Investigation of Conformational Changes Induced by Binding of Pancreatic RNase to Anti-RNase IgG Derived Fab Monomer Using Optical Procedures
H. Younus1*, S. Jamal1, B. Ahmad1, and M. Saleemuddin1,2
1Interdisciplinary Biotechnology Unit and 2Department of Biochemistry, Faculty of Life Sciences, Aligarh Muslim University, Aligarh 202002, India; fax: +91-571-272-1776; E-mail: hinayounus@rediffmail.com; alg_btisamua@sancharnet.in* To whom correspondence should be addressed.
Received June 20, 2005; Revision received September 10, 2005
The conformational changes induced in Fab fragments of polyclonal anti-RNase antibody molecules obtained by digestion with papain as a result of binding of pancreatic RNase have been studied. The RNase-Fab complex (RN-Fab), being soluble, could be subjected to thermodynamic investigations using optical strategies, also because of the absence of tryptophan in RNase. Internalization of the chromophores (tryptophans and tyrosines) of Fab occurs when it binds to RNase, suggesting an increase in the compactness of Fab due to the binding of RNase.
KEY WORDS: antigen-antibody complex, Fab, RNase, conformational changesDOI: 10.1134/S0006297906020155
Abbreviations: RNase) bovine pancreatic ribonuclease A; Fab) antigen binding fragment; RN-Fab) RNase-Fab monomer complex; ELISA) enzyme-linked immunosorbent assay; HRP) horseradish peroxidase; CDRs) complementary determining regions.
Specific protein-protein interactions constitute the basis of many
biologically important recognition processes including specific
combination of antibodies with their protein antigens. Binding to
specific antibodies has been shown to improve the resistance of a
number of enzymes against various forms of inactivation, such as
temperature, pH, denaturants, and proteases [1-3], and is being explored for accomplishing enzyme
stabilization and immobilization for a variety of applications. Among
the principal strategies employed, formation of enzyme antibody adducts
or binding to supports precoupled with antibodies are important [4, 5]. While several polyclonal
and monoclonal antibodies have been shown to stabilize antigenic
enzymes, more recently the potential usefulness of epitope specific
antibodies has been demonstrated [6, 7]. While the majority of reports describe studies on
insoluble enzyme anti-enzyme antibody preparations, we have reported
that complexing of antigenic enzyme with monomeric Fab,
derived from IgG raised in rabbits, results in the improvement of the
stability of the former [8]. Such preparations have
potential in enzyme therapy.
Fab and Fc behave as independent regions within IgG and hence their different functions, i.e., antigen binding and adsorption to receptors/tissue, respectively, can be studied independently using isolated Fab and Fc fragments [9]. Analysis of kinetic data on hapten-antibody reaction suggested that the antibody molecule may undergo a conformational change upon interaction with the hapten [10]. Whether or not an antigen/antibody molecule retains its native conformation in the antigen-antibody complex is still an important but unresolved issue.
Large aggregates of antigen-antibody complexes may result when polyvalent antigen and polyclonal antibody interact [5, 11, 12]. A plethora of literature is now available on the usefulness of antigen binding Fab fragment in the purification of proteins [13] and in clinical use [14-16]. Binding of monomeric Fab fragment to protein does not result in the formation of large and insoluble aggregates due to the monovalent nature of the former. It was therefore envisaged that conformational changes in an antibody molecule upon binding antigen would be better studied using its Fab monomer since this Fab-antigen complex would be soluble and hence facilitate thermodynamics studies. RNase has been used as the antigen due to its unique absence of tryptophan [17]. It was therefore envisaged that tryptophan fluorescence studies of RNase-Fab complex (RN-Fab) would give information only on the conformational change of the Fab. Absorption spectroscopy has also been used to study the changes that occur in the Fab.
MATERIALS AND METHODS
RNase type I-A and papain were purchased from Sigma (USA). Goat anti-rabbit immunoglobulin (IgG)-peroxidase conjugate and tetramethyl benzidine/H2O2 were supplied by Genei Laboratories (India). Microtiter plates were purchased from Granier (USA). Protein A-Sepharose was purchased from Pharmacia Biotech (Sweden). All the other chemicals used were of analytical grade.
RNase was homogenous on the basis of size since it gave a single band in SDS-PAGE. It was therefore used without purification. Protein concentration of RNase samples was determined spectrophotometrically at 278 nm using the molar extinction coefficient of 9800 M-1*cm-1 [3].
Rabbits were immunized with RNase as follows: healthy rabbits were injected subcutaneously with 500 µg of RNase using Freund's adjuvant. The animals were boosted on day 21 and subsequently bled after a week for monitoring the production of RNase specific antibodies.
The generation of RNase specific antibodies was measured in the sera of RNase immunized rabbits by ELISA. Ninety-six-well microtiter plates were coated overnight with 100 µl of RNase (5 µg/ml) in 0.05 M carbonate-bicarbonate buffer, pH 9.6, at 4°C. After extensive washing with PBS-Tween 20, 100 µl of blocking buffer (3% skimmed milk in PBS-Tween 20) was applied to the wells and the plates incubated at 37°C for 90 min. After removal of blocking buffer, serially diluted test and control sera were added and binding was allowed to proceed at 37°C for 2 h. The microtiter plates were washed and incubated with 100 µl of HRP-conjugated goat anti-rabbit IgG at 37°C for 1 h. After the usual washing steps, the peroxidase reaction was initiated by the addition of 100 µl the substrate tetramethyl benzidine (100 µg/ml)/H2O2 (0.035%), arrested by the addition of 100 µl 8 M H2SO4, and absorbance was measured at 450 nm in an ELISA reader.
The IgG was purified from the sera of rabbits immunized with RNase by affinity chromatography following a published procedure [18]. Briefly, the immune sera were allowed to bind to protein A-Sepharose packed in a small column (5 × 1 cm) at pH 8.9 in the presence of 3 M NaCl. The column was washed thoroughly to remove any unbound protein. Finally, the bound IgGs were eluted with 0.1 M glycine-HCl buffer, pH 3.0, and the eluate immediately neutralized with Tris-HCl buffer. The strong binding between the RNase and IgG necessitated the use of acid pH for elution of IgG. The purity of the IgGs was determined by SDS-PAGE (12% separating and 5% stacking gels) [19]. Two bands were visible in the SDS-PAGE of IgG corresponding to the heavy (50 kD) and light chain (25 kD) of IgG. The concentration of the IgG was determined spectrophotometrically by absorbance measurements at 280 nm (A280 of 1 mg/ml IgG = 1.4).
Fab monomer was prepared as follows: purified IgG (10 mg/ml) was dialyzed against 0.1 M sodium phosphate buffer, pH 7.0. Cysteine-HCl (1 mg/10 mg IgG), EDTA (0.5 mg/10 mg IgG), and papain (1 mg/100 mg IgG) were added to the dialyzed IgG and the mixture incubated for 7 h at 37°C. The digest was frozen to inhibit further digestion. RNase specific Fab was purified on an RNase-Sepharose column. RNase was immobilized on CNBr activated Sepharose-4B following a published procedure [20]. The above digest was dialyzed against 0.1 M Tris-HCl, pH 8.9, containing 3 M NaCl and then allowed to bind to RNase-Sepharose packed in a small column (5 × 1 cm). The column was washed thoroughly to remove unbound protein. Finally, the bound Fab was eluted with 0.1 M glycine-HCl buffer, pH 3.0, and the eluate immediately neutralized with Tris-HCl buffer. The purity of Fab was determined by SDS-PAGE under reducing conditions (12% separating and 5% stacking gels) [19]. A single band with molecular mass of 25 kD was obtained in the case of the Fab. Since Fab consists of two chains, each of molecular mass of 25 kD and joined together by a disulfide bond, it is expected to give a single band of 25 kD in SDS-PAGE under reducing conditions. Hence, from the results it was ascertained that the Fab was pure.
Conformational changes induced in the Fab monomer upon binding RNase were studied by absorption spectroscopic measurements using a Hitachi U-1500 spectrophotometer (Hitachi, Japan). RNase and anti-RNase Fab, in molar ratio of 1 : 1, were allowed to bind to each other to form a complex. For this, RNase (70 µg/ml) and the Fab (250 µg/ml) were taken in 0.1 M sodium phosphate buffer, pH 7.0, and incubated at 37°C for 2 h and then at 4°C overnight. Samples of RNase (70 µg/ml) and the Fab (250 µg/ml) were also prepared separately in the same buffer. Absorption spectra of RNase, Fab, and RN-Fab were measured in the wavelength range of 240-330 nm.
Conformational changes induced in Fab monomer upon binding RNase were also studied by fluorescence spectroscopy using a Shimadzu RF-540 spectrofluorimeter (Shimadzu, Japan) and a cuvette of 1 cm path length. Samples of RNase, Fab, and RN-Fab were prepared as described above, excited at 295 nm, and the normalized emission spectra measured at 320-420 nm with a step width of 2 nm.
RESULTS AND DISCUSSION
RNase was immunogenic and readily elicited the formation of antibodies in rabbits. As determined by ELISA, the anti-RNase antibody titer (i.e., the maximum dilution of the antiserum at which the absorbance at 450 nm remains constant) was high (100,000), and this preparation was used in all the experiments.
Anti-RNase IgG and its Fab obtained by papain cleavage of the IgG were also obtained in pure form after purification on protein A-Sepharose and RNase-Sepharose columns, respectively. Conformational changes in Fab on binding RNase were studied by absorption and fluorescence spectroscopy. In these studies, the spectrum of free Fab has been compared with that of the complex of Fab with RNase after subtracting the spectra of RNase from that of the complex. Therefore, by doing so, only the changes that occur in Fab upon binding its specific antigen have been studied.
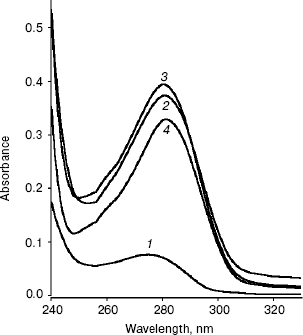
Conformational changes in RNase bound Fab as studied by absorption spectroscopy. The absorption spectra of RNase, Fab, RN-Fab, and (RN-Fab) minus RNase in the wavelength range of 240-330 nm are shown in Fig. 1. The lambdamax of these samples are 276, 280, 279, and 282 nm, respectively. Comparing the absorption spectra of Fab and (RN-Fab) minus RNase, it was observed that there is a red shift (2 nm) in lambdamax and a decrease in absorbance of about 0.05 in case of the latter. This implies that some of the chromophores in the Fab are moving into a hydrophobic environment upon binding the antigen, i.e., internalization of these chromophores is taking place. Hence, perhaps the structure of the Fab becomes more compact on binding the antigen.
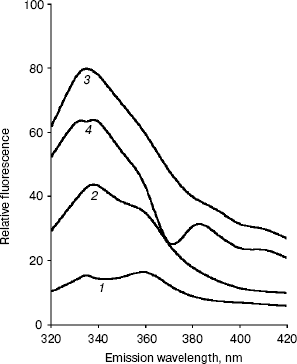
Conformational changes in RNase bound Fab as studied by fluorescence spectroscopy. The fluorescence spectra of RNase, Fab, RN-Fab, and (RN-Fab) minus RNase are shown in Fig. 2. The samples were excited at 295 nm (for tryptophan), and the emission spectra (tryptophan fluorescence) were measured in the wavelength range of 320-420 nm. As expected, the relative fluorescence exhibited by RNase was very low since it lacks tryptophan residues. The emission lambdamax of Fab, RN-Fab, and (RN-Fab) minus RNase are 338, 333, and 333 nm, respectively. Comparing the fluorescence spectra of Fab and (RN-Fab) minus RNase, it was observed that there is a blue shift (5 nm) in emission lambdamax and fluorescence enhancement of about 21 units in case of the latter. This implies that some of the tryptophans in the Fab are moving into a hydrophobic environment, and hence internalization of these tryptophans is taking place. Therefore, it implies that the structure of Fab becomes more compact upon formation of the complex with the antigen.Fig. 1. Absorption spectra of RNase (1), Fab (2), RN-Fab (3), and (RN-Fab) minus RNase (4). The concentration of RNase and Fab were 70 and 250 µg/ml, respectively. The concentrations of these are also the same in the RN-Fab. Samples were prepared in 0.1 M sodium phosphate buffer, pH 7.0.
Since both absorption and fluorescence studies converge to the same interpretation, it seems that the conformational changes that take place in the Fab upon binding the antigen, RNase, are such that they make the structure of the Fab more compact. X-Ray crystallographic analysis of the three-dimensional structure of an anti-lysozyme Fab-lysozyme complex showed that the contact between the antibody combining site and the lysozyme epitope is extensive and involves many residues [21]. The interaction between the two proteins is very tight with no water molecules remaining between the combining site and the epitope. X-Ray studies on the complex of the antigen, hen egg-white lysozyme (HEL) and the FV fragment of the anti-HEL antibody D1.3 also show that there is a concomitant decrease in mobility in the antibody structure upon complex formation [22]. Available data on antibody-antigen complexes show that the binding site in antibody molecules assumes a shape according to the geometry of the antigen [23]. All of the complementary determining regions (CDRs) are important in binding antigen. Amino acid residues at antigen binding sites are expected to vary from one antibody to another. However, in the four antibodies against lysozyme a sizable number (5 to 8) of aromatic amino acid and a few (1 to 3) aspartic acid residues are involved in antigen binding [24-27]. The changes that may occur in the antibody upon binding an antigen consists of combinations of simple side-chain movements, concerted movements of individual CDRs, and displacement of VH relative to VL. The three-dimensional structure of a specific antibody (Fab 17/9) to a peptide immunogen from influenza virus hemagglutinin and two independent crystal complexes of this antibody with bound peptide have been determined by X-ray crystallographic techniques [27]. Comparison of the bound and unbound Fab structures shows that a major rearrangement in the H3 loop accompanies antigen binding. The structures of the free and antigen bound antibodies demonstrate the flexibility of the antibody combining site and provide an example of induced fit as a mechanism for antibody-antigen recognition.Fig. 2. Fluorescence spectra of RNase (1), Fab (2), RN-Fab (3), and (RN-Fab) minus RNase (4). The concentration of RNase and Fab were 70 and 250 µg/ml, respectively. The concentrations of these are also the same in the RN-Fab. Samples were prepared in 0.1 M sodium phosphate buffer, pH 7.0.
The present study suggests that the Fab undergo conformational changes when forming an antigen-Fab complex. Since Fab is a fragment of the antibody molecule, which has the same binding capacity as the antibody molecule, it implies that conformational changes take place in the antibody molecule when the antigen-antibody complex is formed. In the case of RNase and anti-RNase Fab, the structure of the Fab becomes more compact. Therefore, the fit between the two seems to be quite close. The reaction between an antigen and its antibody is highly specific and involves noncovalent interactions [28]. Therefore, a close fit of antigen and antibody molecule is also a prerequisite, since these forces operate over very small distances.
Facilities provided by Aligarh Muslim University are gratefully acknowledged. The work was also supported by the department of Science and Technology, Government of India, under its FIST program, and the University Grants Commission, India, under its special assistance program.
REFERENCES
1.Saleemuddin, M. (1999) Adv. Biochem. Eng.
Biotechnol., 64, 203-206.
2.Ben-Yoseph, Y., Geiger, B., and Arnon, R. (1975)
Immunochemistry, 12, 221-226.
3.Younus, H., Owais, M., Rao, D. N., and Saleemuddin,
M. (2001) Biochim. Biophys. Acta, 1548, 114-120.
4.Khan, S. A., and Iqbal, J. (2000) Biotechnol.
Appl. Biochem., 32, 89-94.
5.Jafri, F., Husain, S., and Saleemuddin, M. (1995)
Biotechnol. Tech., 9, 117-122.
6.Younus, H., Koditz, J., Saleemuddin, M., and
Ulbrich-Hofmann, R. (2002) Biotechnol. Lett., 24,
1821-1826.
7.Turkova, J. (1999) Chromatogr. B. Biomed. Sci.
Appl., 722, 11-31.
8.Gupta, P., Khan, R. H., and Saleemuddin, M. (2003)
Biochim. Biophys. Acta, 1646, 131-135.
9.Vermeer, A. W. P., Norde, W., and Amerongen, A. V.
(2000) Biophys. J., 79, 2150-2154.
10.Pecht, I. (1982) in The Antigens (Sela,
M., ed.) Vol. 6, Academic Press, London, p. 1.
11.Shami, E. Y., Ramjeesingh, M., Rothstein, A., and
Zywulko, M. (1991) Enzyme Microb. Technol., 13,
424-429.
12.Feinstein, R. N., Jaroslow, B. N., Howard, J. B.,
and Faulhaber, J. T. (1971) J. Immunol., 106,
1316-1322.
13.Prisyazhnoy, V. S., Fusek, M., and Alakhov, Y. B.
(1988) Chromatogr. J., 424, 243-253.
14.Reff, M. E., and Heard, C. (2001) Crit. Rev.
Oncol. Hematol., 40, 25-35.
15.Lu, Z., Kopeckova, P., and Kopecek, J. (1999)
Nat. Biotechnol., 17, 1101-1104.
16.Lu, Z., Shiah, J., Kopeckova, P., and Kopecek, J.
(2001) Control. Release, 74, 263-268.
17.Raines, R. T. (1998) Chem. Rev.,
98, 1045-1065.
18.Stults, N. L., Asta, L. M., and Lee, Y. C. (1989)
Analyt. Biochem., 180, 114-119.
19.Laemmli, U. K. (1970) Nature, 227,
680-685.
20.Porath, J., Axen, R., and Ernback, S. (1967)
Nature, 215, 1491-1492.
21.Sheriff, S., Silverton, E. W., Padlan, E. A.,
Cohen, G. H., Smith-Gill, S. J., Finzel, B. C., and Davies, D. R.
(1987) Proc. Natl. Acad. Sci. USA, 84, 8075-8079.
22.Bhat, T. N., Bentley, G. A., Boulot, G., Greene,
M. I., Tello, D., Dall'Acqua, W., Souchon, H., Schwarz, F. P.,
Mariuzza, R. A., and Poljak, R. J. (1994) Proc. Natl. Acad. Sci.
USA, 91, 1089-1093.
23.Ahmad, A., and Salahuddin, A. (1994) Ind. J.
Biochem. Biophys., 31, 156-159.
24.Amit, A. G., Mariuzza, R. A., Phillips, S. E. V.,
and Poljak, R. J. (1986) Science, 233, 747-753.
25.Padlan, E. A., Silverton, E. W., Sheriff, S.,
Cohen, G. H., Smith-Gill, S. J., and Davies, D. R. (1989) Proc.
Natl. Acad. Sci. USA, 86, 5938-5942.
26.Chitarra, V., Alzari, P. M., Bentley, G. A.,
Bhat, T. N., Eisele, J. L., Houduse, A., Lescar, J., Souchon, H., and
Poljak, R. J. (1993) Proc. Natl. Acad. Sci. USA, 90,
7711-7715.
27.Rini, J. M., Schulze-Gahmen, U., and Wilson, I.
A. (1992) Science, 255, 959-965.
28.Van Oss, Carel, J., and Absolom, D. R. (1984) in
Molecular Immunology (Atassi, M. Z., Carel, J., van Oss, and
Absolom, D. R., eds.) Chap. 16, Marcel Dekker, Inc., New York, pp.
337-360.