Codon 12 Region of Mouse K-ras Gene Is the Site for in vitro Binding of Transcription Factors GATA-6 and NF-Y
E. V. Gorshkova*, V. I. Kaledin, V. F. Kobzev, and T. I. Merkulova
Institute of Cytology and Genetics, Siberian Division of the Russian Academy of Sciences, pr. Lavrentieva 10, 630090 Novosibirsk, Russia; fax: (3832) 33-1278; E-mail: schly@mail.ru* To whom correspondence should be addressed.
Received April 18, 2005; Revision received June 16, 2005
Codon 12 of the K-ras gene is a generally recognized example of a mutational hot spot. By the approach of gel retardation and specific antibodies, a double-stranded oligonucleotide corresponding to the codon 12 region of the mouse K-ras gene (from 20 to 50 bp with respect to the exon 1 start) was found to be a site for cooperative binding of the transcription factors GATA-6 and NF-Y. GATA-6 and NF-Y were selectively activated with lung carcinogens 3-methylcholanthrene and nitrosoethylurea in mice of strains susceptible to lung tumorigenesis but not in animals of resistant strains. The interaction of GATA-6 and NF-Y with the codon 12 region of the K-ras gene is suggested to be involved in the mechanism of lung carcinogenesis.
KEY WORDS: K-ras gene, codon 12, transcription factors GATA-6 and NF-Y, lung carcinogenesis
Abbreviations: NU) nitrosoethylurea; 3-MC) 3-methylcholanthrene.
K-ras is a member of the small family of Ras genes
encoding closely cognate proteins H-, N-, and K-Ras which are
low-molecular-weight (~21 kD) GTPases involved in cell growth and
differentiation. These proteins are located on the inner surface of the
plasma membrane and exist in alternating active and inactive states.
The transition of Ras from the inactive GDP-bound state into the active
GTP-bound state is stimulated by guanine nucleotide exchange factors
(GNEFs) and the reverse transition is stimulated by GTPase-activating
proteins (GAPs). And these factors are influenced by a number of
surface cellular receptors, in particular, receptors of growth factors
and cytokines [1]. On activation with external
stimuli, Ras can interact with many effector proteins: Raf kinases, PI3
kinases, some isoforms of protein kinase C, etc., and switch on the
so-called kinase cascades which play an important role in regulation of
gene expression [1, 2].
Mutations in the Ras family genes have been detected in various malignant tumors of humans and experimental animals mainly in codons 12, 13, and 61 [3]. Such mutations are known to decrease the GTPase activity of Ras proteins and disturb their ability for binding GAPs and thus maintain the permanently activated state of Ras that promotes cell malignant transformation [4]. The transition G→A in the second position of codon 12 of the K-ras gene is the most common mutation. The incidence of this and other less common nucleotide substitutions in the codon 12 region of the K-ras gene is up to 75-100% in cases of pancreas cancer, 50-80% in cases of large intestine cancer, and to 25-50% in human lung cancer [5-7].
Studies on the K-ras gene in mice are of interest because various spontaneous and chemically induced tumors are associated with mutations in the same codons as observed in humans. In particular, mutations in codon 12 of the K-ras gene were found in 60-95% of mouse lung tumors induced by chemical carcinogens [8, 9]. Moreover, it is very likely that these mutations are already enough to give rise to tumorigenesis, because using of the Cre-lox mediated somatic recombination to introduce activated K-ras expression in lung epithelium of transgenic mice was sufficient for development of lung adenocarcinoma in 100% of animals [10].
Mechanisms responsible for high incidence of mutations in codon 12 of the K-ras gene in tumors are unclear. On one hand, the presence of an altered Ras seems to provide selective advantages for the cells due to increase in their ability for proliferation and resistance to apoptosis [11]. On the other hand, codon 12 of the K-ras gene is the place of preferential formation of adducts in response to treatment with carcinogens [12, 13]. It was suggested that the selectivity in adduct production in codon 12 should be associated with both specific features of the primary structure of DNA and/or its modifications in the region of this codon and specific features of the local chromatin structure in this region as a result of DNA-protein interactions [13].
Therefore, we have studied the binding of proteins of extracts of lung cell nuclei of mice with an oligonucleotide corresponding to the codon 12 region of the mouse K-ras gene and also the effects of lung carcinogens 3-methylcholanthrene (3-MC) and nitrosoethylurea (NU) on the DNA-binding activity and expression of proteins interacting with this region in mice of strains susceptible and resistant to lung carcinogenesis.
MATERIALS AND METHODS
Reagents. The following reagents were used: [alpha-32P]dATP and ECL kit from Amersham (England); Retina X-ray film (Germany); PVDF Immobilon-P membrane (Millipore, USA); Klenow fragment of DNA polymerase I from E. coli (SibEnzyme, Russia); DEAE-81 paper, Whatman 3MM (Whatman, England); Tris, HEPES, spermine, spermidine, and Bromophenol Blue (Sigma, USA); acrylamide and bis-acrylamide (Reanal, Hungary); SDS (Bio-Rad, USA); EGTA, phenylmethylsulfonyl fluoride (PMSF), and EDTA (Fluka, Switzerland); sucrose and glycerol (Serva, Germany). Other reagents were of domestic production, of special purity and chemical purity.
Animals. Two-week-old mice of the strains A/He, ICR, C3H/A, and C57BL bred in the Institute of Cytology and Genetics, Siberian Division of the Russian Academy of Sciences, were used.
Preparation of extracts from lung cell nuclei and gel retardation of oligonucleotides. Extracts of nuclei were prepared as described in [14] in the modification [15]. All procedures were performed on ice as quickly as possible. To study the effects of carcinogens on the binding of transcriptional factors to DNA, the mice were injected intraperitoneally 12 h before the sacrifice with NU or 3-MC (80 mg per kg body weight) dissolved in 10% PEG-1500 or olive oil, respectively.
Oligonucleotides corresponding to both chains of the region from 20 to 50 bp of exon 1 of the mouse K-ras gene:
KR, 5´- CAGTTGGTGGTTGGAGCTGGTGGCGTAGGCAAGAG-3´
(codon 12 is underlined) and also to binding sites of the transcription factors:
GATA-6, 5´-cagtGATCTCCGGCAACTGATAAGGATTCCCTG-3´ [16];
Ets, 5´-cagtTCGAACTTCCTGCTCGA-3´ [17];
NF-Y, 5´-CAGTAGACCGTACGTGATTGGTTAATCTCTT-3´;
LKLF, 5´-CAGTAATAGGGATGGGCAGAAGGCAG-3´
(sequences NF-Y and LKLF correspond to oligonucleotides for gel shift of Santa Cruz Biotechnology, Inc., USA) were synthesized using an ASM-102I automatic synthesizer (Biosset, Novosibirsk) by the H-phosphonate method [18]. After annealing, the oligonucleotides were labeled with [alpha-32P]dATP using Klenow fragment of DNA polymerase I. The reaction mixture (16 µl) contained 1 ng labeled oligonucleotide, 25 mM HEPES (pH 7.8), 80 mM KCl, 0.2 mM EDTA, 0.2 mM EGTA, 1 mM dithiothreitol, 10% glycerol, 2 µg protein of the nuclear extract preincubated with sheared salmon sperm DNA (1 µg per 7 µg protein) for 10 min at 0°C. The mixture was incubated at 14°C for 15 min and then subjected to electrophoresis in 5% polyacrylamide gel in 0.5-fold TBE buffer (89 mM Tris-borate, 89 mM H3BO3, 2 mM EDTA). When antibodies to GATA-6 and NF-YA (Santa Cruz Biotechnology, Inc.) were used, the extract of nuclei was incubated concurrently with sheared salmon sperm DNA and 1-2 µl of the antibodies for 15 min at 0°C.
Western-blotting. Proteins were separated by electrophoresis by the Laemmli method [19] in 10% polyacrylamide gel, placing onto each lane 5 µg nuclear extract from mouse lung cells. The separated proteins were carried from the gel onto a PVDF Immobilon-P membrane using a TE-70 device (Amersham, USA). The bands were treated with antibodies and detected with an ECL kit (Amersham) according to the manufacturer's instruction.
RESULTS AND DISCUSSION
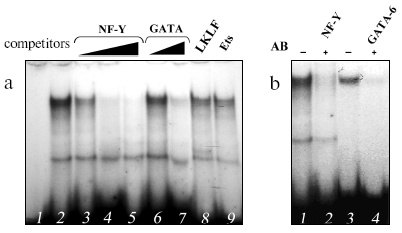
Binding of proteins of extract from mouse lung cell nuclei to the double-chained oligonucleotide corresponding to the codon 12 region of the mouse K-ras gene (from 20 to 50 bp of exon 1, the KR oligonucleotide) was studied using gel retardation. A protein (or proteins) formed a complex with this oligonucleotide (Fig. 1a). To identify this protein, the sequence of the oligonucleotide was searched for regions homologous to sites for binding different transcription factors using the TESS program packet [20]. Regions were found that are similar to the binding sites for transcription factors of the GATA family, the ubiquitous transcription factor NF-Y, and the lung-specific transcription factor LKLF.
Double-stranded oligonucleotides corresponding to the known binding sites for these proteins were used as competitors in experiments with gel retardation of DNA sample. An oligonucleotide corresponding to the binding site of transcription factors for the Ets family was taken as an obvious nonspecific competitor. Two oligonucleotides competing for complexing with KR were found: they contained the binding sites for NF-Y and GATA (Fig. 1a) that suggests the involvement of both nucleotides in the complexing. Experiments with antibodies to one of NF-Y subunits (NF-YA) and GATA-6 (the main representative of the GATA family in lungs [21]) completely verified this suggestion (Fig. 1b). The addition of these antibodies resulted in virtually complete disappearance of the complex; therefore, the transcription factors NF-Y and GATA-6 are likely to cooperatively bind to the codon 12 region of the mouse K-ras gene.Fig. 1. Identification by gel retardation of proteins complexing with the oligonucleotide KR corresponding to the codon 12 region of the K-ras gene. a) Competitive analysis: 1) free probe; 2) binding to proteins of the extract from lung cell nuclei in the absence of competitive oligonucleotides; 3-5) in the presence of 25-, 50-, and 100-fold excess oligonucleotide corresponding to the binding site for NF-Y; 6, 7) in the presence of 50- and 100-fold excess oligonucleotide corresponding to the binding site for GATA; 8, 9) in the presence of 100-fold excess oligonucleotides corresponding to the binding sites for LKLF and Ets. b) Suppression with antibodies of the binding of transcription factors: 1, 3) binding to the extract; 2) binding to the extract in the presence of antibodies to NF-Y; 4) binding to the extract in the presence of antibodies to GATA-6.
NF-Y is known to be a key regulator of the cell cycle [22], and GATA-6 is one of the main factors controlling ontogenesis of pulmonary tissue [21]. Both poor and excess expression of GATA-6 leads to serious disorders in lung morphogenesis [23]. Therefore, it was suggested that binding of the transcription factors NF-Y and GATA-6 to the location of a mutational hot spot should be involved in events initiating tumorigenesis in this organ. Based on this hypothesis, we have studied effects of potent lung carcinogens 3-MC and NU on activities of these factors and their abilities for interaction with the KR oligonucleotide.
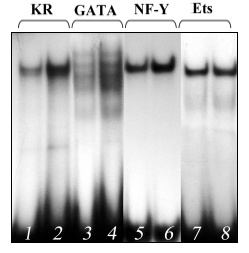
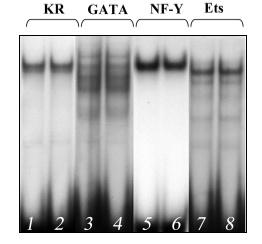
Injection of the carcinogen 3-MC into mice of the ICR strains highly susceptible to lung carcinogenesis [24] was accompanied by a noticeable increase in the DNA-binding activity of GATA-6 and NF-Y of extracts from lung cell nuclei (Fig. 2, lanes 4 and 6) and, respectively, in their interaction with the codon 12 region of the mouse K-ras gene (Fig. 2, lane 2). The carcinogen influenced the binding of NF-Y and GATA-6 to the codon 12 region much more strongly than the oligonucleotides corresponding to the binding sites for these transcription factors, and this also suggested their cooperative interaction with this region. This effect is specific because 3-MC did not alter the DNA-binding activity of the transcription factor Ets, which in this case might be considered to be an internal control (Fig. 2, lane 8). Injection of 3-MC into C57BL mice resistant to lung carcinogenesis [25] did not increase the activities of NF-Y and GATA-6 (Fig. 3). Similar results were obtained on two other strains of mice, A/He and C3H, contrasting in susceptibility to lung cancer [25]. Another lung carcinogen, NU, also increased the DNA-binding activity of the NF-Y and GATA-6 factors in lungs of the tumor susceptible mice (the ICR and A/He strains) but not in the tumor resistant mice (the C57BL and C3H strains) (data not presented).
Fig. 2. Increased binding of proteins of extract from lung cell nuclei of 3-MC-treated ICR mice susceptible to lung carcinogenesis to the following oligonucleotides: 1, 2) oligonucleotide KR; 3, 4) oligonucleotide corresponding to the binding site for GATA; 5, 6) oligonucleotide corresponding to the binding site for NF-Y; 7, 8) oligonucleotide corresponding to the binding site for Ets.
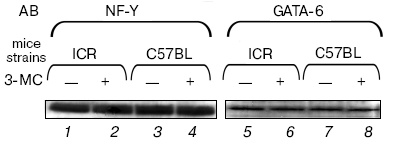
We have shown by Western-blotting hybridization that the observed effect is not associated with changes in concentrations of the transcription factors NF-Y and GATA-6 in the nuclear extracts (Fig. 4). Therefore, it is suggested that the increase in their DNA-binding activities under the influence of carcinogen in the lungs of susceptible mice should be caused by modifications of these proteins and not increase in their concentrations.Fig. 3. Binding of proteins of extract from lung cell nuclei of 3-MC-treated C57BL mice resistant to lung carcinogenesis with the following oligonucleotides: 1, 2) oligonucleotide KR; 3, 4) oligonucleotide corresponding to the binding site for GATA; 5, 6) oligonucleotide corresponding to the binding site for NF-Y; 7, 8) oligonucleotide corresponding to the binding site for Ets.
Thus, studies on the codon 12 region of exon 1 of the K-ras gene revealed the presence of binding sites for the transcription factors GATA-6 and NF-Y and the cooperativity of these factors in the binding to DNA. Thus, this region is suggested to be a composite regulatory element. At present, a number of composite regulatory elements are known which are produced by the binding sites for NF-Y and GATA [26, 27]. In particular, GATA-6 and NF-Y have been shown interact in the binding to the corresponding composite element, and this binding is necessary for expression of the gene encoding the von Willebrand factor in non-endothelial cells [28].Fig. 4. Comparative determination by Western-blotting of concentrations of factors NF-Y (1-4) and GATA-6 (5-8) in extracts from lung cell nuclei of ICR (1, 2, 5, 6) and C57BL mice (3, 4, 7, 8) treated (2, 4, 6, 8) and untreated with 3-MC (1, 3, 5, 7). AB, antibodies; -, control mice; +, mice treated with NU.
The selectivity of production of adducts in codon 12 of the K-ras gene upon treatment with various carcinogens was recently shown to be caused by both specific features of the primary structure of DNA in the region of this codon [12] and yet unknown modifications in the native chromatin structure [13]. Both NF-Y and factors of the GATA family can initiate destruction or remodeling of nucleosomes [29, 30]; therefore, it is suggested that binding of these factors with the mutational hot spot region in the K-ras gene accompanied by local changes in the chromatin structure should play a role in providing the availability of DNA for both reactive carcinogens and modifying enzymes and, thus, contribute to the selectivity of adduct production.
In this connection, we consider as very interesting our data on the increase in the DNA-binding activity of NF-Y and GATA-6 under the influence of lung carcinogens in the nuclei of lung cells of susceptible mice but not of resistant animals and on the sharply increased binding of these factors to the oligonucleotide corresponding to the codon 12 region of the mouse K-ras gene. This seems to be important for mutations in this codon and subsequent development of lung cancer in the susceptible animals.
We will further study the in vivo effect of lung carcinogens on the binding of NF-Y and GATA-6 to the codon 12 region of the K-ras gene in the lungs of susceptible and resistant strains of mice.
The work was supported by the Russian Foundation for Basic Research (the project No. 04-04-48136).
REFERENCES
1.Ellis, C. A., and Clark, G. (2000) Cellular
Signaling, 12, 425-434.
2.Macara, I. G., Lounsbury, K. M., Richards, S. A.,
McKierman, C., and Bar-Sagi, D. (1996) FASEB J., 10,
625-630.
3.Bos, J. L. (1989) Cancer Res., 49,
4682-4689.
4.Trahey, M., and McCormick, F. (1987)
Science, 238, 542-545.
5.Mu, D. Q., Peng, Y. S., and Xu, Q. J. (2004)
World J. Gastroenterol., 10, 471-475.
6.Minamoto, T., Mai, M., and Ronai, Z. (2000)
Cancer Detection and Prevention, 24, 1-12.
7.Johnson, L., Mercer, K., Greenboum, D., Bronson, R.
T., Crowley, D., Tuveson, D. A., and Jacks, T. (2001) Nature,
410, 1111-1115.
8.Ramakrishna, G., Biakovska, A., Perella, C.,
Birely, L., Fornwald, L. W., Diwan, B. A., Schiao, Y.-H., and Anderson,
L. M. (2000) Mol. Carcinogenesis, 28, 156-167.
9.Matzinger, S. A., Chen, B., Wang, Y., Crist, K. A.,
Stoner, G. D., Kellof, G. J., Lubet, R. A., and You, M. (1997)
Gene, 188, 261-269.
10.Meuwissen, R., Linn, S. C., van der Valk, M.,
Mooi, W. J., and Berns, A. (2001) Oncogene, 20,
6551-6558.
11.Koera, K., Nakamura, K., Nakao, K., Miyoshi, J.,
Toyoshima, K., Hatta, T., Otani, H., Aiba, A., and Katsuki, M.
(1997) Oncogene, 15, 1151-1159.
12.Feng, Z., Hu, W., Komissarova, E., Pao, A., Hung,
M. C., Adair, G. M., and Tang, M. S. (2002) J. Biol. Chem.,
277, 12777-12783.
13.Hu, W., Feng, Z., and Tang, M. S. (2003)
Biochemistry, 42, 10012-10023.
14.Gorski, K., Carnero, M., and Schibler, U. (1986)
Cell, 47, 767-776.
15.Shapiro, D. G., Sharp, P. A., Wahli, W. W., and
Keller, M. J. (1988) DNA, 7, 47-55.
16.Yamamoto, M., Ko, L. J., Leonard, M. V., Beug,
H., Orkin, S. H., and Engel, J. (1990) Genes Dev., 4,
1650-1662.
17.Espinas, M. L., Roux, J., Chysdeal, J., Pictet,
R., and Grange, T. (1994) Mol. Cell. Biol., 14,
4116-4125.
18.Kumarev, V. P., Kobzev, V. F., Kuznedelov, K. D.,
and Sredin, Yu. G. (1991) Nucleic Acids Res., 24,
234.
19.Laemmli, U. K. (1970) Nature, 227,
680-685.
20.Schug, J., and Overton, G. C. (1997) Technical
Report CBIL-TR-1997-1001-v 0.0, Comp. Biology and
Informatics Laboratory, School of Medicine, University of
Pennsylvania.
21.Molkentin, J. D. (2000) J. Biol. Chem.,
275, 38949-38952.
22.Elkon, R., Linhart, C., Sharan, R., Shamir, R.,
and Shiloh, Y. (2003) Genome Res., 13, 773-780.
23.Liu, C., Ikegami, M., Stahlman, M. T., Dey, C.
R., and Whitsett, J. A. (2003) Am. J. Physiol. Lung Cell. Mol.
Physiol., 285, 1246-1254.
24.Timofeeva, O. A., Filipenko, M. L., and Kaledin,
V. I. (1999) Genetika, 35, 1309-1312.
25.Shimkin, M. B., and Stoner, G. D. (1975) Adv.
Cancer Res., 21, 1-58.
26.Cassel, D. L., Subudhi, S. K., Surrey, S., and
McKenzie, S. E. (2000) Blood Cells Mol. Dis., 26,
587-597.
27.Huang, D. Y., Kuo, Y. Y., Lai, J. S., Suzuki, Y.,
Sugano, S., and Chang, Z. F. (2004) Nucleic Acids Res.,
32, 3935-3946.
28.Peng, Y., and Jahroudi, N. (2003) J. Biol.
Chem., 278, 8385-8394.
29.Coustry, F., Hu, Q., Crombrugghe, B., and Maity,
S. N. (2001) J. Biol. Chem., 276, 40621-40630.
30.Cirillo, L. N., Lin, F. R., Cuesta, I., Friedman,
D., Jarnik, M., and Zaret, K. S. (2002) Mol. Cell, 9,
279-289.