178-186.
30.Sato, K., Onuma, E., Yocum, R. C., and Ogata, E.
(2003) Semin. Oncol., 30, 167-173.
31.Yamada, G., Ohguro, H., Aketa, K., Itoh, T.,
Shijubo, N., Takahashi, H., Fujiwara, O., Satoh, M., Ohtsuka, K., and
Abe, S. (2003) Hum. Pathol., 34, 717-719.
32.Kim, Y. T., Rha, S. Y., Shim, C. Y., Sohn, J. H.,
Kim, C., Yu, N. C., Chung, H. C., Kim, J. H., Han, D. S., Kim, B. S.,
and Roh, J. K. (2003) Yonsei Med. J., 44, 539-543.
33.Kritchevsky, S. B., Wilcosky, T. C., Morris, D.
L., Truong, K. N., and Tyroler, H. A. (1991) Cancer Res.,
51, 3198-3203.
34.Grosvenor, M., Bulcavage, L., and Chlebowski, R.
T. (1989) Cancer, 63, 330-334.
35.Albert, M. L., and Darnell, R. B. (2004) Nat.
Rev. Cancer, 4, 36-44.
36.Tracey, K. J., Wei, H., Manogue, K. R., Fong, Y.,
Hesse, D. G., Nguyen, H. T., Kuo, G. C., Beutler, B., Cotran, R. S.,
and Cerami, A. (1988) J. Exp. Med., 167, 1211-1227.
37.Beutler, B., and Cerami, A. (1988) Ann. Rev.
Biochem., 57, 505-518.
38.Ou, Y. C., Yang, C. R., Ho, H. C., Cheng, C. L.,
Kao, Y. L., Su, C. K., Chiu, K. Y., and Chen, W. M. (2003) J. Chin.
Med. Assoc., 66, 537-543.
39.Reya, T., Morrison, S. J., Clarke, M. F., and
Weissman, I. L. (2001) Nature, 414, 105-111.
40.Green, D. R., and Evan, G. I. (2002) Cancer
Cell, 1, 19-30.
41.Bodmer, W. F., and Tomlinson, I. (1996) Ciba
Found. Symp., 197, 181-189.
42.Skulachev, V. P. (1999) Biochemistry
(Moscow), 64, 1418-1426.
43.Skulachev, V. P. (2000) IUBMB Life,
49, 365-373.
44.Hamilton, W. D. (1964) J. Theor. Biol.,
7, 1-52.
45.Maynard-Smith, J. (1964) Nature,
201, 1145-1147.
46.Gardner, A., and West, S. A. (2004)
Science, 305, 1413-1414.
47.Thilly, W. G. (2003) Nat. Genet.,
34, 255-259.
48.Turker, M. S. (1998) Semin. Cancer Biol.,
8, 407-419.
49.Johnson, F. B., Sinclair, D. A., and Guarente, L.
(1999) Cell, 96, 291-302.
50.Finkel, T., and Holbrook, N. J. (2000)
Nature, 408, 239-247.
51.Holzenberger, M., Dupont, J., Ducos, B., Leneuve,
P., Geloen, A., Even, P. C., Cervera, P., and Le Bouc, Y. (2003)
Nature, 421, 182-187.
52.Mathon, N. F., and Lloyd, A. C. (2001) Nat.
Rev. Cancer, 1, 203-213.
53.Tuma, R. (2001) Sci. Aging Knowledge
Environ., 2001, oa5.
54.Hussain, S. P., Hofseth, L. J., and Harris, C. C.
(2003) Nat. Rev. Cancer, 3, 276-285.
55.Bergsmedh, A., Szeles, A., Henriksson, M., Bratt,
A., Folkman, M. J., Spetz, A. L., and Holmgren, L. (2001) Proc.
Natl. Acad. Sci. USA, 98, 6407-6411.
56.Bergsmedh, A., Szeles, A., Spetz, A. L., and
Holmgren, L. (2002) Cancer Res., 62, 575-579.
57.Chicurel, M. (2001) Sci. Aging Knowledge
Environ., 2001, oa3.
58.Simpson, A. J. (1997) Adv. Cancer Res.,
71, 209-240.
59.Campisi, J. (1997) J. Am. Geriatr. Soc.,
45, 482-488.
60.DePinho, R. A. (2000) Nature, 408,
248-254.
61.Dolle, M. E., Snyder, W. K., Gossen, J. A.,
Lohman, P. H., and Vijg, J. (2000) Proc. Natl. Acad. Sci. USA,
97, 8403-8408.
62.Tomlinson, I., Sasieni, P., and Bodmer, W. (2002)
Am. J. Pathol., 160, 755-758.
63.Heddle, J. A., and Swiger, R. R. (1996) Mutat.
Res., 365, 107-117.
64.Frank, S. A., and Nowak, M. A. (2003)
Nature, 422, 494.
65.Nowak, M. A., Komarova, N. L., Sengupta, A.,
Jallepalli, P. V., Shih, I., Vogelstein, B., and Lengauer, C. (2002)
Proc. Natl. Acad. Sci. USA, 99, 16226-16231.
66.Loeb, K. R., and Loeb, L. A. (2000)
Carcinogenesis, 21, 379-385.
67.Lichtenstein, A. V., and Potapova, G. I. (2003)
Mol. Biol. (Moscow), 37, 181-193.
68.Takayama, T., Katsuki, S., Takahashi, Y., Ohi,
M., Nojiri, S., Sakamaki, S., Kato, J., Kogawa, K., Miyake, H., and
Niitsu, Y. (1998) N. Engl. J. Med., 339, 1277-1284.
69.Shabad, L. M. (1967) in Precancer in
Experimental and Morphological Aspect [in Russian], Meditsina,
Moscow, pp. 352-373.
70.Braakhuis, B. J. M., Tabor, M. P., Kummer, J. A.,
Leemans, C. R., and Brakenhoff, R. H. (2003) Cancer Res.,
63, 1727-1730.
71.Kuick, R. D., Neel, J. V., Strahler, J. R., Chu,
E. H., Bargal, R., Fox, D. A., and Hanash, S. M. (1992) Proc. Natl.
Acad. Sci. USA, 89, 7036-7040.
72.Aitken, R. J., Koopman, P., and Lewis, S. E. M.
(2004) Nature, 432, 48-52.
73.Cleaver, J. E., Karplus, K., Kashani-Sabet, M.,
and Limoli, C. L. (2001) Mutat. Res., 485, 23-36.
74.Masters, J. R., and Koberle, B. (2003) Nat.
Rev. Cancer, 3, 517-525.
75.Goriely, A., McVean, G. A., Rojmyr, M.,
Ingemarsson, B., and Wilkie, A. O. (2003) Science, 301,
643-646.
76.Tiemann-Boege, I., Navidi, W., Grewal, R., Cohn,
D., Eskenazi, B., Wyrobek, A. J., and Arnheim, N. (2002) Proc. Natl.
Acad. Sci. USA, 99, 14952-14957.
77.Knudson, A. G. (1997) Ann. N. Y. Acad.
Sci., 833, 58-67.
78.Knudson, A. G. (2002) Am. J. Med. Genet.,
111, 96-102.
79.Knudson, A. G. (1993) Proc. Natl. Acad. Sci.
USA, 90, 10914-10921.
80.Kinzler, K. W., and Vogelstein, B. (1997)
Nature, 386, 761-763.
81.Demant, P. (2003) Nat. Rev. Genet.,
4, 721-734.
82.Kuroda, Y., Tsukino, H., Nakao, H., Imai, H., and
Katoh, T. (2003) Cancer Lett., 189, 77-83.
83.Ho, G. Y., Melman, A., Liu, S. M., Li, M., Yu,
H., Negassa, A., Burk, R. D., Hsing, A. W., Ghavamian, R., and Chua, S.
C., Jr. (2003) Br. J. Cancer, 88, 263-269.
84.Peto, J., and Houlston, R. S. (2001) Eur. J.
Cancer, 37, Suppl. 8, S88-S96.
85.Frank, S. A. (2004) Proc. Natl. Acad. Sci.
USA, 101, 8061-8065.
86.Kirkwood, T. B. (2005) Cell, 120,
437-447.
87.Campisi, J. (2003) Nat. Rev. Cancer,
3, 339-349.
88.Coussens, L. M., and Werb, Z. (2002)
Nature, 420, 860-867.
89.Umansky, S. R. (1982) J. Theor. Biol.,
97, 591-602.
90.Camplejohn, R. S., Gilchrist, R., Easton, D.,
McKenzie-Edwards, E., Barnes, D. M., Eccles, D. M., Ardern-Jones, A.,
Hodgson, S. V., Duddy, P. M., and Eeles, R. A. (2003) Br. J.
Cancer, 88, 487-490.
91.Hendrix, M. J., Seftor, E. A., Hess, A. R., and
Seftor, R. E. (2003) Nat. Rev. Cancer, 3, 411-421.
92.Kalluri, R. (2003) Nat. Rev. Cancer,
3, 422-433.
93.Bhowmick, N. A., Chytil, A., Plieth, D., Gorska,
A. E., Dumont, N., Shappell, S., Washington, M. K., Neilson, E. G., and
Moses, H. L. (2004) Science, 303, 848-851.
94.Shyamsundar, R., Kim, Y., Higgins, J.,
Montgomery, K., Jorden, M., Sethuraman, A., van de Rijn, M., Botstein,
D., Brown, P., and Pollack, J. (2005) Genome Biol., 6,
R22.
95.Knudson, A. G. (1996) J. Cancer Res. Clin.
Oncol., 122, 135-140.